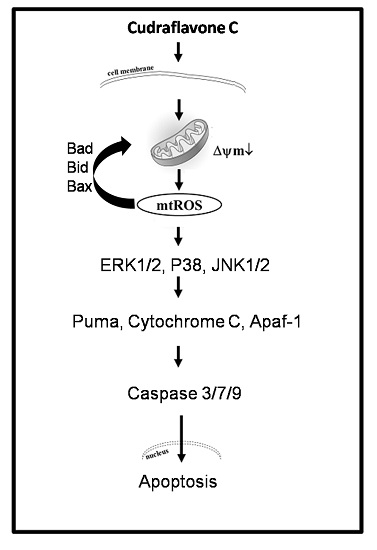
Cudraflavone C Induces Apoptosis of A375.S2 Melanoma Cells through Mitochondrial ROS Production and MAPK Activation


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
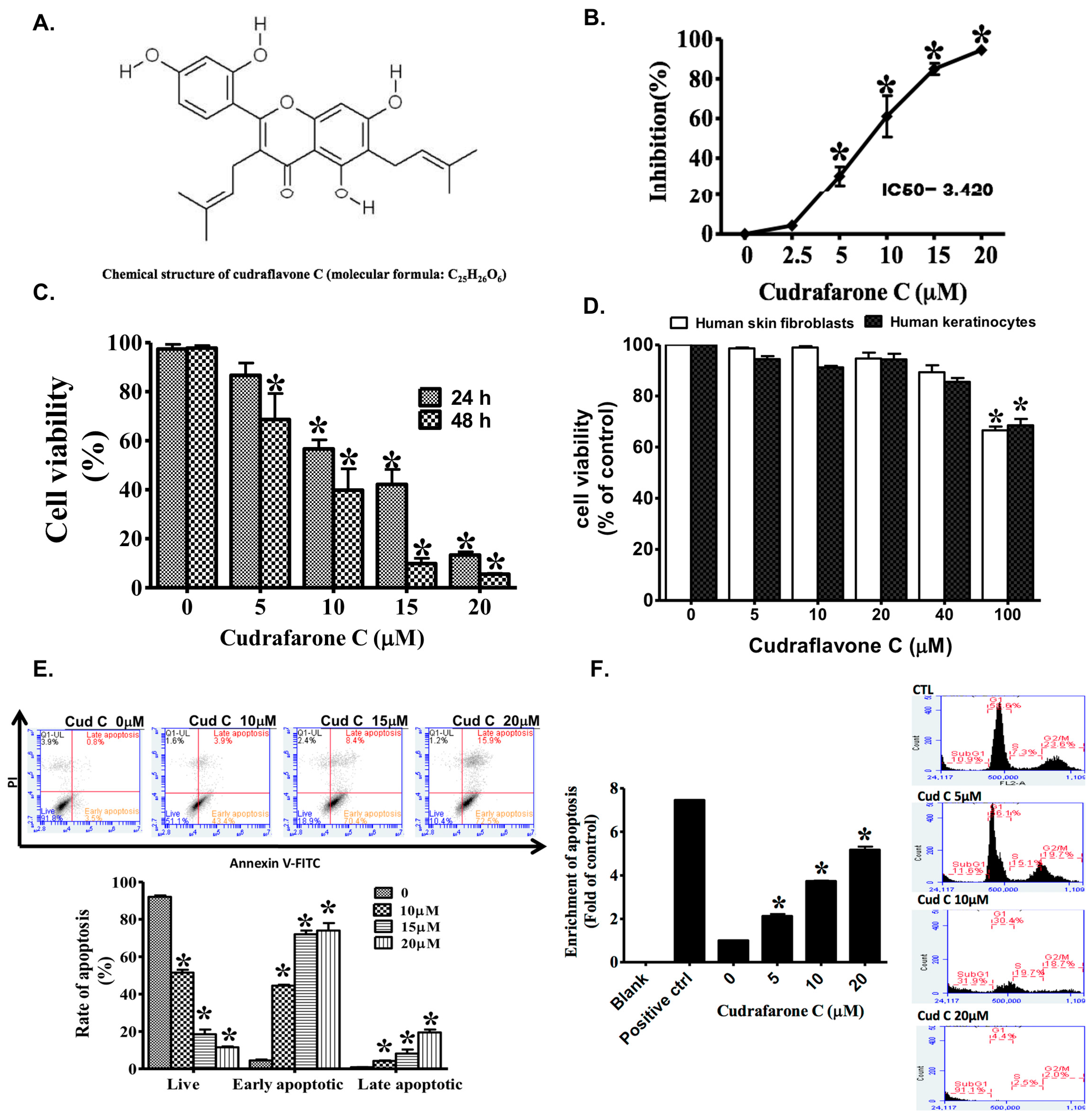
2.1. Cudraflavone C Inhibits Proliferation of A375.S2 Melanoma Cells
2.2. Cudraflavone C Promotes Apoptosis and Cell Cycle Arrest in A375.S2 Melanoma Cells
2.3. Cudraflavone C Promotes Apoptosis in A375.S2 Cells via the Mitochondrial Pathway
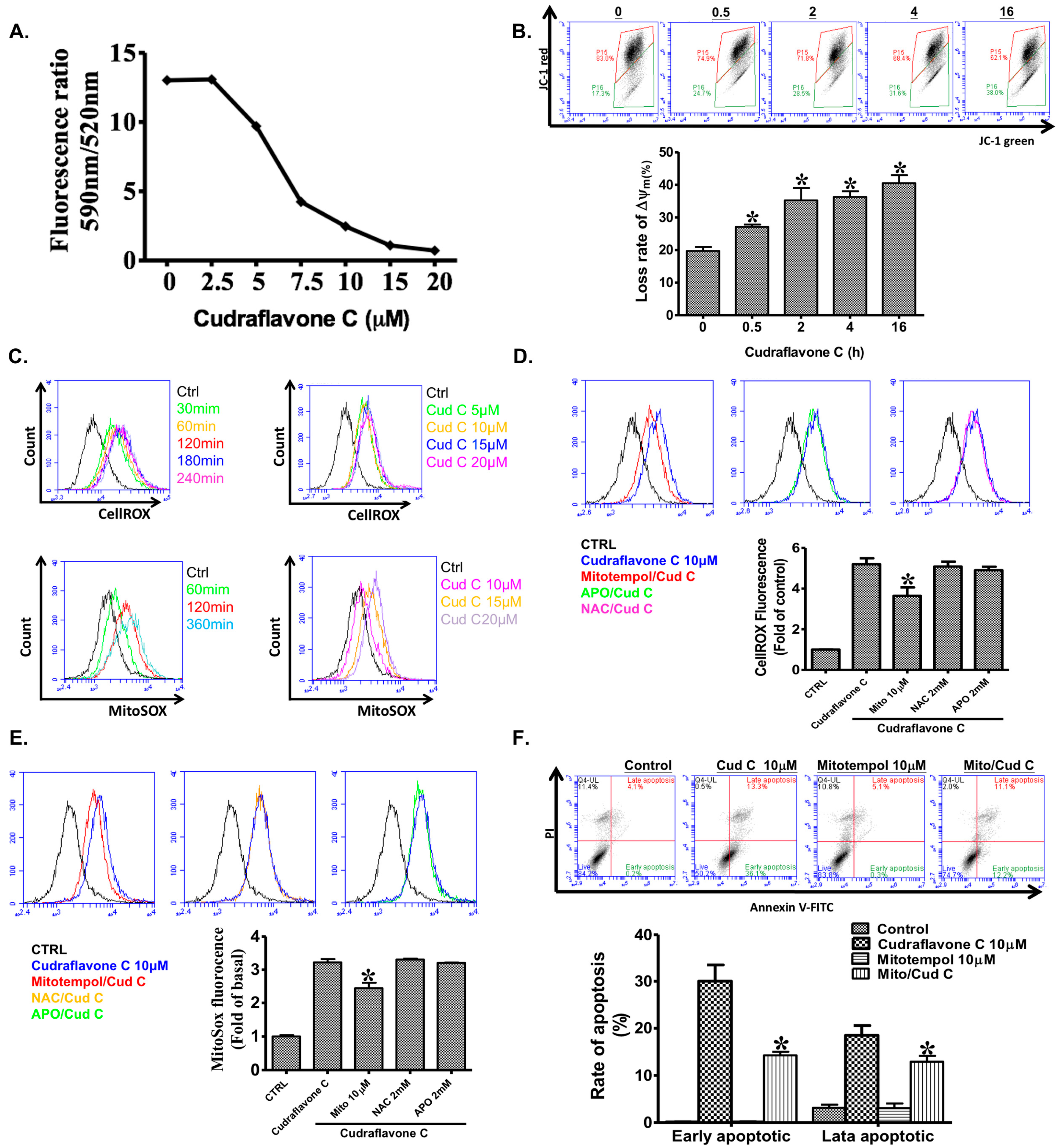
2.4. Cudraflavone C Induces Mitochondrial ROS Production in A375.S2 Cells
2.5. Cudraflavone C Promotes Apoptosis in A375.S2 Cells via Mitochondrial ROS Generation
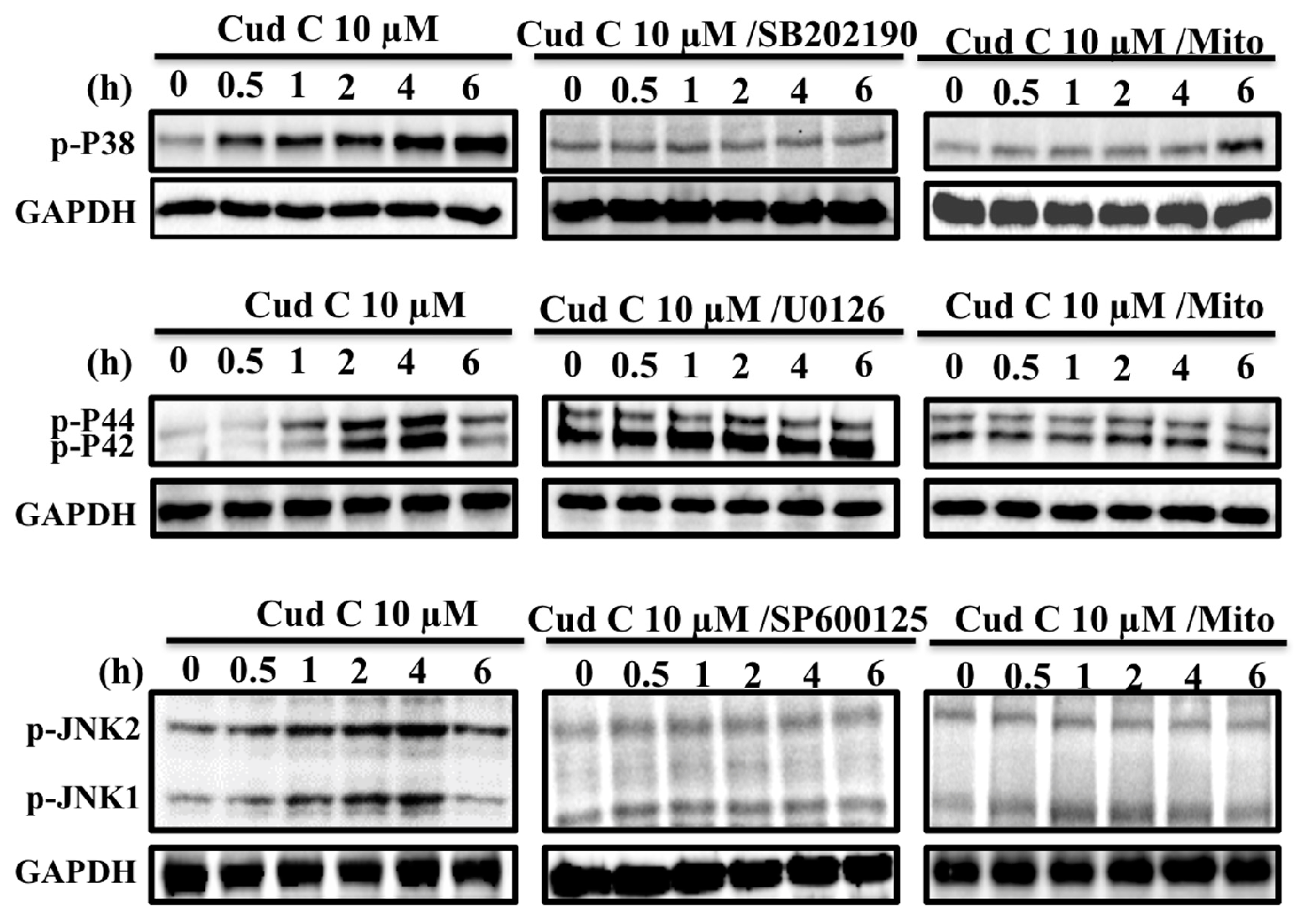
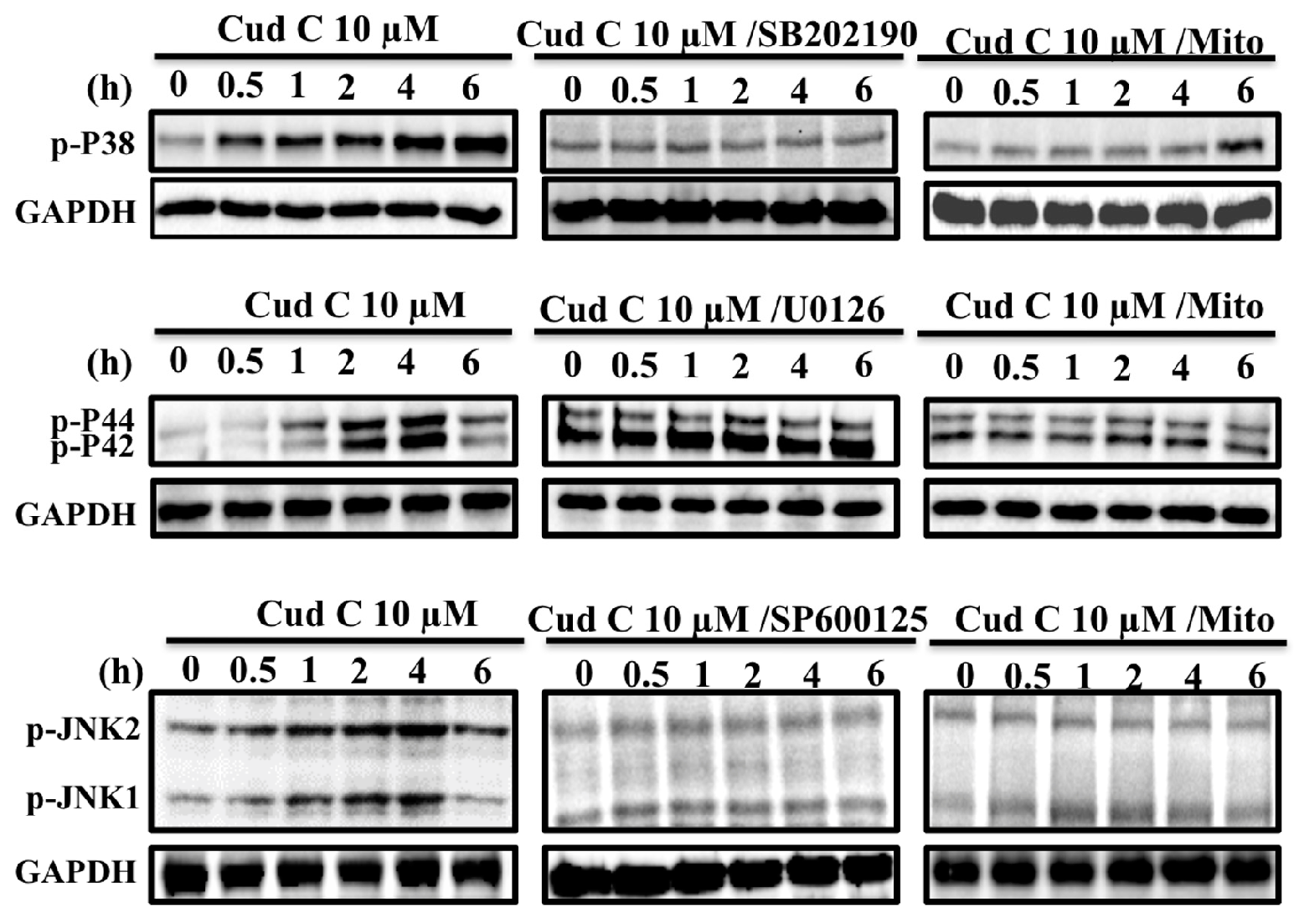
2.6. Cudraflavone C Induces Activation of the MAPK Pathway in A375.S2 Cells via Mitochondrial ROS Generation
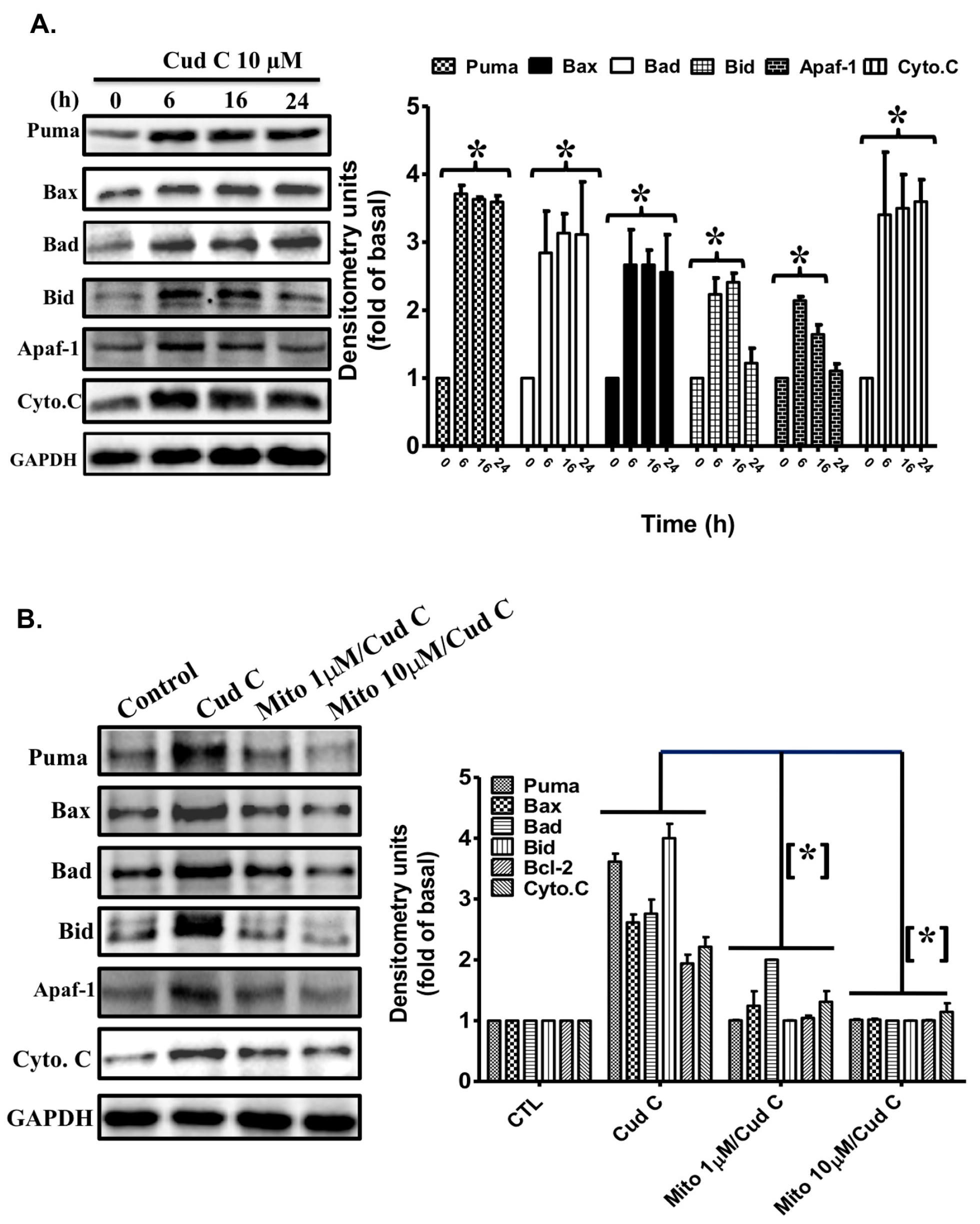
2.7. Cudraflavone C Induces the Expression of Apoptotic Proteins in A375.S2 Cells via Mitochondrial ROS Generation
2.8. Cudraflavone C Induces the Activation of Caspase-3, Caspase-7, and Caspase-9 in A375.S2 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Cell Viability Assay
4.5. Measuring Apoptosis Using Flow Cytometry
4.6. Measuring Apoptosis Using DNA Fragmentation Assay
4.7. Cell Cycle Analysis
4.8. Assessment of Mitochondrial Membrane Potential
4.9. Determination of Mitochondrial Membrane Potential
4.10. Determination of Cellular ROS Production
4.11. Determination of Mitochondrial ROS Production
4.12. Western Blotting
4.13. Determination of Caspase Activities
4.14. Statistical Analysis
Acknowledgments
Author Contributions
Conflict of Interest
References
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Jin, Y.J.; Lin, C.C.; Lu, T.M.; Li, J.H.; Chen, I.S.; Kuo, Y.H.; Ko, H.H. Chemical constituents derived from Artocarpus xanthocarpus as inhibitors of melanin biosynthesis. Phytochemistry 2015, 117, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yan, G.R.; Pan, S.L.; Wang, H.Y.; Hou, A.J. New isoprenylated 2-arylbenzofurans and pancreatic lipase inhibitory constituents from Artocarpus nitidus. Chem. Biodivers. 2009, 6, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Sritularak, B.; Tantrakarnsakul, K.; Lipipun, V.; Likhitwitayawuid, K. Flavonoids with anti-HSV activity from the root bark of Artocarpus lakoocha. Nat. Prod. Commun. 2013, 8, 1079–1080. [Google Scholar] [PubMed]
- Arung, E.T.; Yoshikawa, K.; Shimizu, K.; Kondo, R. Isoprenoid-substituted flavonoids from wood of Artocarpus heterophyllus on B16 melanoma cells: Cytotoxicity and structural criteria. Fitoterapia 2010, 81, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.P.; Qiao, X.; Pan, S.; Shen, H.; Zhu, G.F.; Hou, A.J. New isoprenylated flavonoids and cytotoxic constituents from Artocarpus tonkinensis. J. Asian Nat. Prod. Res. 2010, 12, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Soo, H.C.; Chung, F.F.; Lim, K.H.; Yap, V.A.; Bradshaw, T.D.; Hii, L.W.; Tan, S.H.; See, S.J.; Tan, Y.F.; Leong, C.O.; Mai, C.W. Cudraflavone C Induces Tumor-Specific Apoptosis in Colorectal Cancer Cells through Inhibition of the Phosphoinositide 3-Kinase (PI3K)-AKT Pathway. PLoS ONE 2017, 12, e0170551. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Day, B.J. Antioxidant therapeutics: Pandora’s box. Free Radic. Biol. Med. 2014, 66, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Devasagayam, T.P.; Tilak, J.C.; Boloor, K.K.; Sane, K.S.; Ghaskadbi, S.S.; Lele, R.D. Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Phys. India 2004, 52, 794–804. [Google Scholar]
- Farmer, E.E.; Mueller, M.J. ROS-mediated lipid peroxidation and RES-activated signaling. Annu. Rev. Plant. Biol. 2013, 64, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Zhao, Z.; Goto, S.; Koltai, E. Age-associated neurodegeneration and oxidative damage to lipids, proteins and DNA. Mol. Aspects Med. 2011, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, S.; Ahn, D.U. Protein oxidation: Basic principles and implications for meat quality. Crit.Rev. Food Sci. Nutr. 2013, 53, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Hu, Y.; Lu, W.; Pelicano, H.; Huang, P. Novel action of paclitaxel against cancer cells: Bystander effect mediated by reactive oxygen species. Cancer Res. 2007, 67, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Colin, D.J.; Limagne, E.; Ragot, K.; Lizard, G.; Ghiringhelli, F.; Solary, E.; Chauffert, B.; Latruffe, N.; Delmas, D. The role of reactive oxygen species and subsequent DNA-damage response in the emergence of resistance towards resveratrol in colon cancer models. Cell Death Dis. 2014, 5, e1533. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xu, B.; Beevers, C.S.; Odaka, Y.; Chen, L.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; Huang, S. Curcumin inhibits protein phosphatases 2A and 5, leading to activation of mitogen-activated protein kinases and death in tumor cells. Carcinogenesis 2012, 33, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Harries, M.; Malvehy, J.; Lebbe, C.; Heron, L.; Amelio, J.; Szabo, Z.; Schadendorf, D. Treatment patterns of advanced malignant melanoma (stage III-IV)—A review of current standards in Europe. Eur. J. Cancer 2016, 60, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Svedman, F.C.; Pillas, D.; Taylor, A.; Kaur, M.; Linder, R.; Hansson, J. Stage-specific survival and recurrence in patients with cutaneous malignant melanoma in Europe—A systematic review of the literature. Clin. Epidemiol. 2016, 8, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Adhami, V.M.; Syed, D.N.; Khan, N.; Mukhtar, H. Dietary flavonoid fisetin: A novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem. Pharmacol. 2012, 84, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.E.; Husband, A.J. Flavonoid compounds in the prevention and treatment of prostate cancer. Methods Mol. Med. 2003, 81, 377–394. [Google Scholar] [PubMed]
- Kocic, B.; Kitic, D.; Brankovic, S. Dietary flavonoid intake and colorectal cancer risk: Evidence from human population studies. J. BUON 2013, 18, 34–43. [Google Scholar] [PubMed]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.M. Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr. Cancer Drug. Targets 2008, 8, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Adhami, V.M.; Khan, N.; Khan, M.I.; Mukhtar, H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin. Cancer Biol. 2016, 40–41, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Czemplik, M.; Mierziak, J.; Szopa, J.; Kulma, A. Flavonoid C-glucosides Derived from Flax Straw Extracts Reduce Human Breast Cancer Cell Growth In vitro and Induce Apoptosis. Front. Pharmacol. 2016, 7, 282. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Somasagara, R.R.; Hegde, M.; Nishana, M.; Tadi, S.K.; Srivastava, M.; Choudhary, B.; Raghavan, S.C. Quercetin, a Natural Flavonoid Interacts with DNA, Arrests Cell Cycle and Causes Tumor Regression by Activating Mitochondrial Pathway of Apoptosis. Sci. Rep. 2016, 6, 24049. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hou, M.F.; Tsai, E.M.; Kuo, P.L. Tricetin, a dietary flavonoid, induces apoptosis through the reactive oxygen species/c-Jun NH2-terminal kinase pathway in human liver cancer cells. J. Agric. Food Chem. 2010, 58, 12547–12556. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.; Zhou, D.; Xiong, C.; Cai, Y.; Ruan, J. A novel non-aromatic B-ring flavonoid: Isolation, structure elucidation and its induction of apoptosis in human colon HT-29 tumor cell via the reactive oxygen species-mitochondrial dysfunction and MAPK activation. Food Chem. Toxicol. 2011, 49, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xiao, Y.; Xiong, C.; Wei, A.; Ruan, J. Apoptosis induced by a new flavonoid in human hepatoma HepG2 cells involves reactive oxygen species-mediated mitochondrial dysfunction and MAPK activation. Eur. J. Pharmacol. 2011, 654, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.C.; Lin, C.L.; Cheng, H.M.; Chen, G.S.; Lee, C.W.; Yen, F.L. Artocarpin Induces Apoptosis in Human Cutaneous Squamous Cell Carcinoma HSC-1 Cells and Its Cytotoxic Activity Is Dependent on Protein-Nutrient Concentration. Evid. Based Complement. Altern. Med. 2015, 2015, 236159. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.S.; Hou, A.J.; Zhu, G.F. Isoprenylated xanthones and flavonoids from Cudrania tricuspidata. Chem. Biodivers. 2005, 2, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Han, A.R.; Kang, Y.J.; Windono, T.; Lee, S.K.; Seo, E.K. Prenylated flavonoids from the heartwood of Artocarpus communis with inhibitory activity on lipopolysaccharide-induced nitric oxide production. J. Nat. Prod. 2006, 69, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K.; Sritularak, B.; De-Eknamkul, W. Tyrosinase inhibitors from Artocarpus gomezianus. Planta Med. 2000, 66, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Arung, E.T.; Shimizu, K.; Kondo, R. Artocarpus plants as a potential source of skin whitening agents. Nat.Prod. Commun. 2011, 6, 1397–1402. [Google Scholar] [PubMed]
- Arung, E.T.; Shimizu, K.; Kondo, R. Inhibitory effect of isoprenoid-substituted flavonoids isolated from Artocarpus heterophyllus on melanin biosynthesis. Planta Med. 2006, 72, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Dej-Adisai, S.; Meechai, I.; Puripattanavong, J.; Kummee, S. Antityrosinase and antimicrobial activities from Thai medicinal plants. Arch. Pharm. Res. 2014, 37, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.H.; Zhao, T.; Yan, G.R.; Yang, H.X.; Wang, H.Y.; Hou, A.J. New isoprenylated flavones and stilbene derivative from Artocarpus hypargyreus. Chem. Biodivers. 2012, 9, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chen, J.Y.; Kuo, C.H.; Pai, P.Y.; Ho, T.J.; Chen, T.S.; Tsai, F.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. Mitochondrial ROS-induced ERK1/2 Activation and HSF2-mediated AT1 R Upregulation Are Required for Doxorubicin-induced Cardiotoxicity. J.Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Martinou, J.C. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000, 10, 369–377. [Google Scholar] [CrossRef]
- Adrain, C.; Martin, S.J. The mitochondrial apoptosome: A killer unleashed by the cytochrome seas. Trends Biochem. Sci. 2001, 26, 390–397. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-W.; Yen, F.-L.; Ko, H.-H.; Li, S.-Y.; Chiang, Y.-C.; Lee, M.-H.; Tsai, M.-H.; Hsu, L.-F. Cudraflavone C Induces Apoptosis of A375.S2 Melanoma Cells through Mitochondrial ROS Production and MAPK Activation. Int. J. Mol. Sci. 2017, 18, 1508. https://doi.org/10.3390/ijms18071508
Lee C-W, Yen F-L, Ko H-H, Li S-Y, Chiang Y-C, Lee M-H, Tsai M-H, Hsu L-F. Cudraflavone C Induces Apoptosis of A375.S2 Melanoma Cells through Mitochondrial ROS Production and MAPK Activation. International Journal of Molecular Sciences. 2017; 18(7):1508. https://doi.org/10.3390/ijms18071508
Chicago/Turabian StyleLee, Chiang-Wen, Feng-Lin Yen, Horng-Huey Ko, Shu-Yu Li, Yao-Chang Chiang, Ming-Hsueh Lee, Ming-Horng Tsai, and Lee-Fen Hsu. 2017. "Cudraflavone C Induces Apoptosis of A375.S2 Melanoma Cells through Mitochondrial ROS Production and MAPK Activation" International Journal of Molecular Sciences 18, no. 7: 1508. https://doi.org/10.3390/ijms18071508
APA StyleLee, C.-W., Yen, F.-L., Ko, H.-H., Li, S.-Y., Chiang, Y.-C., Lee, M.-H., Tsai, M.-H., & Hsu, L.-F. (2017). Cudraflavone C Induces Apoptosis of A375.S2 Melanoma Cells through Mitochondrial ROS Production and MAPK Activation. International Journal of Molecular Sciences, 18(7), 1508. https://doi.org/10.3390/ijms18071508