The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology




Abstract
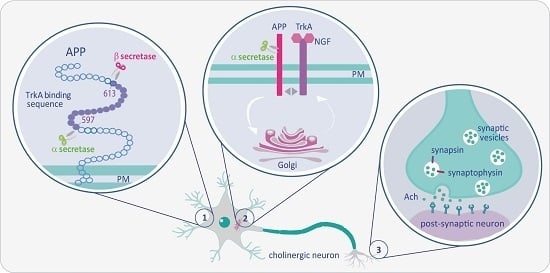
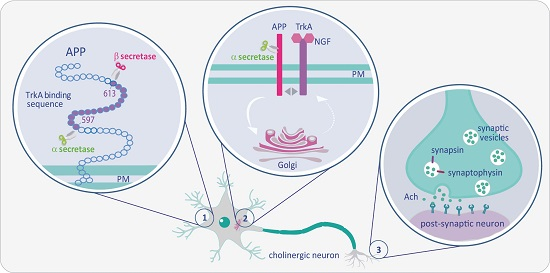
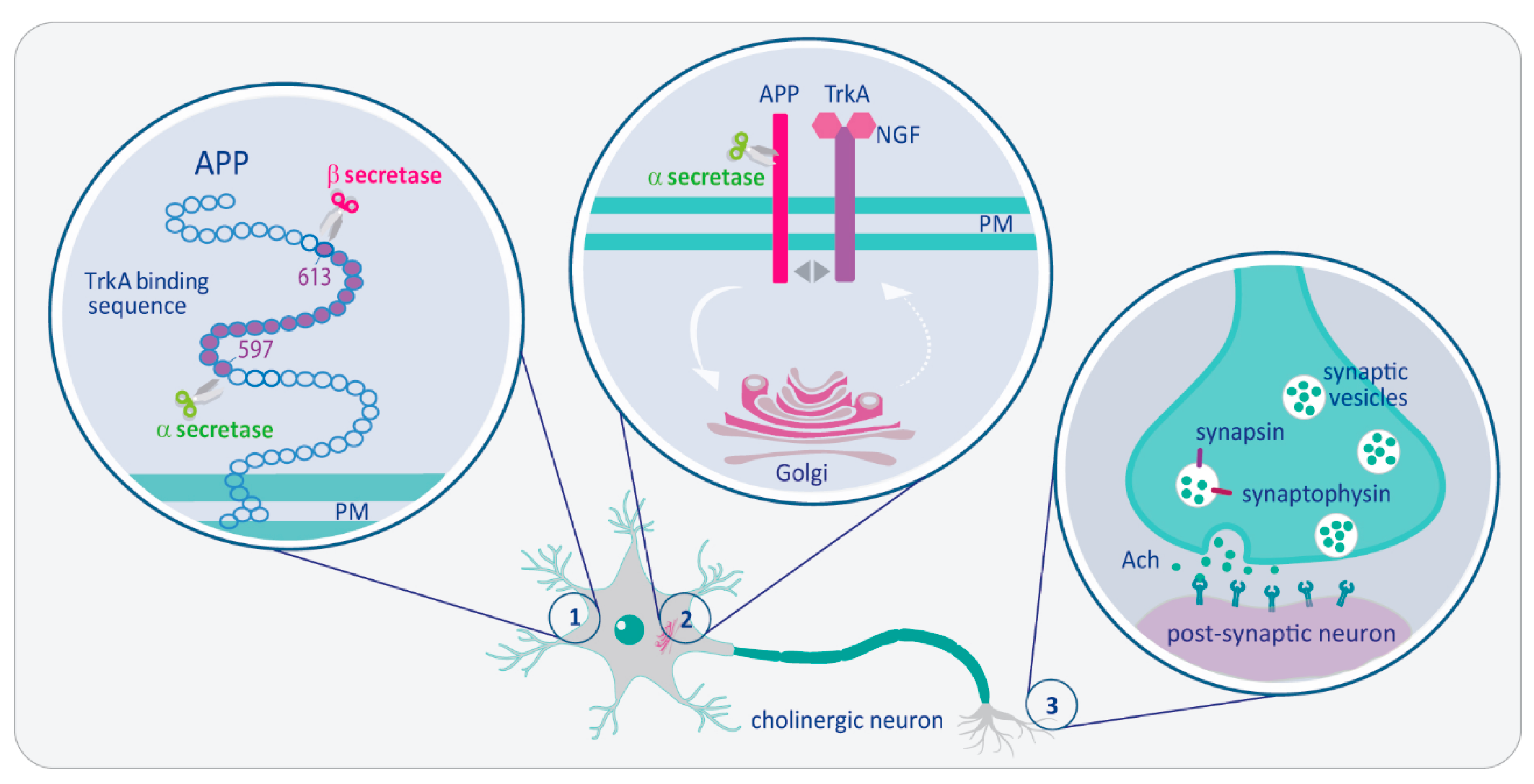
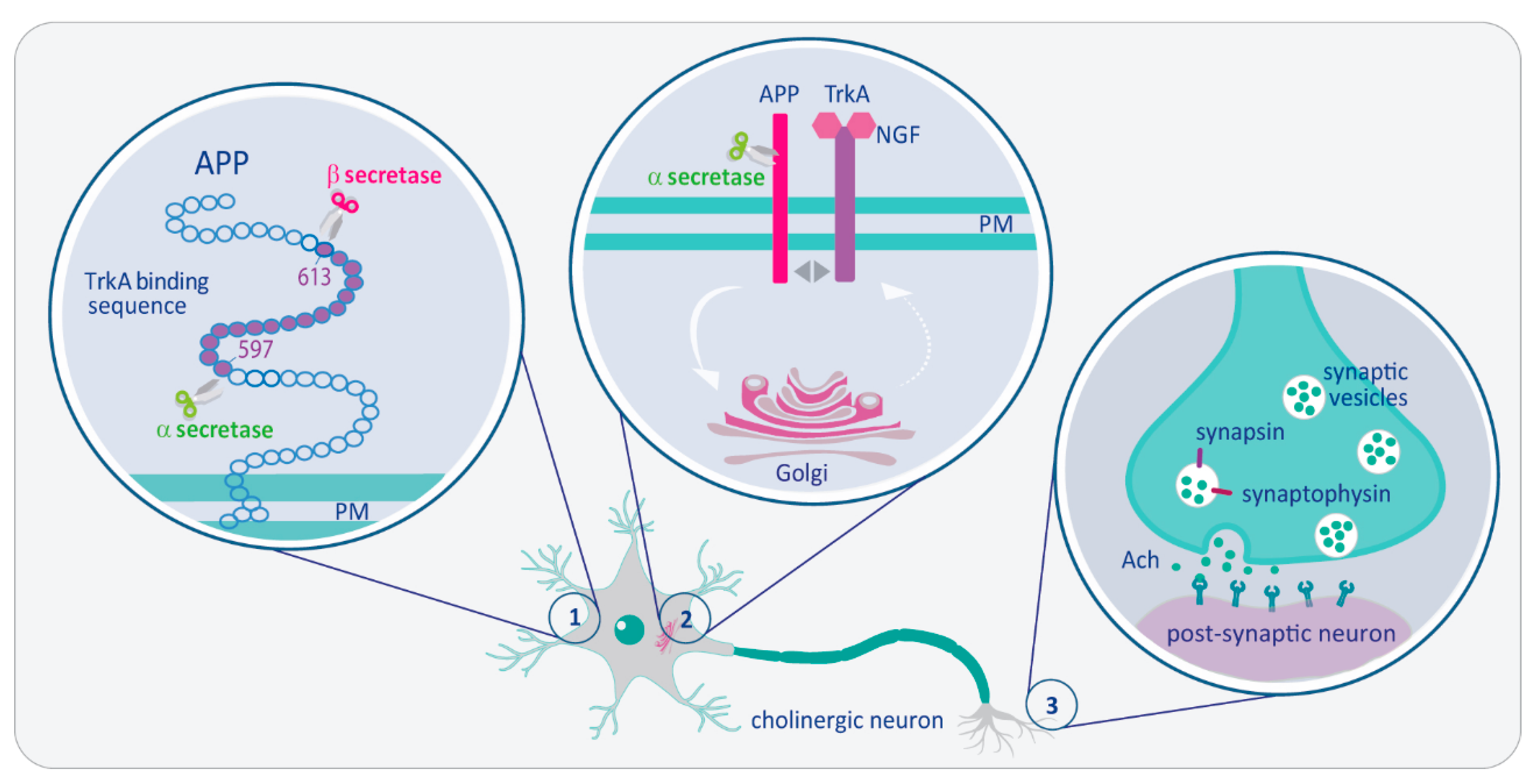
:
{kind=link}
{kind=link}
1. Introduction
2. The Close Association of APP and TrkA
3. The NGF/TrkA-ShcC Pathway Modulates APP Trafficking/Processing by Controlling Its Phosphorylation at the 667VTPEE Domain
4. Impairment of NGF/TrkA Signaling Triggers an Early Activation of “a Dying-Back” Process of Degeneration of Cholinergic Neurons
5. Impaired NGF Signaling in AD Synaptic Failure: Crucial Role of N-Terminal Cleavage of Tau Protein
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| NGF | Nerve growth factor |
| TrkA | High-affinity tropomyosin receptor kinase A |
| p75NTR | Neurotrophin receptor p75 |
| BFCN | Basal forebrain cholinergic neurons |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| MCI | Mild cognitive impairment |
| BiFC | Bimolecular fluorescence complementation |
| PLA | Proximity ligation assay |
| BACE1 | β-Secretase 1 |
| Aβ peptide | Amyloid β peptide |
| JNK | Ser/Thr c-Jun N-terminal kinase |
| ShcC | Sh2 containing sequence C |
| PI3K | Phosphoinositol-3 kinase |
| TGN | Trans-Golgi network |
| ER | Endoplasmic reticulum |
References
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Brückner, M.K.; Bigl, V.; Marcova, L. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. II. Ageing, Korsakoff’s disease, Parkinson’s disease, and Alzheimer’s disease. J. Comp. Neurol. 1995, 351, 189–222. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, G.; Mietelska-Porowska, A.; Mazurkiewicz, M. The cholinergic system, nerve growth factor and the cytoskeleton. Behav. Brain Res. 2011, 221, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Binder, L.; Counts, S.E.; Dekosky, S.T.; Detoledo-Morrell, L.; Ginsberg, S.D.; Ikonomovic, M.D.; Perez, S.E.; Scheff, S.W. Mild cognitive impairment: Pathology and mechanisms. Acta Neuropathol. 2012, 123, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Hempstead, B.L.; Martin-Zanca, D.; Kaplan, D.R.; Parada, L.F.; Chao, M.V. High-affinity NGF binding requires coexpression of the Trk proto-oncogene and the low-affinity NGF receptor. Nature 1991, 350, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Cattaneo, A.; Calissano, P. Nerve growth factor and Alzheimer’s disease: New facts for an old hypothesis. Mol. Neurobiol. 2012, 46, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.L.; Campenot, R.B. Rapid retrograde tyrosine phosphorylation of trkA and other proteins in rat sympathetic neurons in compartmented cultures. J. Cell Biol. 1997, 138, 411–421. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, B.L. Retrograde support of neuronal survival without retrograde transport of nerve growth factor. Science 2002, 295, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.D.; Kaplan, D.R. Trk makes the retrograde. Science 2002, 295, 1471–1473. [Google Scholar] [CrossRef] [PubMed]
- Neet, K.E.; Campenot, R.B. Receptor binding, internalization, and retrograde transport of neurotrophic factors. Cell. Mol. Life Sci. 2001, 58, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Howe, C.L.; Mobley, W.C. Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 2001, 24, 1217–1281. [Google Scholar] [CrossRef] [PubMed]
- Watson, F.L.; Heerssen, H.M.; Moheban, D.B.; Lin, M.Z.; Sauvageot, C.M.; Bhattacharyya, A.; Pomeroy, S.L.; Segal, R.A. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci. 1999, 19, 7889–7900. [Google Scholar] [PubMed]
- Howe, C.L.; Valletta, J.S.; Rusnak, A.S.; Mobley, W.C. NGF signaling from clathrin-coated vesicles: Evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron 2001, 32, 801–814. [Google Scholar] [CrossRef]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Reichardt, L.F.; Mobley, W.C. Going the distance, or not, with neurotrophin signals. Cell 2004, 118, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Lee, R.; Teng, K.K.; Jansen, P. Sortilin is essential for proNGF-induced neuronal cell death. Nature 2004, 427, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; He, B.; Nadeem, M.; Perez, S.E.; Counts, S.E.; Leurgans, S.; Fritz, J.; Lah, J.; Ginsberg, S.D.; Wuu, J.; et al. Hippocampal proNGF signaling pathways and β-amyloid levels in mild cognitive impairment and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Volosin, M.; Song, W.; Almeida, R.D.; Kaplan, D.R.; Hempstead, B.L.; Friedman, W.J. Interaction of survival and death signaling in basal forebrain neurons: Roles of neurotrophins and proneurotrophins. J. Neurosci. 2006, 26, 7756–7766. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V.; Bothwell, M. Neurotrophins: To cleave or not to cleave. Neuron 2002, 33, 9–12. [Google Scholar] [CrossRef]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M.D. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci. 2001, 18, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.; Howe, W.M.; Welchko, R.M.; Naughton, S.X.; D’Amore, D.E.; Han, D.H.; Deo, M.; Turner, D.L.; Sarter, M. Diminished TrkA receptor signaling reveals cholinergic-attentional vulnerability of aging. Eur. J. Neurosci. 2013, 37, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Yegla, B.; Parikh, V. Effects of sustained proNGF blockade on attentional capacities in aged rats with compromised cholinergic system. Neuroscience 2014, 261, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Lima Garcez, M.; Ioppi Zugno, A. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331. [Google Scholar] [CrossRef] [PubMed]
- Rossner, S.; Ueberham, U.; Schliebs, R.; Perez-Polo, J.R.; Bigl, V. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog. Neurobiol. 1998, 56, 541–569. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Che, S.; Wuu, J.; Counts, S.E.; Mufson, E.J. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Nadeem, M.; Wuu, J.; Ginsberg, S.D.; Saragovi, H.U.; Mufson, E.J. Reduction of cortical TrkA but not p75NTR protein in early-stage Alzheimer’s disease. Ann. Neurol. 2004, 56, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Mufson, E.J. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J. Neuropathol. Exp. Neurol. 2005, 64, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.J.; Eriksdotter-Jonhagen, M.; Granholm, A.-C. Nerve growth factor in treatment and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2006, 80, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Price, D.L.; Koliatsos, V.E.; Sisodia, S.S.; Koo, E.H.; Martin, L.J.; Walker, L.C.; Appelgate, M.D.; Cork, L.C. Amyloid-related proteins and nerve growth factor in Alzheimer’s disease and animal models. Clin. Neuropharmacol. 1991, 14, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Crutcher, K.A. Nerve growth factor and Alzheimer’s disease. Rev. Neurosci. 1994, 5, 179–211. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.B.; Siegel, G.J. Effect of PPF and ALCAR on the induction of NGF- and p75-mRNA and on APP processing in Tg2576 brain. Neurochem. Int. 2003, 43, 225–233. [Google Scholar] [CrossRef]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; U, H.S.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 2005, 11, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H.; Yang, J.H.; Barba, D.; U, H.-S.; Bakay, R.A.E.; Pay, M.M.; Masliah, E.; Conner, J.M.; Kobalka, P.; Roy, S.; et al. Nerve growth factor gene therapy: Activation of neuronal responses in Alzheimer disease. JAMA Neurol. 2015, 72, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Huh, C.Y.L.; Danik, M.; Manseau, F.; Trudeau, L.-E.; Williams, S. Chronic exposure to nerve growth factor increases acetylcholine and glutamate release from cholinergic neurons of the rat medial septum and diagonal band of broca via mechanisms mediated by p75NTR. J. Neurosci. 2008, 28, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M. The cholinergic lesion of Alzheimer’s disease: Pivotal factor or side show? Learn. Mem. 2004, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and a production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Di Luzio, A.; Meli, G.; D’Aguanno, S.; Severini, C.; Ciotti, M.T.; Cattaneo, A.; Calissano, P. Activation of the amyloidogenic route by NGF deprivation induces apoptotic death in PC12 cells. J. Alzheimer’s Dis. 2008, 13, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Calissano, P.; Amadoro, G.; Matrone, C.; Ciafrè, S.; Marolda, R.; Corsetti, V.; Ciotti, M.T.; Mercanti, D.; di Luzio, A.; Severini, C.; et al. Does the term “trophic” actually mean anti-amyloidogenic? The case of NGF. Cell Death Differ. 2010, 17, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Luvisetto, S.; la Rosa, L.R.; Tamayev, R.; Pignataro, A.; Canu, N.; Yang, L.; Barbagallo, A.P.M.; Biundo, F.; Lombino, F.; et al. Tyr682 in the Aβ-precursor protein intracellular domain regulates synaptic connectivity, cholinergic function, and cognitive performance. Aging Cell 2012, 11, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Barbagallo, A.P.M.; la Rosa, L.R.; Florenzano, F.; Ciotti, M.T.; Mercanti, D.; Chao, M.V.; Calissano, P.; D’Adamio, L. APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. J. Neurosci. 2011, 31, 11756–11761. [Google Scholar] [CrossRef] [PubMed]
- Triaca, V.; Sposato, V.; Bolasco, G.; Ciotti, M.T.; Pelicci, P.; Bruni, A.C.; Cupidi, C.; Maletta, R.; Feligioni, M.; Nisticò, R.; et al. NGF controls APP cleavage by downregulating APP phosphorylation at Thr668: Relevance for Alzheimer’s disease. Aging Cell 2016, 15, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Canu, N.; Pagano, I.; la Rosa, L.R.; Pellegrino, M.; Ciotti, M.T.; Mercanti, D.; Moretti, F.; Sposato, V.; Triaca, V.; Petrella, C.; et al. Association of TrkA and APP is promoted by NGF and reduced by cell death-promoting agents. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Zhang, B.; Lee, V.M.Y.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Delcroix, J.D.; Swaab, D.F. Alzheimer’s disease and NGF signaling. J. Neural. Transm. 2004, 111, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Boncristiano, S.; Calhoun, M.E.; Kelly, P.H.; Pfeifer, M.; Bondolfi, L.; Stalder, M.; Phinney, A.L.; Abramowski, D.; Sturchler-Pierrat, C.; Enz, A.; et al. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 2002, 22, 3234–3243. [Google Scholar] [PubMed]
- Kim, S.; Sato, Y.; Mohan, P.S.; Peterhoff, C.; Pensalfini, A.; Rigoglioso, A.; Jiang, Y.; Nixon, R.A. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 2016, 21, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Fahnestock, M. ProNGF, but not NGF, switches from neurotrophic to apoptotic activity in response to reductions in TrKA receptor levels. Int. J. Mol. Sci. 2017, 18, 599. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Cuello, A.C. The NGF metabolic pathway in the CNS and its dysregulation in Down syndrome and Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Signal transduction by the neurotrophin receptors. Curr. Opin. Cell Biol. 1997, 9, 213–221. [Google Scholar] [CrossRef]
- Song, W.; Volosin, M.; Cragnolini, A.B.; Hempstead, B.L.; Friedman, W.J. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J. Neurosci. 2010, 30, 15608–15615. [Google Scholar] [CrossRef] [PubMed]
- Calissano, P.; Matrone, C.; Amadoro, G. Nerve growth factor as a paradigm of neurotrophins related to Alzheimer’s disease. Dev. Neurobiol. 2010, 70, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Triaca, V.; Calissano, P. Impairment of the nerve growth factor pathway driving amyloid accumulation in cholinergic neurons: The incipit of the Alzheimer’s disease story? Neural Regen. Res. 2016, 11, 1553–1556. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Nakaya, T. Regulation of amyloid β-protein precursor by phosphorylation and protein interactions. J. Biol. Chem. 2008, 283, 29633–29637. [Google Scholar] [CrossRef] [PubMed]
- Tamayev, R.; Zhou, D.; D’Adamio, L. The interactome of the amyloid β precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 2009, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, J.; Rabizadeh, S.; Banwait, S.; Mehlen, P.; Bredesen, D.E. Selective vulnerability in Alzheimer’s disease: Amyloid precursor protein and p75NTR interaction. Ann. Neurol. 2009, 65, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Chen, Y.; Liu, Y.; Zhao, Y.; Liao, F.F.; Xu, H. APP regulates NGF receptor trafficking and NGF-mediated neuronal differentiation and survival. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.G.; Zhang, Q.; Liu, X.J.; Liu, X.; Jiao, L.; Zhu, W.; Zhang, Z.H.; Zhao, X.L.; He, C. Interaction of Mint2 with TrkA is involved in regulation of nerve growth factor-induced neurite outgrowth. J. Biol. Chem. 2009, 284, 12469–12479. [Google Scholar] [CrossRef] [PubMed]
- Biederer, T.; Cao, X.; Südhof, T.C.; Liu, X. Regulation of APP-dependent transcription complexes by Mint/X11s: Differential functions of Mint isoforms. J. Neurosci. 2002, 22, 7340–7351. [Google Scholar] [PubMed]
- Hill, K.; Li, Y.; Bennett, M.; McKay, M.; Zhu, X.; Shern, J.; Torre, E.; Lah, J.J.; Levey, A.I.; Kahn, R.A. Munc18 interacting proteins: ADP-ribosylation factor-dependent coat proteins that regulate the traffic of β-Alzheimer’s precursor protein. J. Biol. Chem. 2003, 278, 36032–36040. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Madsen, P.; Christensen, E.I.; Nykjær, A.; Gliemann, J.; Kasper, D.; Pohlmann, R.; Petersen, C.M. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001, 20, 2180–2190. [Google Scholar] [CrossRef] [PubMed]
- Vaegter, C.B.; Jansen, P.; Fjorback, A.W.; Glerup, S.; Skeldal, S.; Kjolby, M.; Richner, M.; Erdmann, B.; Nyengaard, J.R.; Tessarollo, L.; et al. Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat. Neurosci. 2011, 14, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Gustafsen, C.; Glerup, S.; Pallesen, L.T.; Olsen, D.; Andersen, O.M.; Nykjaer, A.; Madsen, P.; Petersen, C.M. Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. J. Neurosci. 2013, 33, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Clewes, O.; Fahey, M.S.; Tyler, S.J.; Watson, J.J.; Seok, H.; Catania, C.; Cho, K.; Dawbarn, D.; Allen, S.J. Human ProNGF: Biological effects and binding profiles at TrkA, p75NTR and sortilin. J. Neurochem. 2008, 107, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Al-Shawi, R.; Hafner, A.; Olson, J.; Chun, S.; Raza, S.; Thrasivoulou, C.; Lovestone, S.; Killick, R.; Simons, P.; Cowen, T. Neurotoxic and neurotrophic roles of proNGF and the receptor sortilin in the adult and ageing nervous system. Eur. J. Neurosci. 2008, 27, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Wuu, J.; Counts, S.E.; Nykjaer, A. Preservation of cortical sortilin protein levels in MCI and Alzheimer’s disease. Neurosci. Lett. 2010, 471, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Coulson, E.J.; Nykjaer, A. Up-regulation of sortilin mediated by amyloid-β and p75NTR: Safety lies in the middle course. J. Neurochem. 2013, 127, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kao, S.C.; Lemere, C.A.; Xia, W.; Tseng, H.C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.H. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-A.; Kim, H.-S.; Ha, T.-Y.; Ha, J.-W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.-P.; Park, C.-H.; Kim, S.; Baik, T.-K.; et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell. Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef] [PubMed]
- Shin, R.-W.; Ogino, K.; Shimabuku, A.; Taki, T.; Nakashima, H.; Ishihara, T.; Kitamoto, T. Amyloid precursor protein cytoplasmic domain with phospho-Thr668 accumulates in Alzheimer’s disease and its transgenic models: A role to mediate interaction of Aβ and tau. Acta Neuropathol. 2007, 113, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Ginsberg, S.D. Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer’s disease. Neurochem. Res. 2002, 27, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Lombino, F.; Biundo, F.; Tamayev, R.; Arancio, O.; D’Adamio, L. An intracellular threonine of amyloid-β precursor protein mediates synaptic plasticity deficits and memory loss. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Galvan, V.; Chen, S.; Lu, D.; Logvinova, A.; Goldsmith, P.; Koo, E.H.; Bredesen, D.E. Caspase cleavage of members of the amyloid precursor family of proteins. J. Neurochem. 2002, 82, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Ramelot, T.A.; Nicholson, L.K. Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J. Mol. Biol. 2001, 307, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Iijima, K.I.; Elliott, J.I.; Kirino, Y.; Suzuki, T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of β-amyloid. J. Biol. Chem. 2001, 276, 40353–40361. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Scott, D.A.; Ganguly, A.; Koo, E.H.; Tang, Y.; Roy, S. Activity-induced convergence of app and bace-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 2013, 79, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Willnow, T.E.; Carlo, A.-S.; Rohe, M.; Schmidt, V. SORLA/SORL1, a neuronal sorting receptor implicated in Alzheimer’s disease. Rev. Neurosci. 2010, 21, 315–329. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, L.R.; Perrone, L.; Nielsen, M.S.; Calissano, P.; Andersen, O.M.; Matrone, C. Y682G mutation of amyloid precursor protein promotes endo-lysosomal dysfunction by disrupting APP–SorLA interaction. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Soriano, S.; Hayes, J.D.; Ostaszewski, B.; Xia, W.; Selkoe, D.J.; Chen, X.; Stokin, G.B.; Koo, E.H. Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. J. Biol. Chem. 1999, 274, 18851–18856. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Henderson, J.T.; O’Bryan, J.P.; Elia, A.J.; Saxton, T.M.; Pawson, T. The mammalian ShcB and ShcC phosphotyrosine docking proteins function in the maturation of sensory and sympathetic neurons. Neuron 2000, 28, 819–833. [Google Scholar] [CrossRef]
- Troglio, F.; Echart, C.; Gobbi, A.; Pawson, T.; Pelicci, P.G.; de Simoni, M.G.; Pelicci, G. The Rai (Shc C) adaptor protein regulates the neuronal stress response and protects against cerebral ischemia. Proc. Natl. Acad. Sci. USA 2004, 101, 15476–15481. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, G.; Troglio, F.; Bodini, A.; Melillo, R.M.; Pettirossi, V.; Coda, L.; de Giuseppe, A.; Santoro, M.; Pelicci, P.G. The neuron-specific Rai (ShcC) adaptor protein inhibits apoptosis by coupling Ret to the phosphatidylinositol 3-kinase/Akt signaling pathway. Mol. Cell. Biol. 2002, 22, 7351–7363. [Google Scholar] [CrossRef] [PubMed]
- Muresan, Z. c-Jun NH2-terminal kinase-interacting protein-3 facilitates phosphorylation and controls localization of amyloid-precursor protein. J. Neurosci. 2005, 25, 3741–3751. [Google Scholar] [CrossRef] [PubMed]
- Baker-Nigh, A.; Vahedi, S.; Davis, E.G.; Weintraub, S.; Bigio, E.H.; Klein, W.L.; Geula, C. Neuronal amyloid-β accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 2015, 138, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W.; Styren, S.D. Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration 1996, 5, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Pigino, G.F.; Brady, S.T.; Lazarov, O.; Binder, L.I.; Morfini, G.A. Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp. Neurol. 2013, 246, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; DeKosky, S.T.; Price, D.A. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging 1990, 11, 29–37. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Terry, R.D.; Mallory, M.; Alford, M.; Hansen, L.A. Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am. J. Pathol. 1990, 137, 1293–1297. [Google Scholar] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.C.A.; Sofroniew, M.V.; Cuello, A.C.; Powell, T.P.S.; Eckenstein, F.; Esiri, M.M.; Wilcock, G.K. Persistence of cholinergic neurons in the basal nucleus in a brain with senile dementia of the Alzheimer’s type demonstrated by immunohistochemical staining for choline acetyltransferase. Brain Res. 1983, 289, 375–379. [Google Scholar] [CrossRef]
- Grothe, M.; Heinsen, H.; Teipel, S.J. Atrophy of the cholinergic basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol. Psychiatry 2012, 71, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.; Heinsen, H.; Teipel, S. Longitudinal measures of cholinergic forebrain atrophy in the transition from healthy aging to Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.S.; Claudio Cuello, A. Altered synaptic function in Alzheimer’s disease. Eur. J. Pharmacol. 2006, 545, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Latina, V.; Caioli, S.; Zona, C.; Ciotti, M.T.; Amadoro, G.; Calissano, P. Impaired NGF/TrkA signaling causes early AD-linked presynaptic dysfunction in cholinergic primary neurons. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Hartikka, J.; Hefti, F. Comparison of nerve growth factor’s effects on development of septum, striatum, and nucleus basalis cholinergic neurons in vitro. J. Neurosci. Res. 1988, 21, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Hartikka, J.; Hefti, F. Development of septal cholinergic neurons in culture: Plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J. Neurosci. 1988, 8, 2967–2985. [Google Scholar] [PubMed]
- Oosawa, H.; Fujii, T.; Kawashima, K. Nerve growth factor increases the synthesis and release of acetylcholine and the expression of vesicular acetylcholine transporter in primary cultured rat embryonic septal cells. J. Neurosci. Res. 1999, 57, 381–387. [Google Scholar] [CrossRef]
- Auld, D.S.; Mennicken, F.; Quirion, R. Nerve growth factor rapidly induces prolonged acetylcholine release from cultured basal forebrain neurons: Differentiation between neuromodulatory and neurotrophic influences. J. Neurosci. 2001, 21, 3375–3382. [Google Scholar] [PubMed]
- Araki, W.; Wurtman, R.J. Increased expression of amyloid precursor protein and amyloid precursor-like protein 2 during trophic factor withdrawal-induced death of neuronal PC12 cells. Mol. Brain Res. 1998, 56, 169–177. [Google Scholar] [CrossRef]
- Overk, C.R.; Masliah, E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem. Pharmacol. 2014, 88, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 2003, 24, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.W.; Watson, D.F. Axonal transport in neurological disease. Ann. Neurol. 1988, 23, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.R.; Owen, M.J. Alzheimer’s disease: The amyloid hypothesis on trial. Br. J. Psychiatry 2016, 208, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Chetelat, G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat. Rev. Neurol. 2013, 9, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Chételat, G.; Dickson, D.; Fagan, A.M.; Frisoni, G.B.; Jagust, W.; Mormino, E.C.; Petersen, R.C.; Sperling, R.A.; et al. Suspected non-Alzheimer disease pathophysiology—Concept and controversy. Nat. Rev. Neurol. 2016, 12, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Delacourte, A.; Sergeant, N.; Champain, D.; Wattez, A.; Maurage, C.; Lebert, F.; Pasquier, F.; David, J.-P. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology 2002, 59, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J. Formation of neurofibrillary tangles in P301L Tau transgenic mice induced by Aβ 42 fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004, 43, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Ribé, E.M.; Pérez, M.; Puig, B.; Gich, I.; Lim, F.; Cuadrado, M.; Sesma, T.; Catena, S.; Sánchez, B.; Nieto, M.; et al. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol. Dis. 2005, 20, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Guillozet-Bongaarts, A.L.; Glajch, K.E.; Libson, E.G.; Cahill, M.E.; Bigio, E.; Berry, R.W.; Binder, L.I. Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol. 2007, 113, 513–520. [Google Scholar] [CrossRef] [PubMed]
- García-Sierra, F.; Mondragón-Rodríguez, S.; Basurto-Islas, G. Truncation of tau protein and its pathological significance in Alzheimer’s disease. J. Alzheimers. Dis. 2008, 14, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, P.M. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J. Neurosci. 2004, 24, 7895–7902. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Garg, S.; Mandelkow, E.-M.; Mandelkow, E. Proteolytic processing of tau. Biochem. Soc. Trans. 2010, 38, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed]
- King, M.E.; Kan, H.M.; Baas, P.W.; Erisir, A.; Glabe, C.G.; Bloom, G.S. Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J. Cell Biol. 2006, 175, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau reduction prevents Aβ-induced defects in axonal transport. Science 2010, 330, 198. [Google Scholar] [CrossRef] [PubMed]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.J.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O.; et al. Tau protein is required for amyloid-induced impairment of hippocampal long-term potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-tau antibodies that block tau aggregate seeding invitro markedly decrease pathology and improve cognition in vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.L.; Chen, X.; Kazim, S.F.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J. Neural Transm. 2015, 122, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Savanur, G.; Madhavadas, S. Passive immunization targeting the N-terminal region of phosphorylated tau (residues 68–71) improves spatial memory in okadaic acid induced tauopathy model rats. Biochem. Biophys. Res. Commun. 2017, 483, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Sadot, E.; Heicklen-Klein, A.; Barg, J.; Lazarovici, P.; Ginzburg, I. Identification of a tau promoter region mediating tissue-specific-regulated expression in PC12 cells [published erratum appears in J. Mol. Biol. 1996, 258, 539]. J. Mol. Biol. 1996, 256, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.; Heldman, E.; Gurwitz, D.; Haring, R.; Karton, Y.; Meshulam, H.; Pittel, Z.; Marciano, D.; Brandeis, R.; Sadot, E.; et al. M1 Agonists for the Treatment of Alzheimer’s Disease. Novel Properties and Clinical Update; Wiley: Hoboken, NJ, USA, 1996; Volumn 777. [Google Scholar]
- Nuydens, R.; Dispersyn, G.; de Jong, M.; van den Kieboom, G.; Borgers, M.; Geerts, H. Aberrant tau phosphorylation and neurite retraction during NGF deprivation in PC12 cells. Biochem. Biophys. Res. Commun. 1997, 240, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Shelton, S.B.; Johnson, G.V.W. Tau and HMW tau phosphorylation and compartmentalization in apoptotic neuronal PC12 cells. J. Neurosci. Res. 2001, 66, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Ciotti, M.T.; Florenzano, F.; Capsoni, S.; Amato, G.; Calissano, P. Endogenous Aβ causes cell death via early tau hyperphosphorylation. Neurobiol. Aging 2011, 32, 969–990. [Google Scholar] [CrossRef] [PubMed]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Adalbert, R.; Gilley, J.; Coleman, M.P. Aβ, tau and ApoE4 in Alzheimer’s disease: The axonal connection. Trends Mol. Med. 2007, 13, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Schindowski, K.; Belarbi, K.; Buée, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav. 2008, 7, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Amadoro, G.; Gentile, A.; Capsoni, S.; Ciotti, M.T.; Cencioni, M.T.; Atlante, A.; Canu, N.; Rohn, T.T.; Cattaneo, A.; et al. Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol. Cell. Neurosci. 2008, 38, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: Possible implications for neurodegeneration. J. Alzheimer’s Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Sokolow, S.; Henkins, K.M.; Bilousova, T.; Gonzalez, B.; Vinters, H.V.; Miller, C.A.; Cornwell, L.; Poon, W.W.; Gylys, K.H. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J. Neurochem. 2015, 133, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Sancesario, G.M.; Lubrano, A.; Melchiorri, G.; Bernardini, S.; Calissano, P.; Sancesario, G. Cerebrospinal fluid levels of a 20–22 kDa NH2 fragment of human tau provide a novel neuronal injury biomarker in Alzheimer’s disease and other dementias. J. Alzheimer’s Dis. 2014, 42, 211–226. [Google Scholar] [CrossRef]
- Schroeder, S.K.; Joly-Amado, A.; Gordon, M.N.; Morgan, D. Tau-directed immunotherapy: A promising strategy for treating Alzheimer’s disease and other tauopathies. J. Neuroimmune Pharmacol. 2016, 11, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M. Tau immunotherapy. Neurodegener. Dis. 2016, 16, 34–38. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canu, N.; Amadoro, G.; Triaca, V.; Latina, V.; Sposato, V.; Corsetti, V.; Severini, C.; Ciotti, M.T.; Calissano, P. The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. Int. J. Mol. Sci. 2017, 18, 1319. https://doi.org/10.3390/ijms18061319
Canu N, Amadoro G, Triaca V, Latina V, Sposato V, Corsetti V, Severini C, Ciotti MT, Calissano P. The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology. International Journal of Molecular Sciences. 2017; 18(6):1319. https://doi.org/10.3390/ijms18061319
Chicago/Turabian StyleCanu, Nadia, Giuseppina Amadoro, Viviana Triaca, Valentina Latina, Valentina Sposato, Veronica Corsetti, Cinzia Severini, Maria Teresa Ciotti, and Pietro Calissano. 2017. "The Intersection of NGF/TrkA Signaling and Amyloid Precursor Protein Processing in Alzheimer’s Disease Neuropathology" International Journal of Molecular Sciences 18, no. 6: 1319. https://doi.org/10.3390/ijms18061319