1. Introduction
Mammalian metallothionein-3 isoform (MT3) is an unusual protein with a puzzling role in neurochemistry [
1]. It has the hallmarks of a typical mammalian metallothionein and is comprised of 68 amino acids, 20 of which are cysteines. Hence, it is assumed that, like other metallothioneins (MT), it binds essential metal ions such as zinc (Zn) and copper (Cu) and sequesters toxic metal ions such as cadmium (Cd) and silver (Ag) [
2]. Most mammalian metallothioneins are induced by high levels of zinc by a mechanism initiated by Zn
2+ binding to a metal-responsive transcription factor [
3]. Because toxic metals such as Cd
2+ and Ag
+ displace zinc bound to metallothionein, their presence also indirectly leads to elevated MT levels.
Despite the presence of the canonical 20 cysteines in its structure, MT3 was first identified not as a metallothionein, but as an inhibitor of neuronal growth [
4,
5]. Although the molecular basis of its ability to inhibit neuronal growth is not understood, the effect has been connected to the seven-amino-acid acidic region that is unique to MT3 [
6]. Recently, MT3 has been shown to bind to β-actin and facilitate actin polymerization [
7]. β-Actin polymerization is involved in cytoskeletal growth, fueling speculation that the neuronal growth inhibitor effects of MT3 could occur via an interaction with β-actin [
8].
MT3 is specifically expressed in the normal human brain and is significantly under-expressed in the brains of patients with Alzheimer’s disease [
1,
4,
9]. Little is known about what constitutes “normal” bioinorganic chemistry in the central nervous system [
10], though recent works point to essential roles for zinc and copper ions in the brain and suggest that both ions can function in neuronal signaling [
11]. Therefore, it is noteworthy that MT3 might modulate the bioinorganic chemistry of essential (zinc, copper), therapeutic (lithium) and toxic (lead) metal ions in the brain. MT3 has been speculated to play a special role in mediating the chemistry of copper, which has been found in the brain not only as bound to biomolecules and highly regulated, but also as existing in dynamic, loosely bound pools [
11]. We have speculated that MT3 might be involved in the effect that the exposure to lead (Pb) has on brain development [
12]. It is well known that exposure to lead during childhood leads to irreversible long-lasting developmental effects, including perhaps an increased risk of certain forms of mental illnesses [
13,
14]. Based on a report that lithium chloride treatment leads to decreased levels of
Mt3 in mice brains, we have considered the possibility that MT3 might be involved in the pharmacological benefits of lithium treatment for people suffering from bipolar mood disorder [
15].
In this paper, we explore factors that may potentially impact MT3 gene expression and, indirectly, function. MT3 is known to be abundant in the dentate gyrus (DG) [
16]. Our study of gene expression in the DG of a population of litter-mate matched mice, presented here, revealed significant differences between tested mice in the expression levels of
Mt3, along with a suite of other genes associated with zinc homeostasis. Significantly,
Mt2 was upregulated in one population of mice, while
Mt3 was upregulated in the other population of mice, pointing to a very different mechanism of regulation of these two metallothioneins.
We also examine some inorganic factors that may have an impact on expression levels of metallothioneins in human neuronal cells. There is conflicting published data about the role that metal ions have on the
MT3 levels, pointing to the importance of studying
MT3 expression in multiple cell lines [
17,
18,
19]. We show that, compared to
MT2,
MT3 responds differently to identical metal treatments. The effects that metal ions have on
MT3 levels are far more modest than the effects that these metal ions have on
MT2 levels. This fact, again, is pointing to a possible regulatory mechanism that differentially controls the expression of
MT2 vs.
MT3 in neuronal tissues.
3. Discussion
Metallothioneins are small proteins whose molecular simplicity belies a history replete with efforts to definitively determine their function [
20,
21,
22]. As a member of this protein family known for its confusing biochemical functions, MT3 plays an even more opaque role in mammalian biology. The presented work strives to provide an insight into the biology of MT3 by studying the factors that regulate its expression as well as characterizing the degree of structural changes conferred by the binding of the protein with different metal ions.
Metallothioneins are thought to act as zinc chaperones transporting zinc ions to their target locations and perhaps facilitating their release when warranted [
1]. Consistent with this function, most metallothioneins are upregulated by high levels of zinc. The presence of a metal response element (MRE) in the promoter region of
MT1 and
MT2 causes most metallothioneins to be induced by zinc [
3]. The MRE is activated via interaction with the zinc finger-containing metal transcription factor 1 (MTF1) [
23]. MTF1 is responsible for regulating a number of genes associated with heavy metals and oxidative stress [
23]. Metallothionein is the prototypical gene target for MTF1.
Metallothioneins are also thought to function as “heavy metal sponges,” binding toxic metal ions and potentially sequestering them. Heavy metals, including cadmium and silver, have been repeatedly shown to increase metallothionein levels. However, the effect is an indirect one. Heavy metals affect MT levels because they displace zinc from metallothionein, and the displaced zinc binds to the metal transcription factor, which in turn binds to the MRE and activates the transcription of metallothioneins [
24]. Interestingly, despite the fact that lead is a heavy metal, it has never been clear whether lead affects the levels of any metallothioneins [
25,
26,
27]. We recently showed that lead can displace zinc from Mt3 with an equilibrium constant of about 1 × 10
4 and thus should be able to displace zinc in vivo, leading to the upregulation of MTs by the mechanism described above [
12]. Nevertheless, some published studies show no change in MT levels following lead treatment [
17], while others do show a change [
28].
Metallothioneins have also long been associated with a role in ameliorating oxidative stress [
22]. MTs can release Zn
2+ and two electrons while forming a disulfide bond. This may be relevant to immune responses and inflammation [
22,
29]. MTF1 also binds to the metal response element of other genes involved in cellular redox homeostasis, including selenoproteins H and W [
30,
31]. Thus, it is speculated that metallothioneins and other proteins involved in combatting oxidative stress will be simultaneously overexpressed under conditions of oxidative stress.
Very significantly, however,
MT3 appears to be regulated differently from other mammalian metallothioneins [
18,
32].
MT3 does not appear to respond to zinc levels in either astrocytes or neurons in the same way that other MTs respond to zinc, even though the promoter region of
MT3 has several regions that are similar to the metal response elements that regulate
MT1 and
MT2 levels [
1,
5,
17,
19,
32]. MTF1 does not bind to the MRE sequence in the
MT3 promoter region [
32].
Both our cell culture data and our microarray data are consistent with the assertion that, in the brain, MT3 is regulated differently from other metallothioneins. Our cell culture data shows that, in two different neuronal cell lines, addition of zinc leads to a significant overexpression of MT2, while having essentially no effect on MT3.
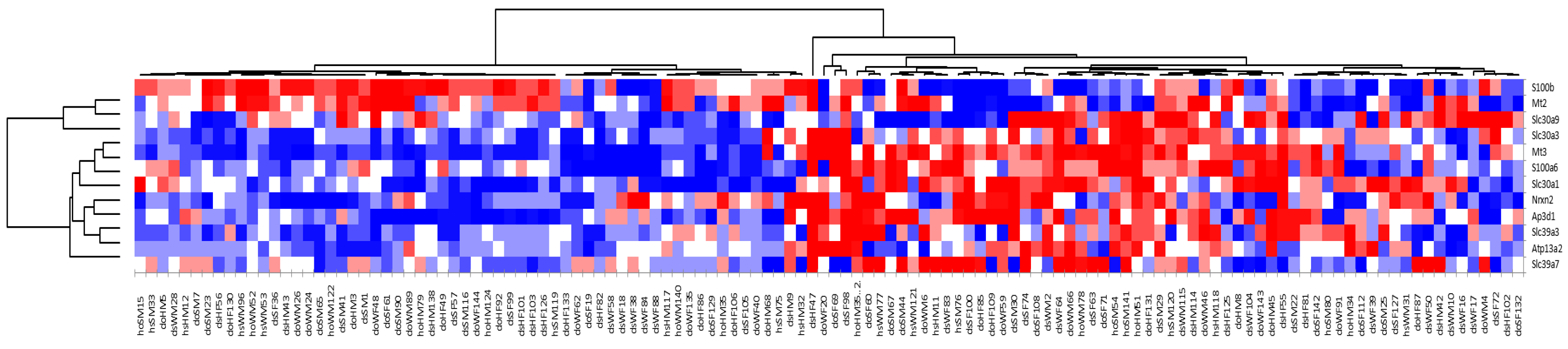
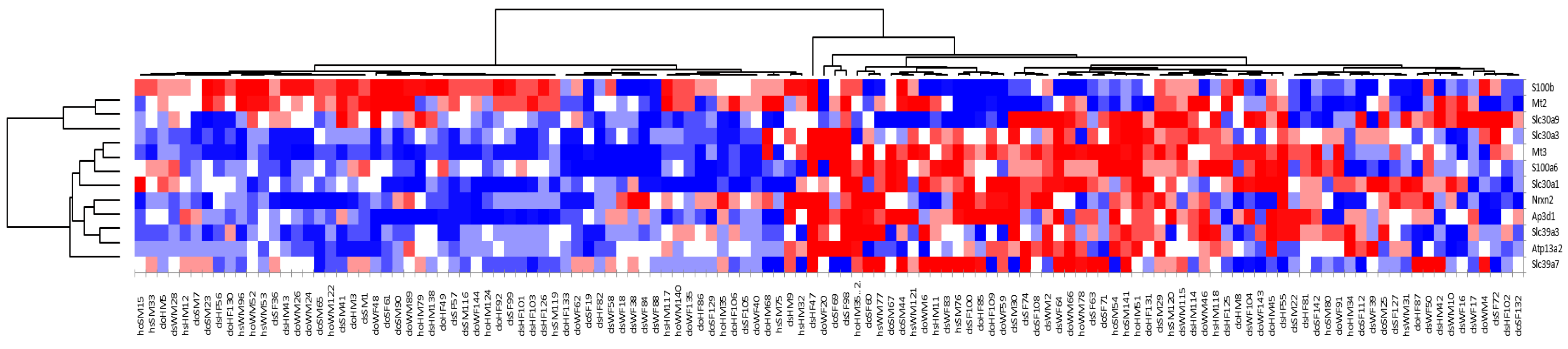
The microarray data demonstrate that, among a group of littermate-matched mice, approximately half the population (Group A) had statistically significant upregulation of Mt2 and S100b, while the other half of the population (Group B) had statistically significant upregulation of Mt3 and a number of known zinc transporters (S100a6, Slc30a1, Slc39a7, Slc30a3, Slc39a3, and Ap3d1), as well as Nrx2 and Atp13a2. The fact that Mt2 was highly expressed in Group A while Mt3 was highly expressed in Group B indicates that these two genes are regulated differently. Furthermore, the observation that Mt3 was upregulated along with a number of other zinc transporters suggests that this protein has a role in zinc homeostasis in the brain.
Of particular note is the differential pattern of coexpression of members of the S100 family.
S100b, which was upregulated together with
Mt2, expresses a calcium- and zinc-binding protein involved in immune responses.
S100a6, which is upregulated with Mt3, expressed a calcium- and zinc-binding protein that has been associated with misregulation of zinc levels in several neurodegenerative diseases [
33].
S100a6 is overexpressed in mouse models of amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease and is speculated to contribute to disease progression via its high affinity for zinc [
34]. It has also been associated with cell proliferation and neuronal degeneration, processes with which
MT3 is also associated [
35,
36,
37,
38]. Other researchers have noted that
S100b and
S100A6 are differentially expressed in human cortical tissue and have speculated that despite being members of the same family of enzymes they play different neurochemical roles [
38].
S100b has been linked to neuronal growth, including the formation of neurite extension [
38], while
S100a6 has been associated with cytoskeletal dynamics (as has
MT3 [
7,
8]) [
39].
S100a6 is known to be upregulated under conditions of oxidative stress [
39].
S100b has been associated with the protective effects of nutritional zinc against methyl mercury exposure [
40], as well as linked to
MT2 expression in bipolar mood disorder [
41]. Additional studies are underway in our laboratories to further explore the molecular mechanisms that link this differential expression of
MT2/
S100b and
MT3/
S100A6.
There are a few factors that have been reported to affect
MT3 expression. These include lithium (downregulation), age (upregulation) and hypoxia (upregulation), but in each case, the molecular mechanism for this regulation is not understood. Lithium was shown to decrease
MT3 expression in mice that were given lithium chloride [
15]. Our cell culture results, however, showed essentially no effect of lithium on
MT3 levels. The antidepressant eugenol has also been shown to induce
Mt3 expression in the hippocampus of mice while having no effect on
Mt1 levels [
42]. Both of these experiments further suggest that MT3 may play a role in aspects of brain chemistry not normally associated with metallothionein function and also point to regulation of MT3 by a non-MRE pathway.
MT3 levels increase with age [
36,
43,
44,
45,
46]. In mice brains, Mt3 was found in higher concentrations after 12 weeks of age [
43]. In rat brains, a very large increase in Mt3 levels was seen when the rats passed middle age (16 months) [
36]. The authors speculate that, because MT3 has more Cu-thionein character, its upregulation could indicate an enhanced role in copper chelation to protect the aging brain from copper redox toxicity. However, it is unclear by what mechanism aging could lead to higher MT3 levels [
36].
MT3 also responds to hypoxia [
45,
46,
47]. In one study of hypoxia,
MT3 was the gene most sensitive to hypoxia, with 3- and 7-fold increases in expression levels after 10 and 30 min treatments, respectively [
45]. In another study,
MT3 levels increased by a factor of more than 600 in 60 min upon exposure to hypoxic conditions [
46]. MT3 gene expression can also reportedly be substantially induced by hypoxia mimetics (CoCl
2, desferrioxamine, dimethyloxalylglycine), which indicates that it is transcriptionally regulated through HIF1 [
46]. Hypoxia has been suggestively linked to an imbalance in zinc homeostasis [
47]. Also, while other metallothioneins have been shown to respond to hypoxia, in the case of other MTs, activation via the MRE was implicated [
48]. Again, in our cell culture experiments, the hypoxia mimic Co
2+ showed essentially no effect on either
MT2 or
MT3 expression levels.
It is challenging to study factors that influence
MT3 levels, and our work illustrates this as well. Few established cell lines express
MT3 to levels comparable to the levels that have been measured in the brain [
31]. In this study, we selected two different neuronal cell lines, both of which were known to have measurable but not extremely high
MT3 levels, making it possible to study treatments that might lead to increased (or decreased) gene and protein expression. Even still, the cell culture work does not appear to capture all of the complexity visible in animal models.
Considering that MT3 is in so many respects similar to MT1 and MT2, how can one explain its strikingly different neurochemistry? Part of the answer presumably rests with the small structural differences between MT3 and other MTs [
49]. The unique N-terminal sequence of MT3 is required for the growth inhibitory activity of MT3 in the neural system [
50]. It has also been shown to be responsible for Cd
2+-induced death of the proximal tubule cell [
50]. These amino acids have also been shown to be responsible for zinc-specific MT3 binding to actin [
7]. Protein–protein interactions, modulated by MT3’s unique structure, may be critical to many of MT3’s neurochemical functions [
9,
49]. The 3D structure of metallothionein is determined by metal binding. Thioneins are unstructured and unfolded in the absence of metal ions and then fold into bilobal proteins upon binding seven divalent metal ions [
51,
52]. Suprametalation has been suggested to cause the protein to rearrange to form a single, more globular structure [
53]. Apo-MT2 and apo-MT3 in solution have practically identical structures with low helical content [
54], supporting the common notion that apo-metallothioneins exhibit little or no secondary structure [
55]. In the presence of metals, however, metallothioneins fold into varied structures. Metallothioneins bind divalent metals such as Zn
2+ and Cd
2+ in tetrahedrally coordinated M
3S
9 and M
4S
11 binding clusters, separated with sections of random coil. These structures are characterized by circular dichroism spectra that show a positive peak at 240 nm [
56].
We and others have speculated that some of the neurotoxic effects of lead exposure might be mediated by differences in metallothionein structure when bound to lead vs. zinc [
12,
57,
58]. Zinc prefers a coordination number of four, corroborated by its tetrahedral coordination with thiol sites [
56]. In contrast to the coordination chemistry of Zn
2+, that of Pb
2+ is much more complex and varied [
59,
60]. Recent evidence indicates few four-coordinate Pb(II) complexes; they are either three-coordinate or five-coordinate [
31]. Possibly, lead’s (potentially) stereochemically active lone pair plays a role in determining its geometric preferences [
30,
32]. He et al. speculate that lead forms a trigonal pyramidal coordination when bound to MT2 with three sulfurs and electron donating oxygens (Pb–S
3O) [
57]. The known differences in metal binding preferences between MT3 and other metallothioneins, including the presence of an 8th metal binding site in MT2, is likely to effect the overall structure of metal-replete MT3 [
54,
61,
62]. Work is ongoing to determine if these structural differences lead to functional differences in the way that MT3 interacts with partner proteins [
9].
Part of the answer also presumably lies with the differential expression of MT3. This work confirms the differential expression of MT3 but does not elucidate what the molecular mechanism is by which it is regulated. Additional work in our labs is also underway to test hypotheses associated with regulatory pathways for MT3.
4. Materials and Methods
4.1. Materials
Metal salts used in this work include: lead acetate (99–103%, Thermo Fisher Scientific, Waltham, MA, USA), zinc sulfate monohydrate (99%, Strem Chemicals, Newburyport, MA, USA), cobalt (II) chloride hexahydrate (98.0–102.0%, Sigma Aldrich, St. Louis, MO, USA), and lithium sulfate monohydrate (99%, Sigma Aldrich).
An ÄKTA Pure 25 L FPLC (GE Healthcare, Pittsburgh, PA, USA) was used for protein purification, and a Forma Anaerobic System 1025 (Thermo Forma, Marietta, OH, USA) was used for anaerobic protein manipulations. Low-speed centrifugation was done with an Allegra X-30R benchtop centrifuge (Beckman Coulter, Indianapolis, IN, USA) and higher speed centrifugation above 10,000 rpm was done with an Avanti J-265 XPI superspeed centrifuge (Beckman Coulter). Sonication was done with a Vibra-Cell VC 505 Ultrasonic Liquid Processor (Sonics & Materials, Inc., Newtown, CT, USA), and lyophilization was done on a FreeZone 2.5 Liter Benchtop Freeze Dry System (Labconco, Kansas City, MO, USA). UV-Vis spectra were taken on a Varian Cary 50 UV-Vis spectrometer (Agilent Technologies, Santa Clara, CA USA).
MES, MOPS, and bis-Tris buffers were purchased in highest purity from Sigma Aldrich (St. Louis, MO, USA), as were metal salts, dithiothreitol (DTT), and 5,5′-dithiobis-(2-nitrobenzoi acid) (DTNB). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Thrombin from bovine plasma was obtained from GE Healthcare Life Sciences (Pittsburgh, PA, USA). GSTrap FF columns, HiTrap desalting columns, and a HiLoad 26/600 75 pg size exclusion column were also purchased from GE Healthcare Life Sciences (Pittsburgh, PA, USA). All buffers were prepared with milli-Q (18.2 MΩ·cm) water, filtered through 0.22 μm filter unit, and degassed under vacuum until no air bubbles were seen. Metal stock solutions were prepared using the appropriate mass of metal salt and then diluted with oxygen-free buffers in the anaerobic chamber.
4.2. Animal Studies Ethics Statement and Animal Use
All animal care and testing protocols were approved by the Institutional Animal Care and Use Committee at Columbia University, animal welfare assurance # A3007-01 (31 July 2013).
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and bred to establish colonies at the Columbia University animal facility (New York City, NY, USA). All mice were group-housed according to institutional guidelines, with water and a commercial mouse diet available ad libitum. Gene expression analysis was performed on age- and sex-matched mice (8- to 12-week-old; 61 females, 63 males).
4.3. Tissue Preparation
The dentate gyrus (DG) was dissected from adult mice hippocampi under a stereomicroscope using an established method [
63]. Brains were collected from deeply anesthetized mice and placed into ice-cold phosphate-buffered saline (PBS). After exposing the medial side of the brain, the DG was isolated and retrieved using forceps. DG tissue was placed directly into cryovials containing RNAlater and kept at 4 °C overnight. The following day, RNAlater was removed, and samples were frozen at −80 °C until further use.
4.4. RNA Extraction, Amplification, and Gene Expression Microarray Experiments
Before tissue homogenization, all surfaces and instruments were wiped clean with an Ambion RNaseZap RNase decontamination solution (Thermo Fisher Scientific). RNA was extracted from DG tissue according to RNeasy Mini Kit instructions (Qiagen, Germantown, MD, USA). DG tissues of individual mice were homogenized using a Powergen 700 homogenizer (Thermo Fisher Scientific) in the kit lysis buffer. Samples were quantified spectrophotometrically using a NanoDrop 2000 (Thermo Fisher Scientific) and further analyzed using a 2100 Bioanalyzer (Agilent Technologies).
10 ng of the extracted RNA were amplified using an Ambion MessageAmp II aRNA Amplification Kit in vitro transcription protocol following the manufacturer instructions (Thermo Fisher Scientific). The resulting aRNA was purified using an Ambion MEGAclear Transcription Clean-Up Kit (Thermo Fisher Scientific), and 2.5 µg of aRNA from each sample were labeled with AlexaFluor647 or AlexaFluor536 using RNA alkylation with monoreactive cisplatin dye complexes from an Invitrogen ULYSIS Nucleic Acid Labeling Kits (Molecular Probes, Thermo Fisher Scientific). To correct for dye-related sensitivity bias in the downstream detection of hybridization signals, the dye selection was balanced between the sample groups across the experiment. Using spectroscopic measurements of the labeled probes, labeling efficiencies were estimated as 0.5–1.5% of the dye with respect to RNA bases. The labeled aRNA samples were hybridized to SurePrint G3 Mouse GE 8x60K Microarray Kit (Agilent) following the manufacturer instructions. After hybridization, the microarray slides were scanned on the two channels using Agilent C scanner (Agilent), and the image segmentation, gridding, and feature extraction were performed using the onboard Scan Control software (v.8.5.1).
4.5. Analysis of Microarray Data
The raw hybridization signal intensities were normalized within the arrays by Agilent software following global normalization using the loess function from the Bioconductor package affy (v.1.42.3) [
64,
65]. Background filtering was performed on Agilent’s “Well Above Background” flag function using score modulation between the relevant groups of the samples. For a gene to be scored as present, the gene expression signals must be flagged “Well Above Background” in more than 50% of samples within a group. Gene function assignments were done using Gene Ontology (AmiGO, v.2.4.26) [
66]. The resulting data were imported into the Spotfire (v.7.6.1) for Functional Genomics Suite (TIBCO, New York City, NY, USA) for data visualization and further review using PCA and hierarchical clustering. Analysis of differential gene expression was performed using a
t-test followed by Benjamini-Hochberg multiple testing correction [
67]. Differentially expressed genes for functional enrichment surveys were selected at FDR < 0.05 and fold change of 1.3. Functional enrichment analysis in differentially expressed genes was carried out with DAVID (v.6.8) [
68].
4.6. General Cell Culture and Metal Treatment
Human neuroblastoma cell lines, BE(2)C and SH-SY5Y (ATCC), were cultured in 1X Dulbecco’s Modified Eagle Media (DMEM) with 4.5 g/L glucose and l-glutamine without sodium pyruvate (Corning Inc., Corning, NY, USA), supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific) using standard mammalian cell culture procedures. Cells were maintained at 37 °C and 5% CO2 in a humidified incubator.
One day prior to metal treatment, BE(2)C and SH-SY5Y cells were seeded at 400,000 BE(2)C cells or 450,000 SH-SY5Y cells per well onto tissue culture treated 6-well plates (2 mL media per well). At designated treatment times, the cell culture media was replaced with 2 mL of fresh DMEM with 10% FBS and 100 mM metal solutions of lead acetate, zinc sulfate, cobalt chloride, or lithium sulfate were added to the media to a final concentration of 100 µM metal ion.
4.7. Total RNA Extraction, cDNA Synthesis, and qPCR
Prior to RNA extraction, metal containing media was aspirated from the cells and the cells were washed with PBS. Total RNA was extracted using the PureLinkTM RNA Mini Kit (Invitrogen) according to manufacturer’s instructions for monolayer cells. The quantity and relative purity of the total RNA was measured by UV-Vis spectroscopy on a Nanodrop 2000 spectrophotometer using the absorbance at 260 nm and the 260/280 nm ratio, respectively. All RNA were stored at −80 °C until further use.
Total cellular messenger RNA were reverse transcribed using the First Strand cDNA Synthesis (Quick Protocol, New England BioLabs, Ipswich, MA, USA). Within each trial, total RNA used for reverse transcription were maximized and standardized within the range of 200–1000 ng of RNA. To amplify only the mRNA transcripts, an anchored poly-DT primer (5 mg/mL, IDT DNA) was used. Complementary DNA (cDNA) synthesis was performed in a thermocycler (Bio-Rad, Hercules, CA, USA) according to the following protocol: 42 °C for 60 min, 80 °C for 10 min. The extracted cDNA was incubated at 4 °C for immediate use and stored at −20 °C for future use.
Real time quantitative PCR (qPCR) was performed using PowerUP SYBR Green Master Mix (Thermo Fisher Scientific) according to the manufacturer’s protocol for a 20 µL reaction using 2 µL of the cDNA product as the PCR template.
MT3,
MT2, and the housekeeping gene
HSP90AB1 were quantified (in triplicate) for each metal treatment condition as well as the non-treatment control. The primers used for quantitative PCR amplification of
MT2,
MT3, and
HSP90AB1 cDNA are listed in
Table 5. A LightCycler 480 instrument (Roche Molecular Diagnostics, Pleasanton, CA, USA) was used for qPCR amplification and monitoring, according to the PowerUp SYBR Green Master Mix thermal cycling protocol guidelines. Based on the primers used, the annealing temperature for the qPCR was set at 58 °C. qPCR data was analyzed using the ΔΔ
CP method [
69]. The presence of MT3 in the cell lines was confirmed by western blot analysis and by SDS-PAGE gels with purified Mt3 as a control.
4.8. Protein Purification
Recombinant mouse Zn
7-Mt3 was purified by following a modified protocol from our previous study [
12,
70,
71]. Cells were induced at OD
600 ~ 0.7 by adding 1 mM isopropyl β-
d-1-thiogalactopyranoside (IPTG). After incubation at room temperature and 250 rpm for 1 h, ZnCl
2 was added to a final concentration of 0.5 mM, and the cultures were incubated at 37 °C and 250 rpm for 16–18 h.
Cells were thawed, separated into approximately 15 g portions, and each portion was suspended in a solution of 1.248 g sucrose in 15 mL PBS (140 mM NaCl, 2.7 mM KCl 10 mM Na2HPO4, 1.8 mM KH2PO4). Argon was bubbled through the mixture before centrifuging at 8000× g for 30 min at 4 °C and collecting the pellets. To a suspension of the pellet in 15 mL PBS, 5.7 mM β-mercaptoethanol (BME) was added. After bubbling with argon, the suspension was centrifuged at 8000× g for 30 min at 4 °C. The pellets were collected and resuspended in PBS to a total volume of 15 mL. Just before sonicating the solution for 7 min, with 10 s rest every 5 s, 1 mM DTT and 0.1 mM phenylmetylsulfonyl fluoride (PMSF) were added. The lysed cells were then centrifuged at 8000× g for 30 min at 4 °C. The supernatant was collected and centrifuged at 35,000 rpm for 30 min to remove any particulate matter. A Bradford standard assay using bovine serum albumin (BSA) was used to determine the protein concentration of the cell lysate.
Using previously published protocols, GST-tagged Mt3 protein was separated via GSTrap FF columns and cleaved using bovine plasma thrombin [
12]. Mt3 was isolated via HiLoad 26/600 75 pg size exclusion column. Mt3 concentration was determined by measuring the absorbance of the samples in 1 mM HCl at 220 nm and calculating using an extinction coefficient of 53,000 M
−1·cm
−1 [
62]. The presence of Mt3 was confirmed by SDS-PAGE on a 15% polyacrylamide gel, either with silver or Coomassie staining and florescence imaging on bromobimane-modified MT3 samples [
72].
Mt3 was concentrated and buffer-exchanged into Milli Q water using 3 kDa Amicon centrifugal filter units. The concentrate was lyophilized overnight, and reconstituted in Tris-HClO4 buffer to maintain the pH at 7.0. Mt3 concentration was determined both by using a DTNB assay measuring the concentration of free thiols (20 free thiols per protein) and by measuring the absorbance at 220 nm in 1 mM HCl as described above.




{kind=link}
{kind=link}
{kind=link}