
Molecular Basis of Alcohol-Related Gastric and Colon Cancer

Abstract
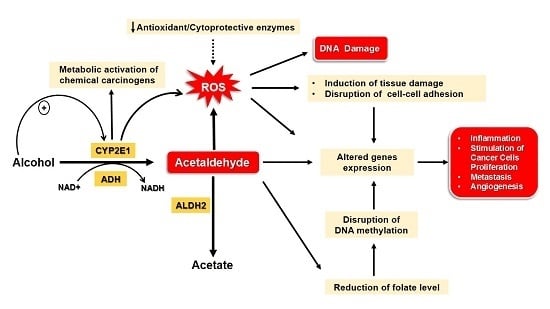
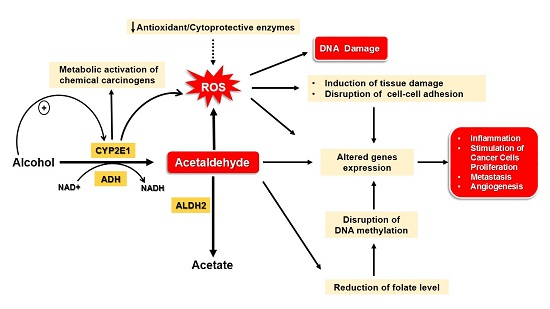
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
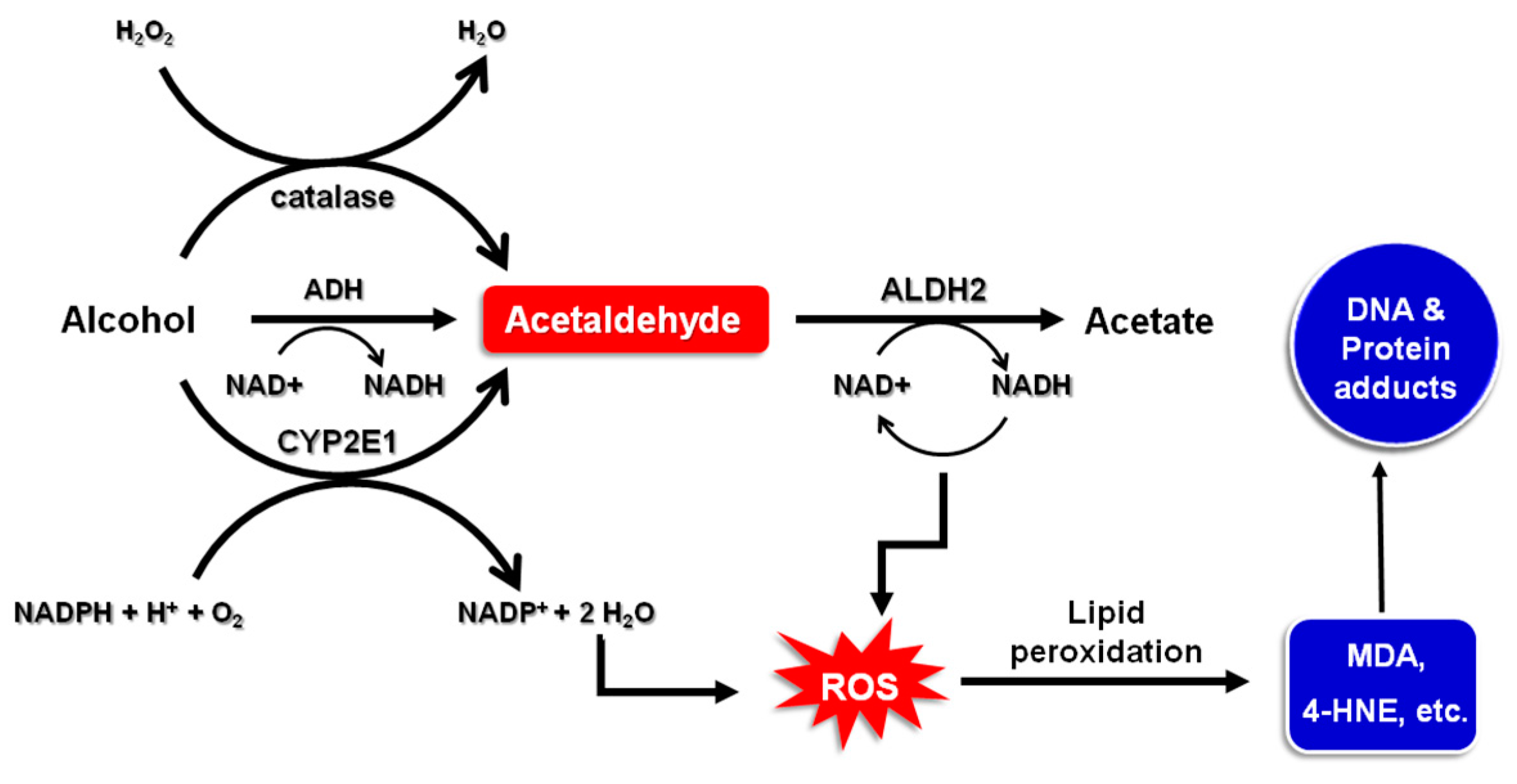
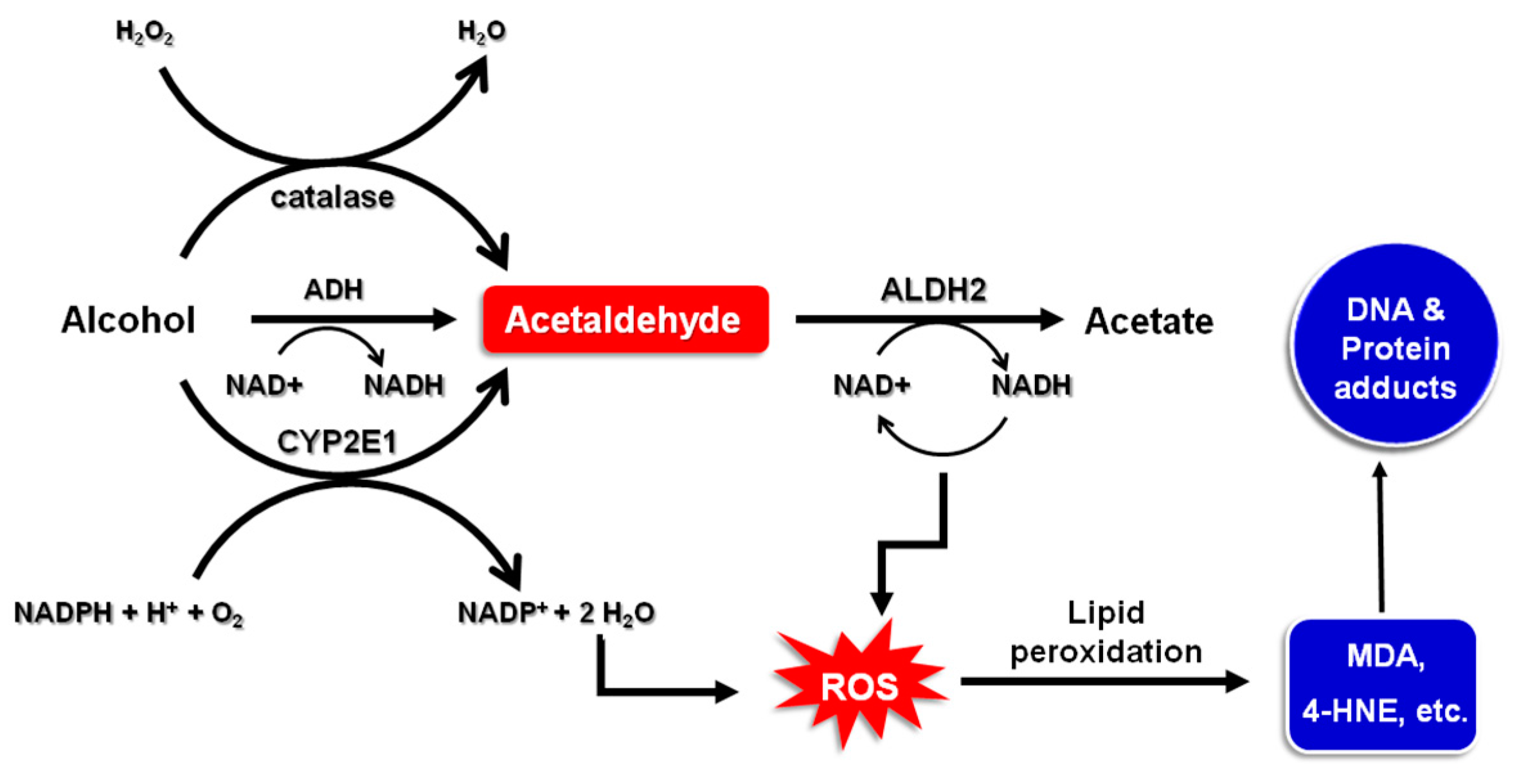
2. Alcohol Metabolism
3. Genetic Polymorphism of Alcohol Metabolizing Enzymes
3.1. Polymorphism Associated with Gastric Cancer
3.2. Polymorphism Associated with Colon Cancer
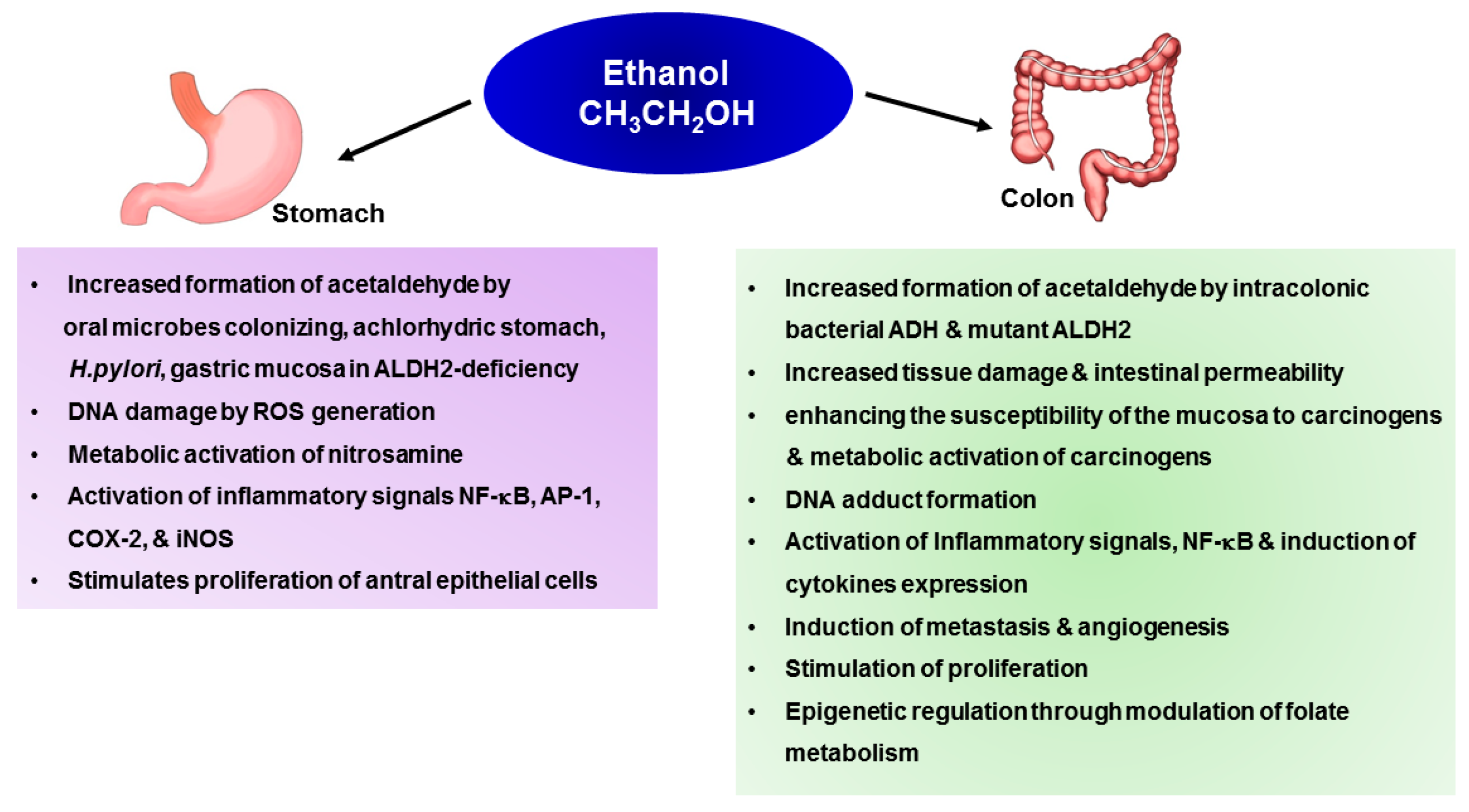
4. Mechanisms Underlying Alcohol-Induced Gastric and Colonic Carcinogenesis
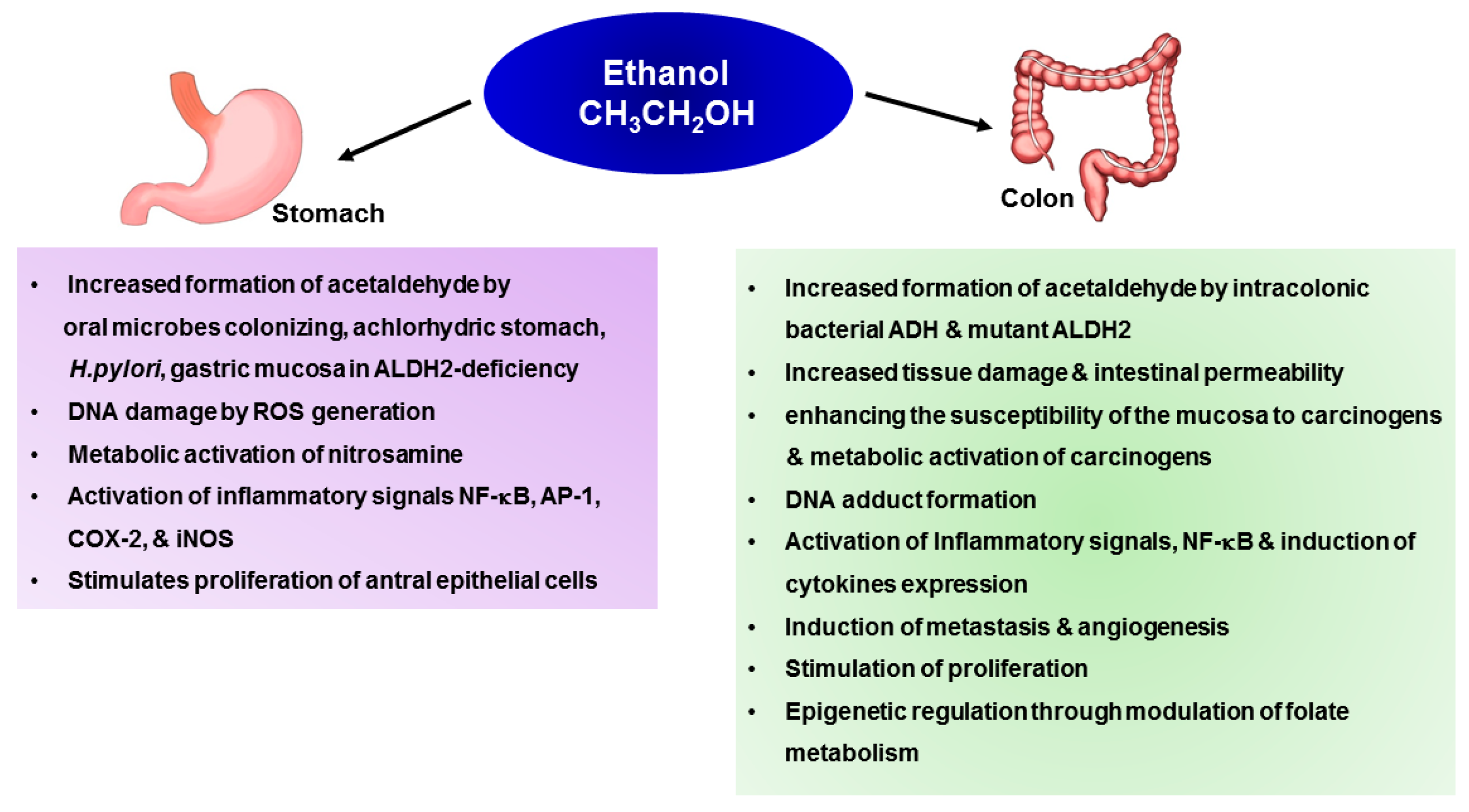
4.1. Gastric Cancer
4.1.1. Generation of Acetaldehyde by Microbiome
4.1.2. Inflammation Induced by Alcohol
4.1.3. Metabolic Activation of Carcinogens and Their DNA Adduct Formation
4.2. Colon Cancer
4.2.1. Generation of Acetaldehyde by Microbiome
4.2.2. Increased Uptake of Carcinogens
4.2.3. Metabolic Activation of Carcinogens and Their DNA Adduct Formation
4.2.4. Inflammation-Associated Tumor Promotion
4.2.5. Invasion and Metastasis
4.2.6. Induction of Angiogenesis
4.2.7. Stimulation of Cancer Cell Proliferation
4.2.8. Epigenetic Mechanism
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GI | Gastrointestinal |
| CYP2E1 | Cytochrome P4502E1 |
| CRC | Colorectal cancer |
| ADH | Alcohol dehydrogenases |
| ALDH | Aldehyde dehydrogenase |
| ROS | Reactive oxygen species |
| 4-HNE | Hydroxynonenal |
| H. pylori | Helicobacter pylori |
| O6-MeG | O6-methylguanine |
| iNOS | Inducible nitric oxide synthase |
| MAM | Methylazoxymethanol |
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| AOM | Azoxymethane |
| DSS | Dextran sulfate sodium |
| MCP-1 | Monocyte chemoattractant protein-1 |
| HUVECs | Human umbilical vein endothelial cells |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| TJ | Tight junction |
| ZO-1 | Zonula occludens-1 |
| MTHFR | Methylenetetrahydrofolate reductase |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Status Report on Alcohol and Health 2014; WHO: Geneva, Switzerland, 2014; pp. 14–376. [Google Scholar]
- Boffetta, P.; Hashibe, M. Alcohol and cancer. Lancet Oncol. 2006, 7, 149–156. [Google Scholar] [CrossRef]
- Bagnardi, V.; Blangiardo, M.; La Vecchia, C.; Corrao, G. A meta-analysis of alcohol drinking and cancer risk. Br. J. Cancer 2001, 85, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- La Vecchia, C.; Negri, E.; Cavalieri d’Oro, L.; Franceschi, S. Liver cirrhosis and the risk of primary liver cancer. Eur. J. Cancer Prev. 1998, 7, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis b virus and hepatitis c virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.S.; Jung, Y.K.; Kim, Y.S.; Kim, S.G.; Kim, Y.S.; Lee, J.I.; Lee, J.W.; Kim, Y.S.; Chun, B.C.; Kim, J.H. Effect of alcohol on the development of hepatocellular carcinoma in patients with hepatitis B virus-related cirrhosis: A cross-sectional case-control study. Korean J. Hepatol. 2010, 16, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Millonig, G.; Seitz, H.K. Alcoholic liver disease and hepatitis C: A frequently underestimated combination. World J. Gastroenterol. 2009, 15, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.H.; Jung, W.; Weiderpass, E.; Jang, J.; Hwang, Y.; Ahn, C.; Ko, K.P.; Chang, S.H.; Shin, H.R.; Yoo, K.Y.; et al. Impact of alcohol drinking on gastric cancer development according to Helicobacter pylori infection status. Br. J. Cancer 2015, 113, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Baloch, Z.; He, T.T.; Xia, X. Alcohol consumption and gastric cancer risk: A meta-analysis. Med. Sci. Monit. 2017, 23, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Ferronha, I.; Bastos, A.; Lunet, N. Prediagnosis lifestyle exposures and survival of patients with gastric cancer: systematic review and meta-analysis. Eur. J. Cancer Prev. 2012, 21, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Won, Y.J.; Oh, C.M.; Kong, H.J.; Lee, D.H.; Lee, K.H. Cancer statistics in korea: Incidence, mortality, survival, and prevalence in 2012. Cancer Res. Treat. 2017, 49, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Boutron, M.C.; Faivre, J.; Dop, M.C.; Quipourt, V.; Senesse, P. Tobacco, alcohol, and colorectal tumors: A multistep process. Am. J. Epidemiol. 1995, 141, 1038–1046. [Google Scholar] [PubMed]
- Kune, G.A.; Bannerman, S.; Watson, L.F. Attributable risk for diet, alcohol, and family history in the melbourne colorectal cancer study. Nutr. Cancer 1992, 18, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Kune, G.A.; Vitetta, L. Alcohol consumption and the etiology of colorectal cancer: A review of the scientific evidence from 1957 to 1991. Nutr. Cancer 1992, 18, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Shin, A.; Park, S.K.; Shin, H.R.; Chang, S.H.; Yoo, K.Y. Alcohol drinking, cigarette smoking and risk of colorectal cancer in the korean multi-center cancer cohort. J. Cancer Prev. 2015, 20, 147–152. [Google Scholar] [CrossRef] [PubMed]
- American Institute for Cancer Research; World Cancer Research Fund. Food, Nutrition, Physical Activity and the Prevention of Cancer: A Global Perspective: A Project of World Cancer Research Fund International; American Institute for Cancer Research: Washington, DC, USA, 2007; pp. 157–171. [Google Scholar]
- Park, S.; Shin, H.R.; Lee, B.; Shin, A.; Jung, K.W.; Lee, D.H.; Jee, S.H.; Cho, S.I.; Park, S.K.; Boniol, M.; et al. Attributable fraction of alcohol consumption on cancer using population-based nationwide cancer incidence and mortality data in the republic of korea. BMC Cancer 2014, 14, 420. [Google Scholar] [CrossRef] [PubMed]
- Ehrig, T.; Bosron, W.F.; Li, T.K. Alcohol and aldehyde dehydrogenase. Alcohol Alcohol. 1990, 25, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Hurley, T.D.; Edenberg, H.J. Genes encoding enzymes involved in ethanol metabolism. Alcohol Res. 2012, 34, 339–344. [Google Scholar] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Personal habits and indoor combustions. The Role of Acetaldehyde in Alcohol-Induced Carcinogenesis. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100E, 471. [Google Scholar]
- Salaspuro, M. Acetaldehyde and gastric cancer. J. Dig. Dis. 2011, 12, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Huang, I.Y.; Ikawa, M. Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in orientals. Proc. Natl. Acad. Sci. USA 1984, 81, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Luczak, S.E.; Glatt, S.J.; Wall, T.L. Meta-analyses of ALDH2 and ADH1B with alcohol dependence in Asians. Psychol. Bull. 2006, 132, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Yabushita, H.; Kanaly, R.A.; Shibutani, S.; Yokoyama, A. Increased DNA damage in ALDH2-deficient alcoholics. Chem. Res. Toxicol. 2006, 19, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol Res. 2013, 35, 6–16. [Google Scholar] [PubMed]
- Albano, E. Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Mol. Aspects Med. 2008, 29, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S.; Kumar, S. Chronic effects of ethanol and/or darunavir/ritonavir on U937 monocytic cells: Regulation of cytochrome P450 and antioxidant enzymes, oxidative stress, and cytotoxicity. Alcohol. Clin. Exp. Res. 2016, 40, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol. 2014, 3, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer. 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Van Engeland, M.; Weijenberg, M.P.; Roemen, G.M.; Brink, M.; de Bruine, A.P.; Goldbohm, R.A.; van den Brandt, P.A.; Baylin, S.B.; de Goeij, A.F.; Herman, J.G. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: The Netherlands cohort study on diet and cancer. Cancer. Res. 2003, 63, 3133–3137. [Google Scholar] [PubMed]
- Parlesak, A.; Billinger, M.H.; Bode, C.; Bode, J.C. Gastric alcohol dehydrogenase activity in man: Influence of gender, age, alcohol consumption and smoking in a caucasian population. Alcohol Alcohol. 2002, 37, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, A.; Sasazuki, S.; Matsuo, K.; Ito, H.; Sawada, N.; Shimazu, T.; Yamaji, T.; Iwasaki, M.; Inoue, M.; Tsugane, S.; et al. Genetic polymorphisms of ADH1B, ADH1C and ALDH2, alcohol consumption, and the risk of gastric cancer: The Japan Public Health Center-based prospective study. Carcinogenesis 2015, 36, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.Y. Cancer prevention in the Asia Pacific region. Asian Pac. J. Cancer Prev. 2010, 11, 839–844. [Google Scholar] [PubMed]
- Wang, H.L.; Zhou, P.Y.; Liu, P.; Zhang, Y. ALDH2 and ALDH1 genetic polymorphisms may contribute to the risk of gastric cancer: A meta-analysis. PLoS ONE 2014, 9, e88779. [Google Scholar]
- Nagayoshi, H.; Matsumoto, A.; Nishi, R.; Kawamoto, T.; Ichiba, M.; Matsuda, T. Increased formation of gastric N2-ethylidene-2′-deoxyguanosine DNA adducts in aldehyde dehydrogenase-2 knockout mice treated with ethanol. Mutat. Res. 2009, 673, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, T.; Salaspuro, M. Aldehyde dehydrogenases of the rat colon: Comparison with other tissues of the alimentary tract and the liver. Alcohol. Clin. Exp. Res. 1996, 20, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Muramatsu, T.; Ohmori, T.; Yokoyama, T.; Okuyama, K.; Takahashi, H.; Hasegawa, Y.; Higuchi, S.; Maruyama, K.; Shirakura, K.; et al. Alcohol-related cancers and aldehyde dehydrogenase-2 in japanese alcoholics. Carcinogenesis 1998, 19, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Crous-Bou, M.; Rennert, G.; Cuadras, D.; Salazar, R.; Cordero, D.; Saltz Rennert, H.; Lejbkowicz, F.; Kopelovich, L.; Monroe Lipkin, S.; Bernard Gruber, S.; et al. Polymorphisms in alcohol metabolism genes ADH1B and ALDH2, alcohol consumption and colorectal cancer. PLoS ONE 2013, 8, e80158. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Wakai, K.; Hirose, K.; Ito, H.; Saito, T.; Suzuki, T.; Kato, T.; Hirai, T.; Kanemitsu, Y.; Hamajima, H.; et al. A gene-gene interaction between ALDH2 Glu487lys and ADH2 His47arg polymorphisms regarding the risk of colorectal cancer in Japan. Carcinogenesis 2006, 27, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Khalsa, J.; Serrano, J. Mechanisms of alcohol-associated cancers: Introduction and summary of the symposium. Alcohol 2005, 35, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Simanowski, U.A.; Garzon, F.T.; Rideout, J.M.; Peters, T.J.; Koch, A.; Berger, M.R.; Einecke, H.; Maiwald, M. Possible role of acetaldehyde in ethanol-related rectal cocarcinogenesis in the rat. Gastroenterology 1990, 98, 406–413. [Google Scholar] [CrossRef]
- Cuomo, R.; Andreozzi, P.; Zito, F.P. Alcoholic beverages and carbonated soft drinks: Consumption and gastrointestinal cancer risks. Cancer Treat. Res. 2014, 159, 97–120. [Google Scholar] [PubMed]
- Maejima, R.; Iijima, K.; Kaihovaara, P.; Hatta, W.; Koike, T.; Imatani, A.; Shimosegawa, T.; Salaspuro, M. Effects of ALDH2 genotype, PPI treatment and l-cysteine on carcinogenic acetaldehyde in gastric juice and saliva after intragastric alcohol administration. PLoS ONE 2015, 10, e0120397. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Roine, R.P.; Salmela, K.S.; Hook-Nikanne, J.; Kosunen, T.U.; Salaspuro, M. Alcohol dehydrogenase mediated acetaldehyde production by Helicobacter pylori—A possible mechanism behind gastric injury. Life Sci. 1992, 51, 1333–1337. [Google Scholar] [CrossRef]
- Salaspuro, M. Interrelationship between alcohol, smoking, acetaldehyde and cancer. Novartis Found. Symp. 2007, 285, 80–89. [Google Scholar] [PubMed]
- Seeman, J.I.; Dixon, M.; Haussmann, H.J. Acetaldehyde in mainstream tobacco smoke: Formation and occurrence in smoke and bioavailability in the smoker. Chem. Res. Toxicol. 2002, 15, 1331–1350. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.X.; Liang, H.; Chai, J.; Cheng, J.; Feng, R.; Chen, P.L.; Geng, Q.Q.; Shen, X.R.; Wang, D.B. Association of risk of gastric cancer and consumption of tobacco, alcohol and tea in the Chinese population. Asian Pac. J. Cancer Prev. 2014, 15, 8765–8774. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Oh, T.Y.; Kim, Y.K.; Baik, J.H.; Therefore, S.; Hahm, K.B.; Surh, Y.J. Protective effects of green tea polyphenol extracts against ethanol-induced gastric mucosal damages in rats: Stress-responsive transcription factors and MAP kinases as potential targets. Mutat. Res. 2005, 579, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Vakevainen, S.; Mentula, S.; Nuutinen, H.; Salmela, K.S.; Jousimies-Somer, H.; Farkkila, M.; Salaspuro, M. Ethanol-derived microbial production of carcinogenic acetaldehyde in achlorhydric atrophic gastritis. Scand. J. Gastroenterol. 2002, 37, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Yokoyama, T.; Omori, T.; Matsushita, S.; Mizukami, T.; Takahashi, H.; Higuchi, S.; Maruyama, K.; Ishii, H.; Hibi, T. Helicobacter pylori, chronic atrophic gastritis, inactive aldehyde dehydrogenase-2, macrocytosis and multiple upper aerodigestive tract cancers and the risk for gastric cancer in alcoholic Japanese men. J. Gastroenterol. Hepatol. 2007, 22, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.M.; Souliotis, V.L.; Chhabra, S.K.; Moskal, T.J.; Harbaugh, S.D.; Kyrtopoulos, S.A. N-nitrosodimethylamine-derived O6-methylguanine in DNA of monkey gastrointestinal and urogenital organs and enhancement by ethanol. Int. J. Cancer 1996, 66, 130–134. [Google Scholar] [CrossRef]
- Iishi, H.; Tatsuta, M.; Baba, M.; Taniguchi, H. Promotion by ethanol of gastric carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine in Wistar rats. Br. J. Cancer 1989, 59, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Jokelainen, K.; Roine, R.P.; Vaananen, H.; Farkkila, M.; Salaspuro, M. In Vitro acetaldehyde formation by human colonic bacteria. Gut 1994, 35, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M. Bacteriocolonic pathway for ethanol oxidation: Characteristics and implications. Ann. Med. 1996, 28, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Jokelainen, K.; Siitonen, A.; Jousimies-Somer, H.; Nosova, T.; Heine, R.; Salaspuro, M. In Vitro alcohol dehydrogenase-mediated acetaldehyde production by aerobic bacteria representing the normal colonic flora in man. Alcohol. Clin. Exp. Res. 1996, 20, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Tsuruya, A.; Kuwahara, A.; Saito, Y.; Yamaguchi, H.; Tenma, N.; Inai, M.; Takahashi, S.; Tsutsumi, E.; Suwa, Y.; Totsuka, Y.; et al. Major Anaerobic Bacteria Responsible for the Production of Carcinogenic Acetaldehyde from Ethanol in the Colon and Rectum. Alcohol Alcohol. 2016, 51, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Homann, N.; Tillonen, J.; Salaspuro, M. Microbially produced acetaldehyde from ethanol may increase the risk of colon cancer via folate deficiency. Int. J. Cancer 2000, 86, 169–173. [Google Scholar] [CrossRef]
- Nosova, T.; Jousimies-Somer, H.; Kaihovaara, P.; Jokelainen, K.; Heine, R.; Salaspuro, M. Characteristics of alcohol dehydrogenases of certain aerobic bacteria representing human colonic flora. Alcohol. Clin. Exp. Res. 1997, 21, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Amin, P.B.; Diebel, L.N.; Liberati, D.M. Dose-dependent effects of ethanol and E. coli on gut permeability and cytokine production. J. Surg. Res. 2009, 157, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Wang, X.D. The role of cytochrome P450 2E1 in ethanol-mediated carcinogenesis. Subcell. Biochem. 2013, 67, 131–143. [Google Scholar] [PubMed]
- Hakkak, R.; Korourian, S.; Ronis, M.J.; Ingelman-Sundberg, M.; Badger, T.M. Effects of diet and ethanol on the expression and localization of cytochromes P450 2E1 and P450 2C7 in the colon of male rats. Biochem. Pharmacol. 1996, 51, 61–69. [Google Scholar] [CrossRef]
- Heit, C.; Dong, H.; Chen, Y.; Shah, Y.M.; Thompson, D.C.; Vasiliou, V. Transgenic mouse models for alcohol metabolism, toxicity, and cancer. Adv. Exp. Med. Biol. 2015, 815, 375–387. [Google Scholar] [PubMed]
- Hayashi, N.; Tsutsumi, M.; Fukura, M.; Yano, H.; Tsuchishima, M.; Takase, S. Effect of chronic dietary ethanol consumption on colonic cancer in rats induced by 1,1-dimethylhydrazine. Alcohol. Clin. Exp. Res. 2007, 31, S72–S76. [Google Scholar] [CrossRef] [PubMed]
- Sohn, O.S.; Fiala, E.S.; Requeijo, S.P.; Weisburger, J.H.; Gonzalez, F.J. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 2001, 61, 8435–8440. [Google Scholar] [PubMed]
- Megaraj, V.; Ding, X.; Fang, C.; Kovalchuk, N.; Zhu, Y.; Zhang, Q.Y. Role of hepatic and intestinal p450 enzymes in the metabolic activation of the colon carcinogen azoxymethane in mice. Chem. Res. Toxicol. 2014, 27, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Tanaka, T.; Sugie, S.; Shinoda, T.; Kato, K.; Tamaya, T.; Mori, H. Enhancing effect of ethanol or sake on methylazoxymethanol acetate-initiated large bowel carcinogenesis in ACI/N rats. Nutr. Cancer. 1991, 15, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Chaudhry, K.K.; Mir, H.; Gangwar, R.; Yadav, N.; Manda, B.; Meena, A.S.; Rao, R. Chronic ethanol feeding promotes azoxymethane and dextran sulfate sodium-induced colonic tumorigenesis potentially by enhancing mucosal inflammation. BMC Cancer 2016, 16, 189. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, S.; Qin, J.; Lv, Y.; Ma, X.; Liu, C. Ethanol upregulates iNOS expression in colon through activation of nuclear factor-κB in rats. Alcohol. Clin. Exp. Res. 2010, 34, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.C.; Vaz, N.M.; Faria, A.M. Ethanol-induced colitis prevents oral tolerance induction in mice. Braz. J. Med. Biol. Res. 2003, 36, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hitron, J.A.; Wise, J.T.; Son, Y.O.; Roy, R.V.; Kim, D.; Dai, J.; Pratheeshkumar, P.; Zhang, Z.; Xu, M.; et al. Ethanol enhances arsenic-induced cyclooxygenase-2 expression via both NFAT and NF-κB signalings in colorectal cancer cells. Toxicol. Appl. Pharmacol. 2015, 288, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Qi, Y.; Chen, L.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Shi, X.; Luo, J. Role of MCP-1 in alcohol-induced aggressiveness of colorectal cancer cells. Mol. Carcinog. 2016, 55, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miki, C.; Okugawa, Y.; Toiyama, Y.; Inoue, Y.; Kusunoki, M. Decreased expression of monocyte chemoattractant protein-1 predicts poor prognosis following curative resection of colorectal cancer. Dis. Colon Rectum 2008, 51, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Mestdagt, M.; Polette, M.; Buttice, G.; Noel, A.; Ueda, A.; Foidart, J.M.; Gilles, C. Transactivation of MCP-1/CCL2 by β-catenin/TCF-4 in human breast cancer cells. Int. J. Cancer 2006, 118, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Keshavarzian, A. Alcohol stimulates activation of Snail, epidermal growth factor receptor signaling, and biomarkers of epithelial-mesenchymal transition in colon and breast cancer cells. Alcohol. Clin. Exp. Res. 2010, 34, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.K. Commentary: Acetaldehyde and epithelial-to-mesenchymal transition in colon. Alcohol. Clin. Exp. Res. 2014, 38, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.; Cullen, J.P.; Cahill, P.A.; Redmond, E.M. Ethanol stimulates endothelial cell angiogenic activity via a Notch- and angiopoietin-1-dependent pathway. Cardiovasc. Res. 2008, 79, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Son, Y.O.; Ding, S.; Wang, X.; Hitron, J.A.; Budhraja, A.; Lee, J.C.; Lin, Q.; Poyil, P.; Zhang, Z.; et al. Ethanol enhances tumor angiogenesis in vitro induced by low-dose arsenic in colon cancer cells through hypoxia-inducible factor 1 α pathway. Toxicol. Sci. 2012, 130, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Ammendola, M.; Patruno, R.; Sacco, R.; Marech, I.; Sammarco, G.; Zuccala, V.; Luposella, M.; Zizzo, N.; Gadaleta, C.; Porcelli, M.; et al. Mast cells positive to tryptase and tumour-associated macrophages correlate with angiogenesis in locally advanced colorectal cancer patients undergone to surgery. Expert Opin. Ther. Targets 2016, 20, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Wimberly, A.L.; Forsyth, C.B.; Khan, M.W.; Pemberton, A.; Khazaie, K.; Keshavarzian, A. Ethanol-induced mast cell-mediated inflammation leads to increased susceptibility of intestinal tumorigenesis in the APC Delta468 min mouse model of colon cancer. Alcohol. Clin. Exp. Res. 2013, 37, E199–E208. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Simanowski, U.A.; Homann, N.; Waldherr, R. Cell proliferation and its evaluation in the colorectal mucosa: Effect of ethanol. Z. Gastroenterol. 1998, 36, 645–655. [Google Scholar] [PubMed]
- Vincon, P.; Wunderer, J.; Simanowski, U.A.; Koll, M.; Preedy, V.R.; Peters, T.J.; Werner, J.; Waldherr, R.; Seitz, H.K. Inhibition of alcohol-associated colonic hyperregeneration by α-tocopherol in the rat. Alcohol. Clin. Exp. Res. 2003, 27, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Simanowski, U.A.; Stickel, F.; Maier, H.; Gartner, U.; Seitz, H.K. Effect of alcohol on gastrointestinal cell regeneration as a possible mechanism in alcohol-associated carcinogenesis. Alcohol 1995, 12, 111–115. [Google Scholar] [CrossRef]
- Roy, H.K.; Gulizia, J.M.; Karolski, W.J.; Ratashak, A.; Sorrell, M.F.; Tuma, D. Ethanol promotes intestinal tumorigenesis in the MIN mouse. Multiple intestinal neoplasia. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1499–1502. [Google Scholar]
- Aradottir, S.; Moller, K.; Alling, C. Phosphatidylethanol formation and degradation in human and rat blood. Alcohol Alcohol. 2004, 39, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974. [Google Scholar] [PubMed]
- Pannequin, J.; Delaunay, N.; Darido, C.; Maurice, T.; Crespy, P.; Frohman, M.A.; Balda, M.S.; Matter, K.; Joubert, D.; Bourgaux, J.F.; et al. Phosphatidylethanol accumulation promotes intestinal hyperplasia by inducing ZONAB-mediated cell density increase in response to chronic ethanol exposure. Mol. Cancer Res. 2007, 5, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.T.; Stover, P.J. Folate-mediated one-carbon metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [PubMed]
- Coppede, F.; Migheli, F.; Lopomo, A.; Failli, A.; Legitimo, A.; Consolini, R.; Fontanini, G.; Sensi, E.; Servadio, A.; Seccia, M.; et al. Gene promoter methylation in colorectal cancer and healthy adjacent mucosa specimens: Correlation with physiological and pathological characteristics, and with biomarkers of one-carbon metabolism. Epigenetics 2014, 9, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Kardon, T.; Noel, G.; Vertommen, D.; Schaftingen, E.V. Identification of the gene encoding hydroxyacid-oxoacid transhydrogenase, an enzyme that metabolizes 4-hydroxybutyrate. FEBS Lett. 2006, 580, 2347–2350. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Saito, K.; Lwin, H.; Yoshiike, N.; Yamamoto, A.; Matsushita, Y.; Date, C.; Tanaka, H. Epidemiological evidence that acetaldehyde plays a significant role in the development of decreased serum folate concentration and elevated mean corpuscular volume in alcohol drinkers. Alcohol. Clin. Exp. Res. 2005, 29, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E. Alcohol, one-carbon metabolism, and colorectal cancer: Recent insights from molecular studies. J. Nutr. 2004, 134, 2475S–2481S. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, H.-K.; Lee, J.Y. Molecular Basis of Alcohol-Related Gastric and Colon Cancer. Int. J. Mol. Sci. 2017, 18, 1116. https://doi.org/10.3390/ijms18061116
Na H-K, Lee JY. Molecular Basis of Alcohol-Related Gastric and Colon Cancer. International Journal of Molecular Sciences. 2017; 18(6):1116. https://doi.org/10.3390/ijms18061116
Chicago/Turabian StyleNa, Hye-Kyung, and Ja Young Lee. 2017. "Molecular Basis of Alcohol-Related Gastric and Colon Cancer" International Journal of Molecular Sciences 18, no. 6: 1116. https://doi.org/10.3390/ijms18061116
APA StyleNa, H.-K., & Lee, J. Y. (2017). Molecular Basis of Alcohol-Related Gastric and Colon Cancer. International Journal of Molecular Sciences, 18(6), 1116. https://doi.org/10.3390/ijms18061116