
It Is Imperative to Establish a Pellucid Definition of Chimeric RNA and to Clear Up a Lot of Confusion in the Relevant Research

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
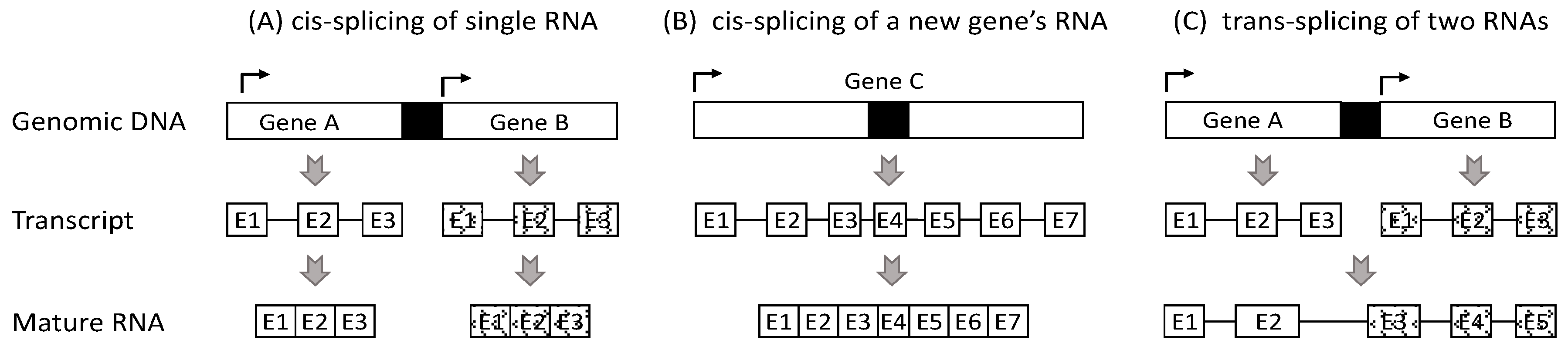
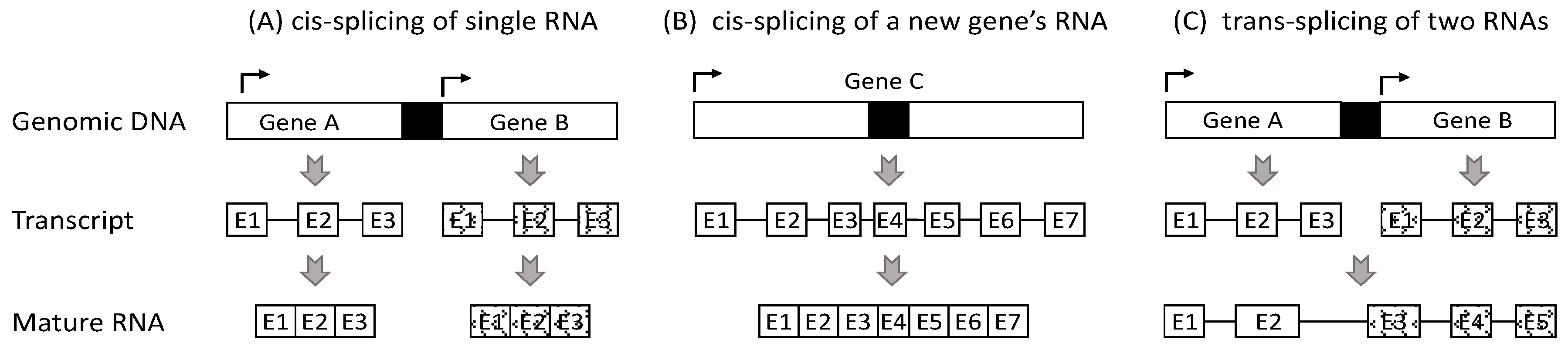
2. Currently, Chimeric RNAs Are Thought to Be Derived from Three Mechanisms
3. Most RNAs from Two Neighboring Genes Should Not Be Deemed as Chimeras
4. There Are Different Ways to Catalog Chimeric RNAs
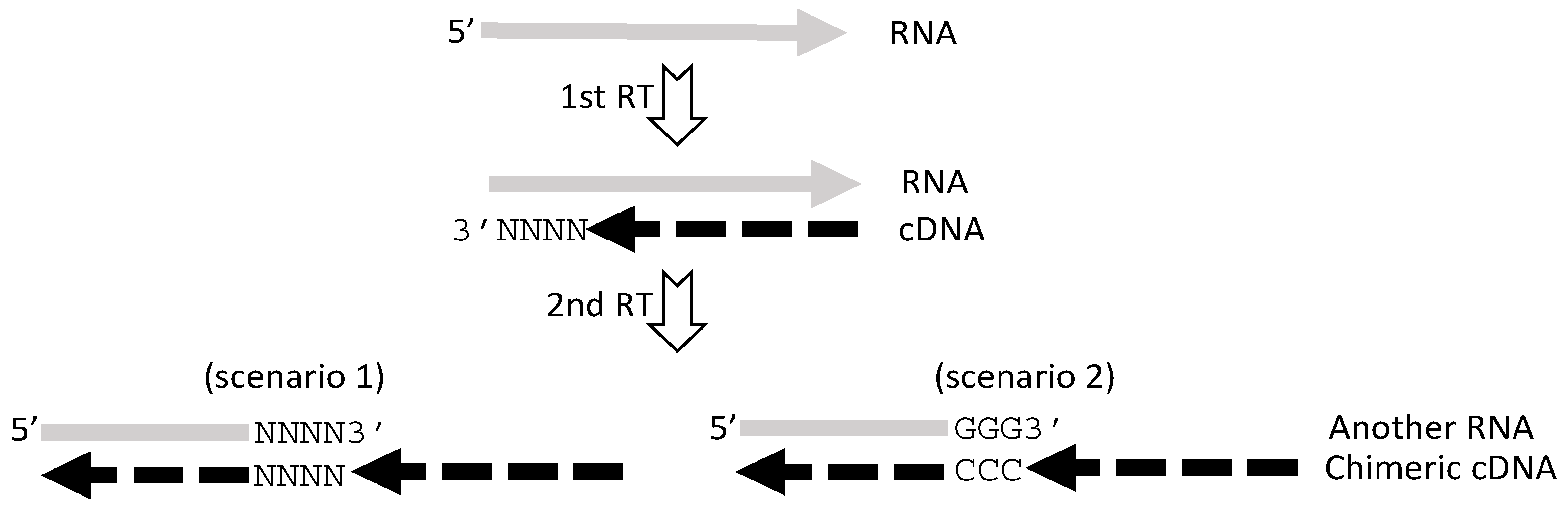
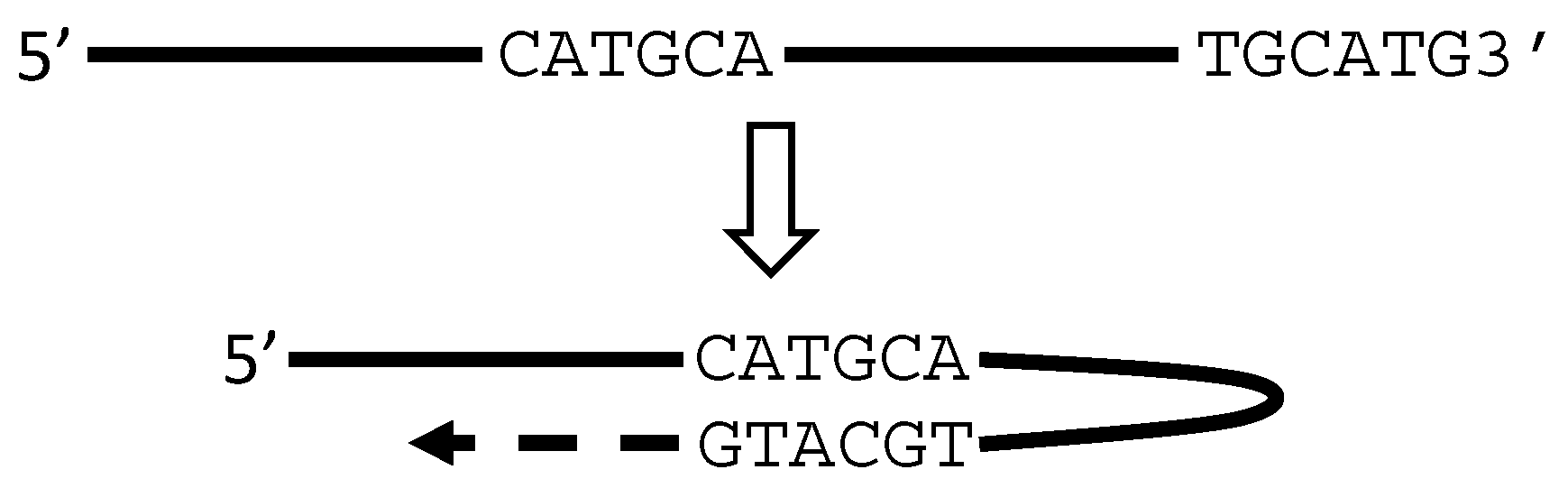
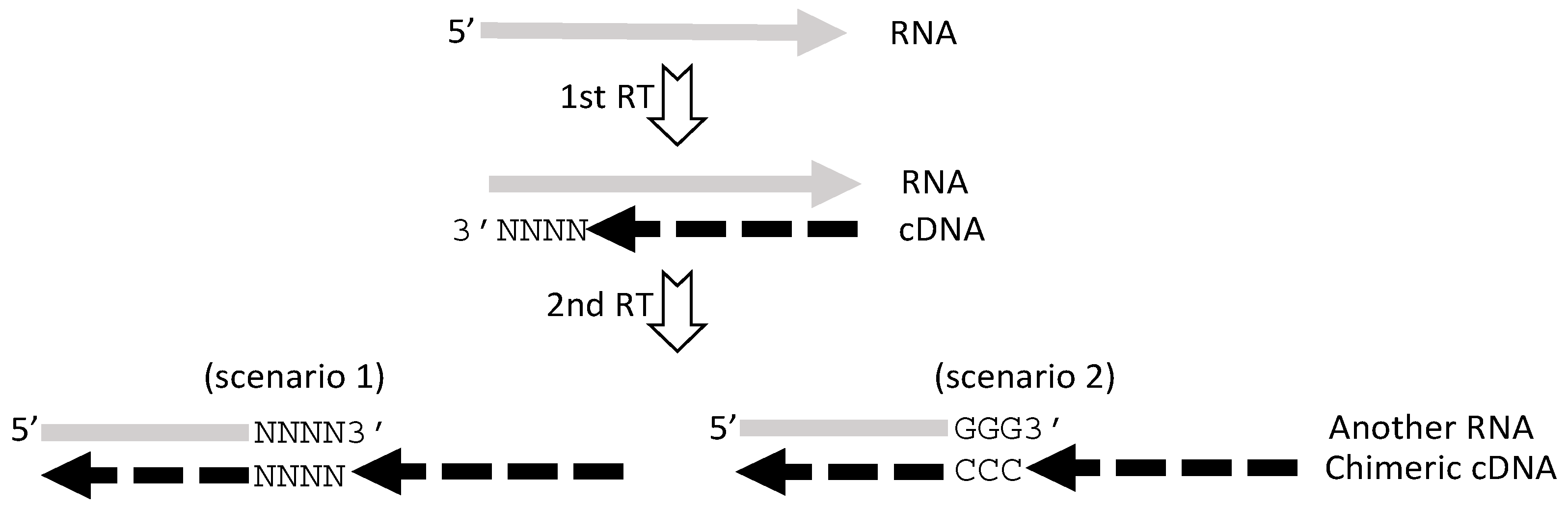
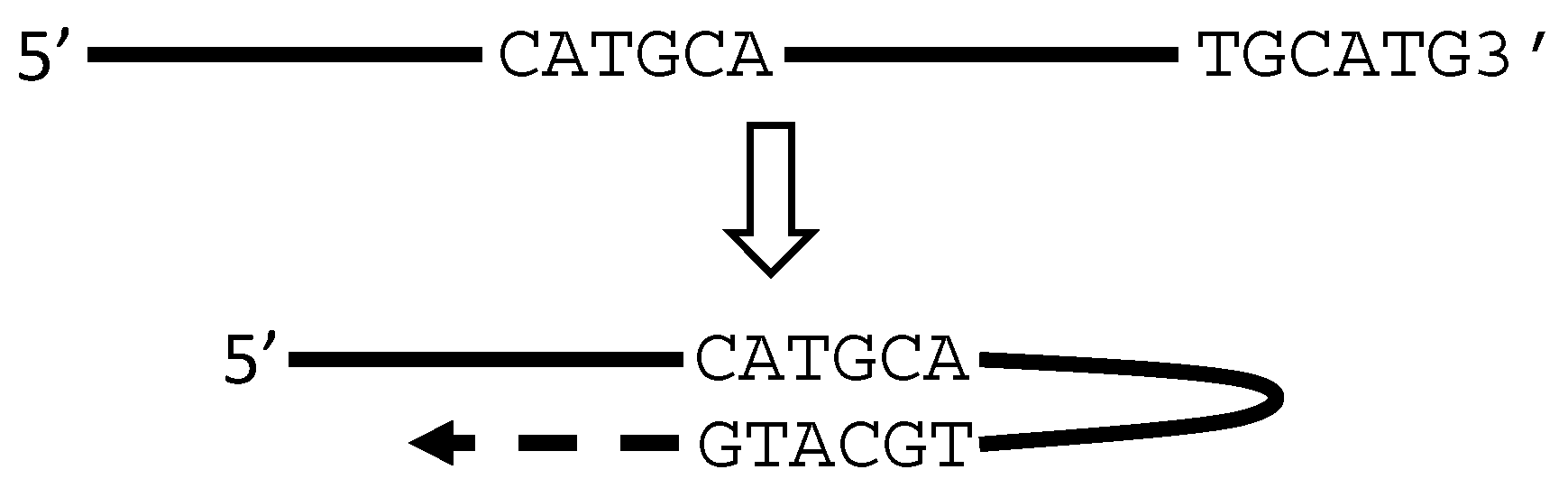
5. RT or PCR Creates Many Artifacts that Fabricate “Trans-Splicing”
6. There Are Other Artifacts with Unknown Mechanisms
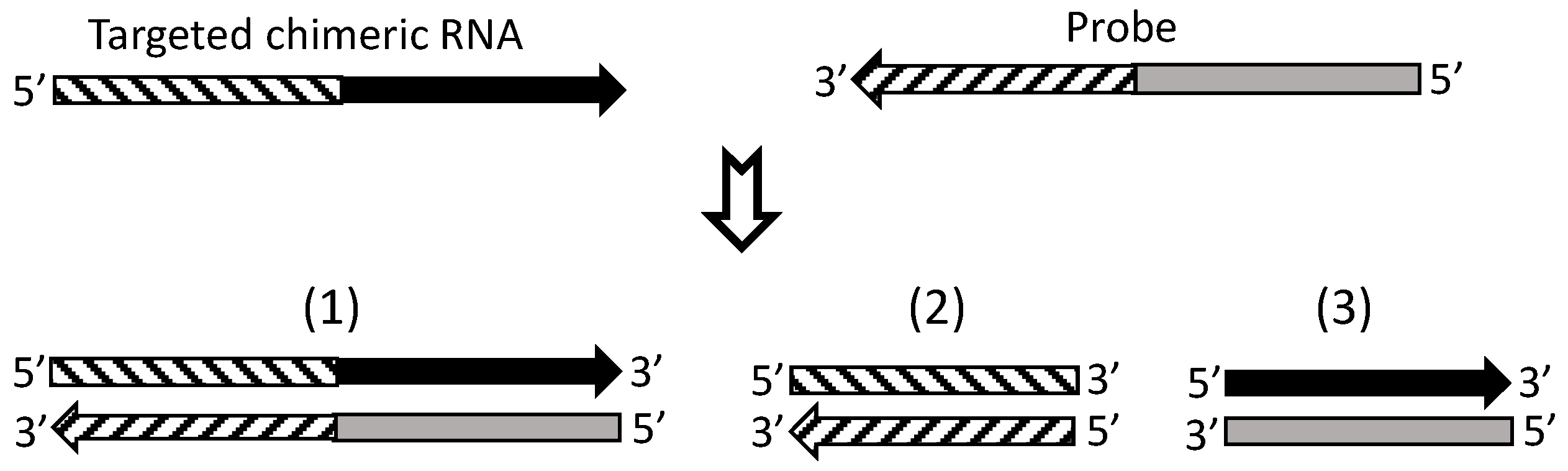
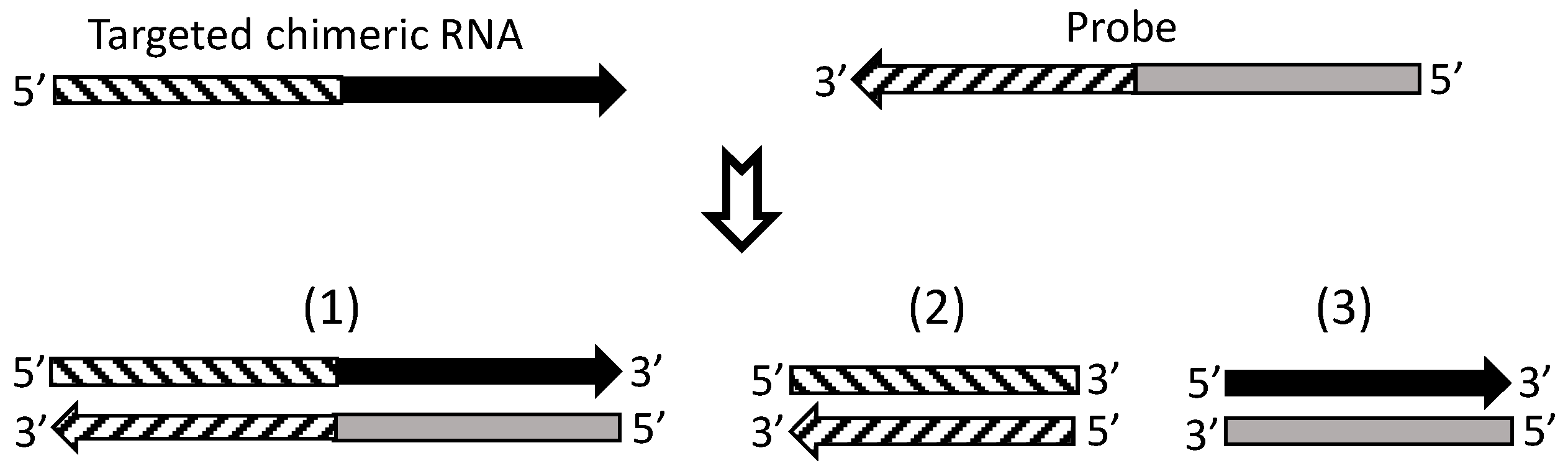
7. Currently, cDNA Protection Assay Is the Best Approach for Verification of Chimeric RNAs
8. We Propose New Criteria to Classify Chimeric RNAs
9. There Are Other Flaws, Constraints and Understudied Tasks in Chimeric RNA Research
- There are still thousands of putative chimeric RNAs that contain sequences of two genes on two different chromosomes [27]. For most of them, it is still unclear whether they have a fusion gene as the genomic basis, and thus it is still unclear whether they are genuine chimeric RNAs by our definition.
- Many RNAs are claimed to be derived from a readthrough mechanism but actually lack concrete experimental evidence proving the readthrough, since detection of a long pre-RNA transcript is one thing but causally-linking it to the mature RNA that contains two-gene sequences is another thing. Therefore, the possibility for them to be derived from a trans-splicing mechanism and thus to be genuine chimeric RNAs still exists, which needs to be ruled in or out.
- Some chimeric RNAs have been reported to be recurrent, usually in cancer. However, often it is not clear whether the “recurrence” means that it is exactly the same chimeric sequence that appears repeatedly, or it means that the same two genes produce chimeric RNAs that are highly similar but still differ slightly in sequence. This question is raised because we sometimes have inquired of peers about the “recurrent” chimeric RNAs they observed and knew that the repeatedly detected chimeras differed slightly among each other in sequence. The reason for the small difference in sequence should be determined.
- Technical detail is insufficiently discussed in many published studies of chimeric RNAs, especially about how possible it is that the chimeras are spurious. In our opinion, when describing a new chimeric RNA, more-than-usual technical detail needs to be furnished and, moreover, it needs to address: (1) whether it has a fusion gene as a genomic basis; and (2) whether it contains an SHS or a gap sequence that highly indicates an artifact.
- Some chimeras encode proteins that share part of the sequence with the proteins produced from one or both of the parental genes. Determining whether these chimeras have protein products is technically difficult in most cases, due to the lack of fusion-protein specific primary antibodies, which in turn is due to a technical constraint in raising such antibodies. Antibodies raised via a traditional approach will likely recognize proteins from one or both parental genes. Theoretically, antibodies for the proteins from one of the two parental genes may be available and be used in western blotting to distinguish fusion-proteins from the proteins of the parental genes by their difference in molecular weight. However, since most genes produce multiple protein isoforms [62], as one of us has shown for protein products of many genes [133,134,135,136,137,138], in practice it is difficult to use these antibodies to corroborate the true existence of protein product(s) of a chimeric RNA, especially with an immunohistochemical staining approach that does not allow us to distinguish one protein from the others by their molecular weights.
10. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jia, Y.; Chen, L.; Ma, Y.; Zhang, J.; Xu, N.; Liao, D.J. To Know How a Gene Works, We Need to Redefine It First but then, More Importantly, to Let the Cell Itself Decide How to Transcribe and Process Its RNAs. Int. J. Biol. Sci. 2015, 11, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wu, J.M.; Bi, A.D.; Ou-Yang, Y.C.; Shen, H.H.; Chirn, G.W.; Zhou, J.H.; Weiss, E.; Holman, E.P.; Liao, D.J. Possible Formation of Mitochondrial-RNA Containing Chimeric or Trimeric RNA Implies a Post-Transcriptional and Post-Splicing Mechanism for RNA Fusion. PLoS ONE 2013, 8, e77016. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yuan, C.; Zellmer, L.; Liu, S.; Xu, N.; Liao, D.J. Hypothesis: Artifacts, Including Spurious Chimeric RNAs with a Short Homologous Sequence, Caused by Consecutive Reverse Transcriptions and Endogenous Random Primers. J. Cancer 2015, 6, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Yang, W.; Ouyang, Y.; Chen, L.; Jiang, H.; Liao, Y.; Liao, D.J. Two RNAs or DNAs May Artificially Fuse Together at a Short Homologous Sequence (SHS) during Reverse Transcription or Polymerase Chain Reactions, and Thus Reporting an SHS-Containing Chimeric RNA Requires Extra Caution. PLoS ONE 2016, 11, e0154855. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liao, J.; Zheng, X.; Shen, H. Chimeric RNAs as potential biomarkers for tumor diagnosis. BMB Rep. 2012, 45, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yang, S.; Zhao, W.; Tang, Z.; Zhang, T.; Li, K. Identification and analysis of pig chimeric mRNAs using RNA sequencing data. BMC Genom. 2012, 13, 429. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA trans-Splicing in Vertebrates. Genome Biol. Evol. 2016, 8, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, L.; Jiang, H.; Wang, W. Short homologous sequences are strongly associated with the generation of chimeric RNAs in eukaryotes. J. Mol. Evol. 2009, 68, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Palanisamy, N.; Brenner, J.C.; Cao, X.; Kalyana-Sundaram, S.; Luo, S.; Khrebtukova, I.; Barrette, T.R.; Grasso, C.; Yu, J.; et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 12353–12358. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Belizario, J.E. The humankind genome: From genetic diversity to the origin of human diseases. Genome 2013, 56, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Genomics. ENCODE project writes eulogy for junk DNA. Science 2012, 337, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Skipper, M.; Dhand, R.; Campbell, P. Presenting ENCODE. Nature 2012, 489, 45. [Google Scholar] [CrossRef] [PubMed]
- Finta, C.; Warner, S.C.; Zaphiropoulos, P.G. Intergenic mRNAs. Minor gene products or tools of diversity? Histol. Histopathol. 2002, 17, 677–682. [Google Scholar] [PubMed]
- Gerstein, M.B.; Bruce, C.; Rozowsky, J.S.; Zheng, D.; Du, J.; Korbel, J.O.; Emanuelsson, O.; Zhang, Z.D.; Weissman, S.; Snyder, M. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007, 17, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Portin, P. The elusive concept of the gene. Hereditas 2009, 146, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, L.; Jia, Q.; Dou, X.; Xu, N.; Liao, D.J. The well-accepted notion that gene amplification contributes to increased expression still remains, after all these years, a reasonable but unproven assumption. J. Carcinog. 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Hungerford, D.A. The philadelphia chromosome and some others. Ann. Intern. Med. 1964, 61, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut 1962, 8, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Koretzky, G.A. The legacy of the Philadelphia chromosome. J. Clin. Investig. 2007, 117, 2030–2032. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.H.D. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497. [Google Scholar]
- Bennour, A.; Ouahchi, I.; Moez, M.; Elloumi, M.; Khelif, A.; Saad, A.; Sennana, H. Comprehensive analysis of BCR/ABL variants in chronic myeloid leukemia patients using multiplex RT-PCR. Clin. Lab. 2012, 58, 433–439. [Google Scholar] [PubMed]
- Ma, W.; Kantarjian, H.; Yeh, C.H.; Zhang, Z.J.; Cortes, J.; Albitar, M. BCR-ABL truncation due to premature translation termination as a mechanism of resistance to kinase inhibitors. Acta Haematol. 2009, 121, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Tayebwa, J. Evolving techniques for gene fusion detection in soft tissue tumours. Histopathology 2014, 64, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Ernst, T.; Fiebig, A.; Collins, A.; Grand, F.; Erben, P.; Reiter, A.; Schreiber, S.; Cross, N.C. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica 2010, 95, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ise, T.; Ha, D.; Saint Fleur, A.; Hahn, Y.; Liu, X.F.; Nagata, S.; Lee, B.; Bera, T.K.; Pastan, I. Evolution and expression of chimeric POTE-actin genes in the human genome. Proc. Natl. Acad. Sci. USA 2006, 103, 17885–17890. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Bera, T.K.; Liu, L.J.; Pastan, I. A primate-specific POTE-actin fusion protein plays a role in apoptosis. Apoptosis 2009, 14, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Ise, T.; Das, S.; Nagata, S.; Maeda, H.; Lee, Y.; Onda, M.; Anver, M.R.; Bera, T.K.; Pastan, I. Expression of POTE protein in human testis detected by novel monoclonal antibodies. Biochem. Biophys. Res. Commun. 2008, 365, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Babushok, D.V.; Ohshima, K.; Ostertag, E.M.; Chen, X.; Wang, Y.; Mandal, P.K.; Okada, N.; Abrams, C.S.; Kazazian, H.H., Jr. A novel testis ubiquitin-binding protein gene arose by exon shuffling in hominoids. Genome Res. 2007, 17, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Igarashi, K. Inference for the initial stage of domain shuffling: Tracing the evolutionary fate of the PIPSL retrogene in hominoids. Mol. Biol. Evol. 2010, 27, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Gingeras, T.R. Implications of chimaeric non-co-linear transcripts. Nature 2009, 461, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Lasda, E.L.; Blumenthal, T. Trans-splicing. Wiley Interdiscip. Rev. RNA 2011, 2, 417–434. [Google Scholar] [CrossRef] [PubMed]
- Glanz, S.; Kuck, U. Trans-splicing of organelle introns—A detour to continuous RNAs. Bioessays 2009, 31, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Glanz, S.; Bunse-Grassmann, A.; Kruse, O.; Kuck, U. RNA trans-splicing: Identification of components of a putative chloroplast spliceosome. Eur. J. Cell Biol. 2010, 89, 932–939. [Google Scholar] [CrossRef]
- Rowley, J.D.; Blumenthal, T. Medicine. The cart before the horse. Science 2008, 321, 1302–1304. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, J.; Ma, X.; Sklar, J. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle 2009, 8, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Wei, Y.; Kang, Y.; Landweber, L.F. Detection of a common chimeric transcript between human chromosomes 7 and 16. Biol. Direct 2012, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Lehman, M.L.; Dinger, M.E.; Hendy, S.C.; Mercer, T.R.; Seim, I.; Lawrence, M.G.; Mattick, J.S.; Clements, J.A.; Nelson, C.C. A variant of the KLK4 gene is expressed as a cis sense-antisense chimeric transcript in prostate cancer cells. RNA 2010, 16, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, X.N.; Yang, L.; Hu, G.J.; Lu, M.; Xiong, Y.; Yang, X.Y.; Chang, C.C.; Song, B.L.; Chang, T.Y.; et al. RNA secondary structures located in the interchromosomal region of human ACAT1 chimeric mRNA are required to produce the 56-kDa isoform. Cell Res. 2008, 18, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.J.; Chen, J.; Zhao, X.N.; Xu, J.J.; Guo, D.Q.; Lu, M.; Zhu, M.; Xiong, Y.; Li, Q.; Chang, C.C.; et al. Production of ACAT1 56-kDa isoform in human cells via trans-splicing involving the ampicillin resistance gene. Cell Res. 2013, 23, 1007–1024. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, H.; Xin, D.; Cheng, H.; Zhou, R. A novel ncRNA gene from mouse chromosome 5 trans-splices with Dmrt1 on chromosome 19. Biochem. Biophys. Res. Commun. 2010, 400, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Noda, T. Genomic organization of the mouse Msh4 gene producing bicistronic, chimeric and antisense mRNA. Gene 2004, 342, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Gabler, M.; Volkmar, M.; Weinlich, S.; Herbst, A.; Dobberthien, P.; Sklarss, S.; Fanti, L.; Pimpinelli, S.; Kress, H.; Reuter, G.; et al. Trans-splicing of the mod(mdg4) complex locus is conserved between the distantly related species Drosophila melanogaster and D. virilis. Genetics 2005, 169, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Labrador, M.; Mongelard, F.; Plata-Rengifo, P.; Baxter, E.M.; Corces, V.G.; Gerasimova, T.I. Protein encoding by both DNA strands. Nature 2001, 409, 1000. [Google Scholar] [CrossRef] [PubMed]
- Janz, S.; Potter, M.; Rabkin, C.S. Lymphoma- and leukemia-associated chromosomal translocations in healthy individuals. Genes Chromosom. Cancer 2003, 36, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.S.; Chakalova, L.; Mitchell, J.A.; Horton, A.; Wood, A.L.; Bolland, D.J.; Corcoran, A.E.; Fraser, P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007, 5, e192. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.R.; Mushinski, E.B.; Williams, J.A.; Hausner, P.F. Immunoglobulin/Myc recombinations in murine Peyer’s patch follicles. Genes Chromosom. Cancer 1997, 20, 1–8. [Google Scholar] [CrossRef]
- Horiuchi, T.; Aigaki, T. Alternative trans-splicing: A novel mode of pre-mRNA processing. Biol. Cell 2006, 98, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Zaphiropoulos, P.G. Trans-splicing in Higher Eukaryotes: Implications for Cancer Development? Front. Genet. 2011, 2, 92. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, L.; Yu, B.; Zellmer, L.; Xu, N.; Liao, D.J. Learning about the Importance of Mutation Prevention from Curable Cancers and Benign Tumors. J. Cancer 2016, 7, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lou, XM.; Jin, LY.; Zhou, R.; Liu, S.; Xu, N.; Liao, D.J. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: Clearance of a few misconceptions. Oncoscience 2014, 1, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lou, X.; Zellmer, L.; Liu, S.; Xu, N.; Liao, D.J. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience 2014, 1, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Akiva, P.; Toporik, A.; Edelheit, S.; Peretz, Y.; Diber, A.; Shemesh, R.; Novik, A.; Sorek, R. Transcription-mediated gene fusion in the human genome. Genome Res. 2006, 16, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Pflueger, D.; Moss, B.; van Doren, V.E.; Chen, C.X.; Kuefer, R.; Tewari, A.K.; Setlur, S.R.; Demichelis, F.; et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009, 69, 2734–2738. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Yang, W.; Guan, Z.; Yu, W.; Liao, D.J. Protein multiplicity can lead to misconduct in western blotting and misinterpretation of immunohistochemical staining results, creating much conflicting data. Prog. Histochem. Cytochem. 2016, 51, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; An, J.; Seim, I.; Walpole, C.; Hoffman, A.; Moya, L.; Srinivasan, S.; Perry-Keene, J.L.; Australian Prostate Cancer Bioresource; Wang, C.; et al. Erratum to: Fusion transcript loci share many genomic features with non-fusion loci. BMC Genom. 2016, 17, 424. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; An, J.; Seim, I.; Walpole, C.; Hoffman, A.; Moya, L.; Srinivasan, S.; Perry-Keene, J.L.; Australian Prostate Cancer Bioresource; Wang, C.; et al. Fusion transcript loci share many genomic features with non-fusion loci. BMC Genom. 2015, 16, 1021. [Google Scholar] [CrossRef] [PubMed]
- Georgomanolis, T.; Sofiadis, K.; Papantonis, A. Cutting a Long Intron Short: Recursive Splicing and Its Implications. Front. Physiol. 2016, 7, 598. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wu, J.; Wu, S.H.; Bi, A.D.; Liao, D.J. Splicing of mouse p53 pre-mRNA does not always follow the “first come, first served” principle and may be influenced by cisplatin treatment and serum starvation. Mol. Biol. Rep. 2012, 39, 9247–9256. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; de Vega, M. Protein-Primed Replication of Bacteriophage Phi29 DNA. Enzymes 2016, 39, 137–167. [Google Scholar] [PubMed]
- Salas, M.; Holguera, I.; Redrejo-Rodriguez, M.; de Vega, M. DNA-Binding Proteins Essential for Protein-Primed Bacteriophage Phi29 DNA Replication. Front. Mol. Biosci. 2016, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Harada, F.; Sawyer, R.C.; Dahlberg, J.E. A primer ribonucleic acid for initiation of in vitro Rous sarcarcoma virus deoxyribonucleic acid synthesis. J. Biol. Chem. 1975, 250, 3487–3497. [Google Scholar] [PubMed]
- Harada, F.; Peters, G.G.; Dahlberg, J.E. The primer tRNA for Moloney murine leukemia virus DNA synthesis. Nucleotide sequence and aminoacylation of tRNAPro. J. Biol. Chem. 1979, 254, 10979–10985. [Google Scholar] [PubMed]
- Peters, G.; Harada, F.; Dahlberg, J.E.; Panet, A.; Haseltine, W.A.; Baltimore, D. Low-molecular-weight RNAs of Moloney murine leukemia virus: Identification of the primer for RNA-directed DNA synthesis. J. Virol. 1977, 21, 1031–1041. [Google Scholar] [PubMed]
- Lerat, H.; Berby, F.; Trabaud, M.A.; Vidalin, O.; Major, M.; Trepo, C.; Inchauspe, G. Specific detection of hepatitis C virus minus strand RNA in hematopoietic cells. J. Clin. Investig. 1996, 97, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Tuiskunen, A.; Leparc-Goffart, I.; Boubis, L.; Monteil, V.; Klingstrom, J.; Tolou, H.J.; Lundkvist, A.; Plumet, S. Self-priming of reverse transcriptase impairs strand-specific detection of dengue virus RNA. J. Gen. Virol. 2010, 91, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Adrover, M.F.; Munoz, M.J.; Baez, M.V.; Thomas, J.; Kornblihtt, A.R.; Epstein, A.L.; Jerusalinsky, D.A. Characterization of specific cDNA background synthesis introduced by reverse transcription in RT-PCR assays. Biochimie 2010, 92, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Frech, B.; Peterhans, E. RT-PCR: ‘background priming’ during reverse transcription. Nucleic Acids Res. 1994, 22, 4342–4343. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Qin, A.X.; Bodell, P.W.; Zhang, L.Y.; Guo, H.; Giger, J.M.; Baldwin, K.M. Regulation of antisense RNA expression during cardiac MHC gene switching in response to pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2351–H2361. [Google Scholar] [CrossRef] [PubMed]
- Moison, C.; Arimondo, P.B.; Guieysse-Peugeot, A.L. Commercial reverse transcriptase as source of false-positive strand-specific RNA detection in human cells. Biochimie 2011, 93, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, A.; Hakansson, J.; Xian, X.; Semb, H.; Kubista, M. Properties of the reverse transcription reaction in mRNA quantification. Clin. Chem. 2004, 50, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Liu, Y.; Yang, M.; Liao, D.J. New methods as alternative or corrective measures for the pitfalls and artifacts of reverse transcription and polymerase chain reactions (RT-PCR) in cloning chimeric or antisense-accompanied RNA. RNA Biol. 2013, 10, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Gubler, U. Second-strand cDNA synthesis: mRNA fragments as primers. Methods Enzymol. 1987, 152, 330–335. [Google Scholar] [PubMed]
- Hanaki, K.; Odawara, T.; Muramatsu, T.; Kuchino, Y.; Masuda, M.; Yamamoto, K.; Nozaki, C.; Mizuno, K.; Yoshikura, H. Primer/template-independent synthesis of poly d(A-T) by Taq polymerase. Biochem. Biophys. Res. Commun. 1997, 238, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hanaki, K.; Odawara, T.; Nakajima, N.; Shimizu, Y.K.; Nozaki, C.; Mizuno, K.; Muramatsu, T.; Kuchino, Y.; Yoshikura, H. Two different reactions involved in the primer/template-independent polymerization of dATP and dTTP by Taq DNA polymerase. Biochem. Biophys. Res. Commun. 1998, 244, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Hanaki, K.; Nakatake, H.; Yamamoto, K.; Odawara, T.; Yoshikura, H. DNase I activity retained after heat inactivation in standard buffer. Biotechniques 2000, 29, 38–40, 42. [Google Scholar] [PubMed]
- Hanaki, K.; Nishihara, T.; Odawara, T.; Nakajima, N.; Yamamoto, K.; Yoshikura, H. RNAse A treatment of Taq and Tth DNA polymerases eliminates primer/template-independent poly(dA-dT) synthesis. Biotechniques 2001, 31, 734, 736, 738. [Google Scholar] [PubMed]
- Nakajima, N.; Hanaki, K.; Shimizu, Y.K.; Ohnishi, S.; Gunji, T.; Nakajima, A.; Nozaki, C.; Mizuno, K.; Odawara, T.; Yoshikura, H. Hybridization-AT-tailing (HybrAT) method for sensitive and strand-specific detection of DNA and RNA. Biochem. Biophys. Res. Commun. 1998, 248, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.Y.; Gomez-Sanchez, C.E. Universal TA cloning. Curr. Issues Mol. Biol. 2000, 2, 1–7. [Google Scholar] [PubMed]
- Alldred, M.J.; Che, S.; Ginsberg, S.D. Terminal continuation (TC) RNA amplification without second strand synthesis. J. Neurosci. Methods 2009, 177, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Che, S.; Ginsberg, S.D. Amplification of RNA transcripts using terminal continuation. Lab. Investig. 2004, 84, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Che, S. RNA amplification in brain tissues. Neurochem. Res. 2002, 27, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Wellenreuther, R.; Schupp, I.; Poustka, A.; Wiemann, S. SMART amplification combined with cDNA size fractionation in order to obtain large full-length clones. BMC Genom. 2004, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Gubler, U. Second-strand cDNA synthesis: Classical method. Methods Enzymol. 1987, 152, 325–329. [Google Scholar] [PubMed]
- Wang, E.; Miller, L.D.; Ohnmacht, G.A.; Liu, E.T.; Marincola, F.M. High-fidelity mRNA amplification for gene profiling. Nat. Biotechnol. 2000, 18, 457–459. [Google Scholar] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Galan, M.; Razzauti, M.; Bard, E.; Bernard, M.; Brouat, C.; Charbonnel, N.; Dehne-Garcia, A.; Loiseau, A.; Tatard, C.; Tamisier, L.; et al. 16S rRNA Amplicon Sequencing for Epidemiological Surveys of Bacteria in Wildlife. mSystems 2016, 1, e00032-16. [Google Scholar] [CrossRef] [PubMed]
- Garrett, R.A. A backward view from 16S rRNA to archaea to the universal tree of life to progenotes: Reminiscences of Carl Woese. RNA Biol. 2014, 11, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, K.M.; Engel, P. Beyond 16S rRNA Community Profiling: Intra-Species Diversity in the Gut Microbiota. Front. Microbiol. 2016, 7, 1475. [Google Scholar] [CrossRef] [PubMed]
- Logares, R.; Sunagawa, S.; Salazar, G.; Cornejo-Castillo, F.M.; Ferrera, I.; Sarmento, H.; Sarmento, H.; Hingamp, P.; Ogata, H.; de Vargas, C.; et al. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 2014, 16, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Quince, C.; Pijl, A.S.; de Hollander, M.; Kowalchuk, G.A. A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS ONE 2012, 7, e30600. [Google Scholar] [CrossRef] [PubMed]
- Acinas, S.G.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M.F. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 2005, 71, 8966–8969. [Google Scholar] [CrossRef] [PubMed]
- Ashelford, K.E.; Chuzhanova, N.A.; Fry, J.C.; Jones, A.J.; Weightman, A.J. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 2005, 71, 7724–7736. [Google Scholar] [CrossRef] [PubMed]
- Ashelford, K.E.; Chuzhanova, N.A.; Fry, J.C.; Jones, A.J.; Weightman, A.J. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl. Environ. Microbiol. 2006, 72, 5734–5741. [Google Scholar] [CrossRef] [PubMed]
- De Santis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.; Faulkner, G.; Hugenholtz, P. Bellerophon: A program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 2004, 20, 2317–2319. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, P.; Huber, T. Chimeric 16S rDNA sequences of diverse origin are accumulating in the public databases. Int. J. Syst. Evol. Microbiol. 2003, 53, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Lahr, D.J.; Katz, L.A. Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase. Biotechniques 2009, 47, 857–866. [Google Scholar] [PubMed]
- Quince, C.; Lanzen, A.; Curtis, T.P.; Davenport, R.J.; Hall, N.; Head, I.M.; Read, L.F.; Sloan, W.T. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods 2009, 6, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.R.; Marcelino, L.A.; Polz, M.F. Heteroduplexes in mixed-template amplifications: Formation, consequence and elimination by “reconditioning PCR”. Nucleic Acids Res. 2002, 30, 2083–2088. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.C.; Wang, Y. Frequency of formation of chimeric molecules as a consequence of PCR coamplification of 16S rRNA genes from mixed bacterial genomes. Appl. Environ. Microbiol. 1997, 63, 4645–4650. [Google Scholar] [PubMed]
- Burzio, V.A.; Villota, C.; Villegas, J.; Landerer, E.; Boccardo, E.; Villa, L.L.; Martínez, R.; Lopez, C.; Gaete, F.; Toro, V.; et al. Expression of a family of noncoding mitochondrial RNAs distinguishes normal from cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9430–9434. [Google Scholar] [CrossRef] [PubMed]
- Landerer, E.; Villegas, J.; Burzio, V.A.; Oliveira, L.; Villota, C.; Lopez, C.; Restovic, F.; Martinez, R.; Castillo, O.; Burzio, L.O. Nuclear localization of the mitochondrial ncRNAs in normal and cancer cells. Cell Oncol. 2011, 34, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Rivas, A.; Burzio, V.; Landerer, E.; Borgna, V.; Gatica, S.; Avila, R.; Lopez, C.; Villota, C.; de la Fuente, R.; Echenique, J.; et al. Determination of the differential expression of mitochondrial long non-coding RNAs as a noninvasive diagnosis of bladder cancer. BMC Urol. 2012, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Vidaurre, S.; Fitzpatrick, C.; Burzio, V.A.; Briones, M.; Villota, C.; Villegas, J.; Echenique, J.; Oliveira-Cruz, L.; Araya, M.; Borgna, V.; et al. Down-regulation of the antisense mitochondrial non-coding RNAs (ncRNAs) is a unique vulnerability of cancer cells and a potential target for cancer therapy. J. Biol. Chem. 2014, 289, 27182–27198. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.; Zarraga, A.M.; Muller, I.; Montecinos, L.; Werner, E.; Brito, M.; Meneses, A.M.; Burzio, L.O. A novel chimeric mitochondrial RNA localized in the nucleus of mouse sperm. DNA Cell Biol. 2000, 19, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.; Araya, P.; Bustos-Obregon, E.; Burzio, L.O. Localization of the 16S mitochondrial rRNA in the nucleus of mammalian spermatogenic cells. Mol. Hum. Reprod. 2002, 8, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.; Burzio, V.; Villota, C.; Landerer, E.; Martinez, R.; Santander, M.; Martinez, R.; Pinto, R.; Vera, M.I.; Boccardo, E.; et al. Expression of a novel non-coding mitochondrial RNA in human proliferating cells. Nucleic Acids Res. 2007, 35, 7336–7347. [Google Scholar] [CrossRef] [PubMed]
- Villota, C.; Campos, A.; Vidaurre, S.; Oliveira-Cruz, L.; Boccardo, E.; Burzio, V.A.; Varas, M.; Villegas, J.; Villa, L.L.; Valenzuela, P.D.; et al. Expression of mitochondrial non-coding RNAs (ncRNAs) is modulated by high risk human papillomavirus (HPV) oncogenes. J. Biol. Chem. 2012, 287, 21303–21315. [Google Scholar] [CrossRef] [PubMed]
- Lobos-Gonzalez, L.; Silva, V.; Araya, M.; Restovic, F.; Echenique, J.; Oliveira-Cruz, L.; Fitzpatrick, C.; Briones, M.; Villegas, J.; Villota, C.; et al. Targeting antisense mitochondrial ncRNAs inhibits murine melanoma tumor growth and metastasis through reduction in survival and invasion factors. Oncotarget 2016, 7, 58331–58350. [Google Scholar] [CrossRef] [PubMed]
- Shepard, P.J.; Choi, E.A.; Lu, J.; Flanagan, L.A.; Hertel, K.J.; Shi, Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 2011, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Slomovic, S.; Laufer, D.; Geiger, D.; Schuster, G. Polyadenylation and degradation of human mitochondrial RNA: The prokaryotic past leaves its mark. Mol. Cell. Biol. 2005, 25, 6427–6435. [Google Scholar] [CrossRef] [PubMed]
- Le Grice, S.F. “In the beginning”: Initiation of minus strand DNA synthesis in retroviruses and LTR-containing retrotransposons. Biochemistry 2003, 42, 14349–14355. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.B.; Ross, S.L.; Marchetti, S.E.; Kennell, J.C. Relaxed primer specificity associated with reverse transcriptases encoded by the pFOXC retroplasmids of Fusarium oxysporum. Eukaryot. Cell 2004, 3, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.; Lim, D.; Kim, S. Compiling Multicopy Single-Stranded DNA Sequences from Bacterial Genome Sequences. Genom. Inform. 2016, 14, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Lanchy, J.M.; Isel, C.; Keith, G.; Le Grice, S.F.; Ehresmann, C.; Ehresmann, B.; Marquet, R. Dynamics of the HIV-1 reverse transcription complex during initiation of DNA synthesis. J. Biol. Chem. 2000, 275, 12306–12312. [Google Scholar] [CrossRef] [PubMed]
- Isel, C.; Lanchy, J.M.; Le Grice, S.F.; Ehresmann, C.; Ehresmann, B.; Marquet, R. Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNA3Lys. EMBO J. 1996, 15, 917–924. [Google Scholar] [PubMed]
- Marquet, R.; Isel, C.; Ehresmann, C.; Ehresmann, B. tRNAs as primer of reverse transcriptases. Biochimie 1995, 77, 113–124. [Google Scholar] [CrossRef]
- Mak, J.; Kleiman, L. Primer tRNAs for reverse transcription. J. Virol. 1997, 71, 8087–8095. [Google Scholar] [PubMed]
- Arts, E.J.; Le Grice, S.F. Interaction of retroviral reverse transcriptase with template-primer duplexes during replication. Prog. Nucleic Acid. Res. Mol. Biol. 1998, 58, 339–393. [Google Scholar] [PubMed]
- Wakefield, J.K.; Wolf, A.G.; Morrow, C.D. Human immunodeficiency virus type 1 can use different tRNAs as primers for reverse transcription but selectively maintains a primer binding site complementary to tRNA(3Lys). J. Virol. 1995, 69, 6021–6029. [Google Scholar] [PubMed]
- Falvey, A.K.; Weiss, G.B.; Krueger, L.J.; Kantor, J.A.; Anderson, W.F. Transcription of single base oligonucleotides by ribonucleic acid-directed deoxyribonucleic acid polymerase. Nucleic Acids Res. 1976, 3, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.G.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535. [Google Scholar] [CrossRef] [PubMed]
- Laurila, M.R.; Salgado, P.S.; Stuart, D.I.; Grimes, J.M.; Bamford, D.H. Back-priming mode of phi6 RNA-dependent RNA polymerase. J. Gen. Virol. 2005, 86, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Unneberg, P.; Claverie, J.M. Tentative mapping of transcription-induced interchromosomal interaction using chimeric EST and mRNA data. PLoS ONE 2007, 2, e254. [Google Scholar] [CrossRef] [PubMed]
- Bollig-Fischer, A.; Thakur, A.; Sun, Y.; Wu, J.-S.; Liao, D.J. The predominant proteins that react to the MC-20 estrogen receptor alpha antibody differ in molecular weight between the mammary gland and uterus in the mouse and rat. Int. J. Biomed. Sci. 2012, 8, 51–63. [Google Scholar] [PubMed]
- Liao, D.J.; Natarajan, G.; Deming, S.L.; Jamerson, M.H.; Johnson, M.; Chepko, G.; Dickson, R.B. Cell cycle basis for the onset and progression of c-Myc-induced, TGFα-enhanced mouse mammary gland carcinogenesis. Oncogene 2000, 19, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.Z.; Pantazis, C.G.; Hou, X.; Li, S.A. Promotion of estrogen-induced mammary gland carcinogenesis by androgen in the male Noble rat: Probable mediation by steroid receptors. Carcinogenesis 1998, 19, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cao, S.; Yang, M.; Wu, S.; Wang, Z.; Lin, X.; Song, X.; Liao, D.J. Basic anatomy and tumor biology of the RPS6KA6 gene that encodes the p90 ribosomal S6 kinase-4. Oncogene 2013, 32, 1794–1810. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lou, X.; Yang, M.; Yuan, C.; Ma, L.; Xie, B.K.; Wu, J.M.; Yang, W.; Shen, S.X.; Xu, N.; et al. Cyclin-dependent kinase 4 may be expressed as multiple proteins and have functions that are independent of binding to CCND and RB and occur at the S and G 2/M phases of the cell cycle. Cell Cycle 2013, 12, 3512–3525. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Sun, Y.; Ma, L.; Wang, C.; Wu, J.M.; Bi, A.; Liao, D.J. Complex alternative splicing of the SMARCA2 gene suggests the importance of SMARCA2-B variants. J. Cancer 2011, 2, 386–400. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, C.; Han, Y.; Zellmer, L.; Yang, W.; Guan, Z.; Yu, W.; Huang, H.; Liao, D.J. It Is Imperative to Establish a Pellucid Definition of Chimeric RNA and to Clear Up a Lot of Confusion in the Relevant Research. Int. J. Mol. Sci. 2017, 18, 714. https://doi.org/10.3390/ijms18040714
Yuan C, Han Y, Zellmer L, Yang W, Guan Z, Yu W, Huang H, Liao DJ. It Is Imperative to Establish a Pellucid Definition of Chimeric RNA and to Clear Up a Lot of Confusion in the Relevant Research. International Journal of Molecular Sciences. 2017; 18(4):714. https://doi.org/10.3390/ijms18040714
Chicago/Turabian StyleYuan, Chengfu, Yaping Han, Lucas Zellmer, Wenxiu Yang, Zhizhong Guan, Wenfeng Yu, Hai Huang, and D. Joshua Liao. 2017. "It Is Imperative to Establish a Pellucid Definition of Chimeric RNA and to Clear Up a Lot of Confusion in the Relevant Research" International Journal of Molecular Sciences 18, no. 4: 714. https://doi.org/10.3390/ijms18040714
APA StyleYuan, C., Han, Y., Zellmer, L., Yang, W., Guan, Z., Yu, W., Huang, H., & Liao, D. J. (2017). It Is Imperative to Establish a Pellucid Definition of Chimeric RNA and to Clear Up a Lot of Confusion in the Relevant Research. International Journal of Molecular Sciences, 18(4), 714. https://doi.org/10.3390/ijms18040714