KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation



Abstract
:1. Introduction
2. Kruppel-Like Factor
3. Kruppel Like Factor 2
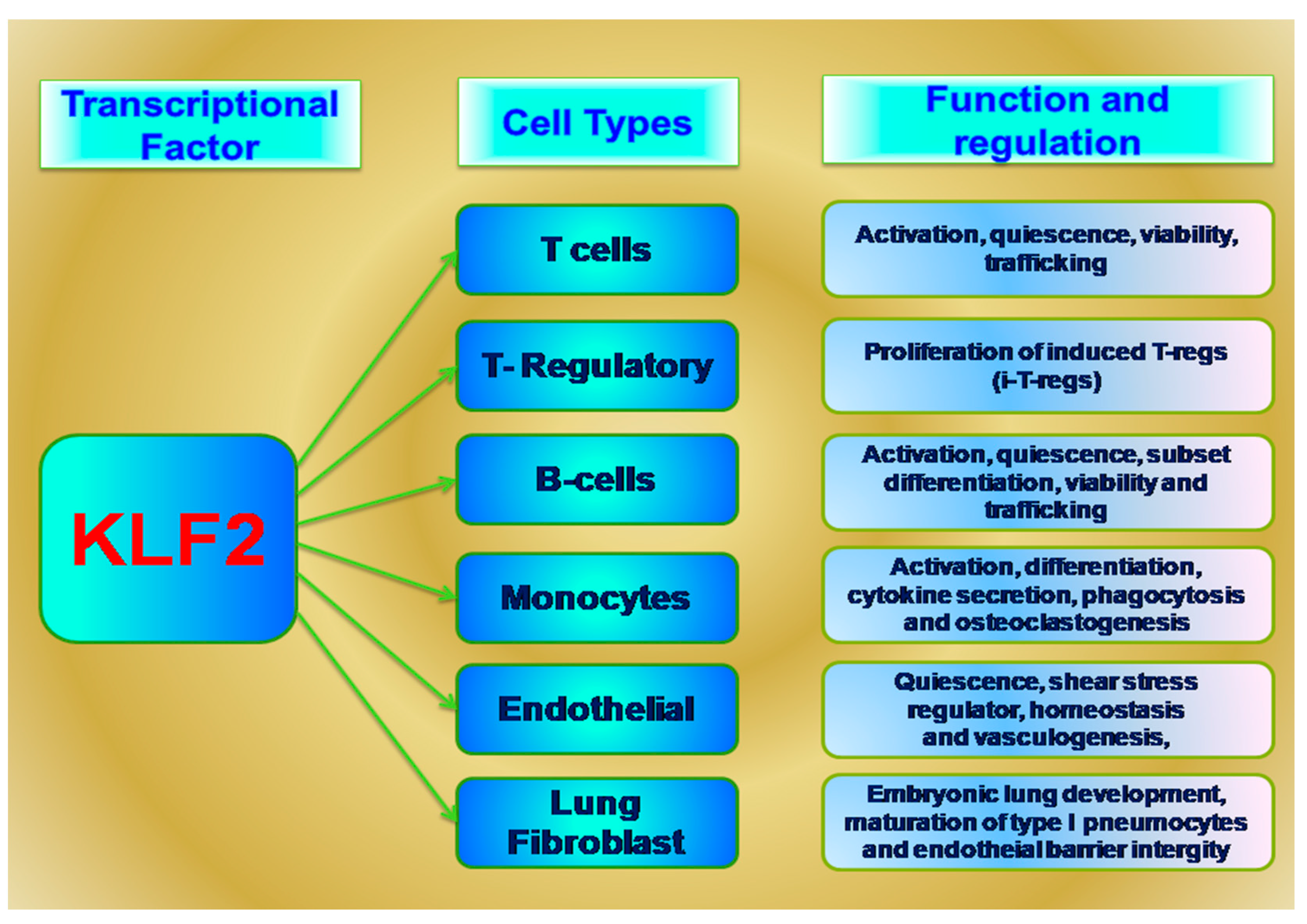
3.1. KLF2 in Lungs
3.2. KLF2 in T Cells
3.3. KLF2 in T-Regulatory Cells
4. KLF2 in B Cells
4.1. KLF2 in Endothelial Cells
4.2. KLF2 in Monocytes
5. Inflammation Master Regulator NF-κB
6. NF-κB Protein Family
7. Structure, Function and Regulation of NF-κB
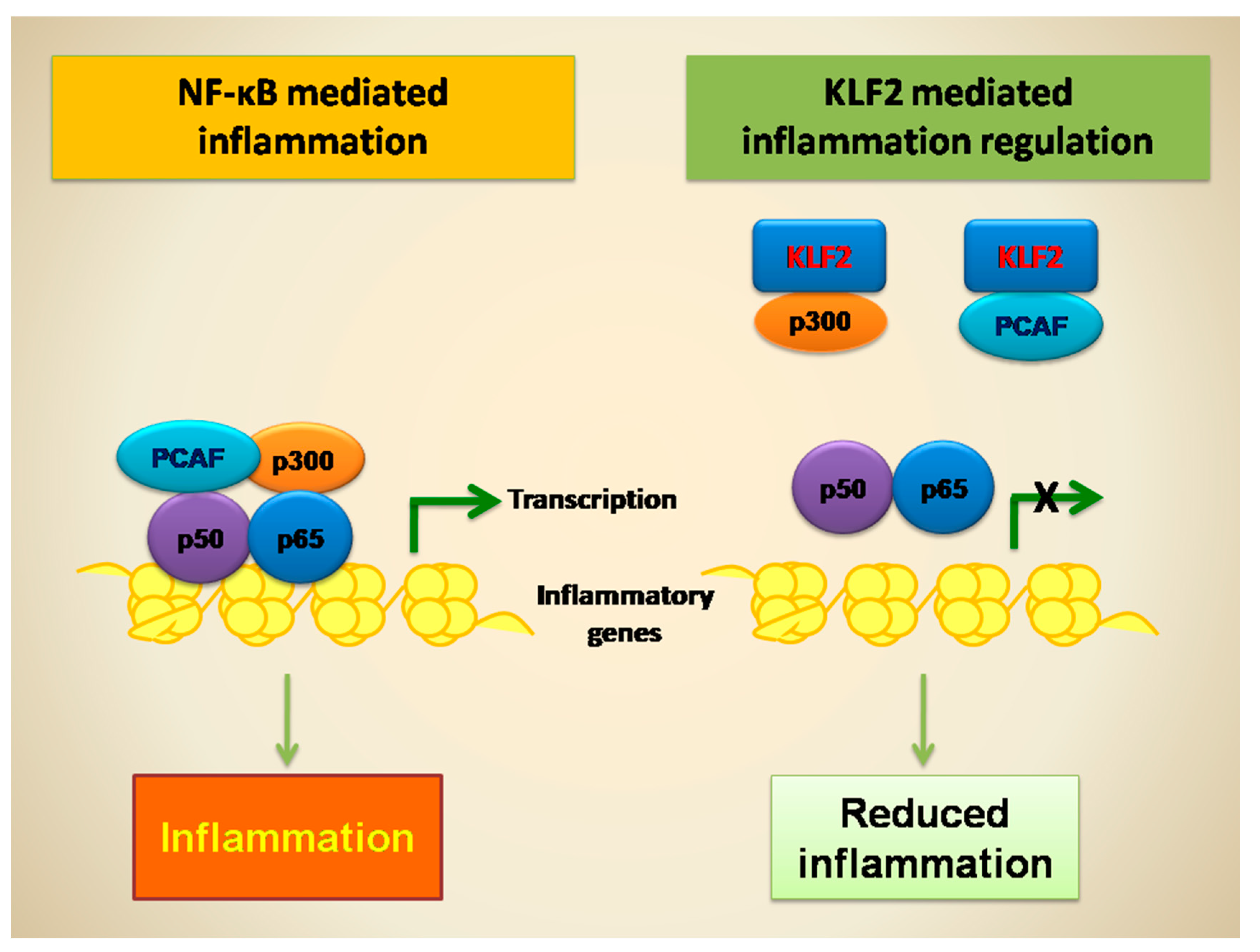
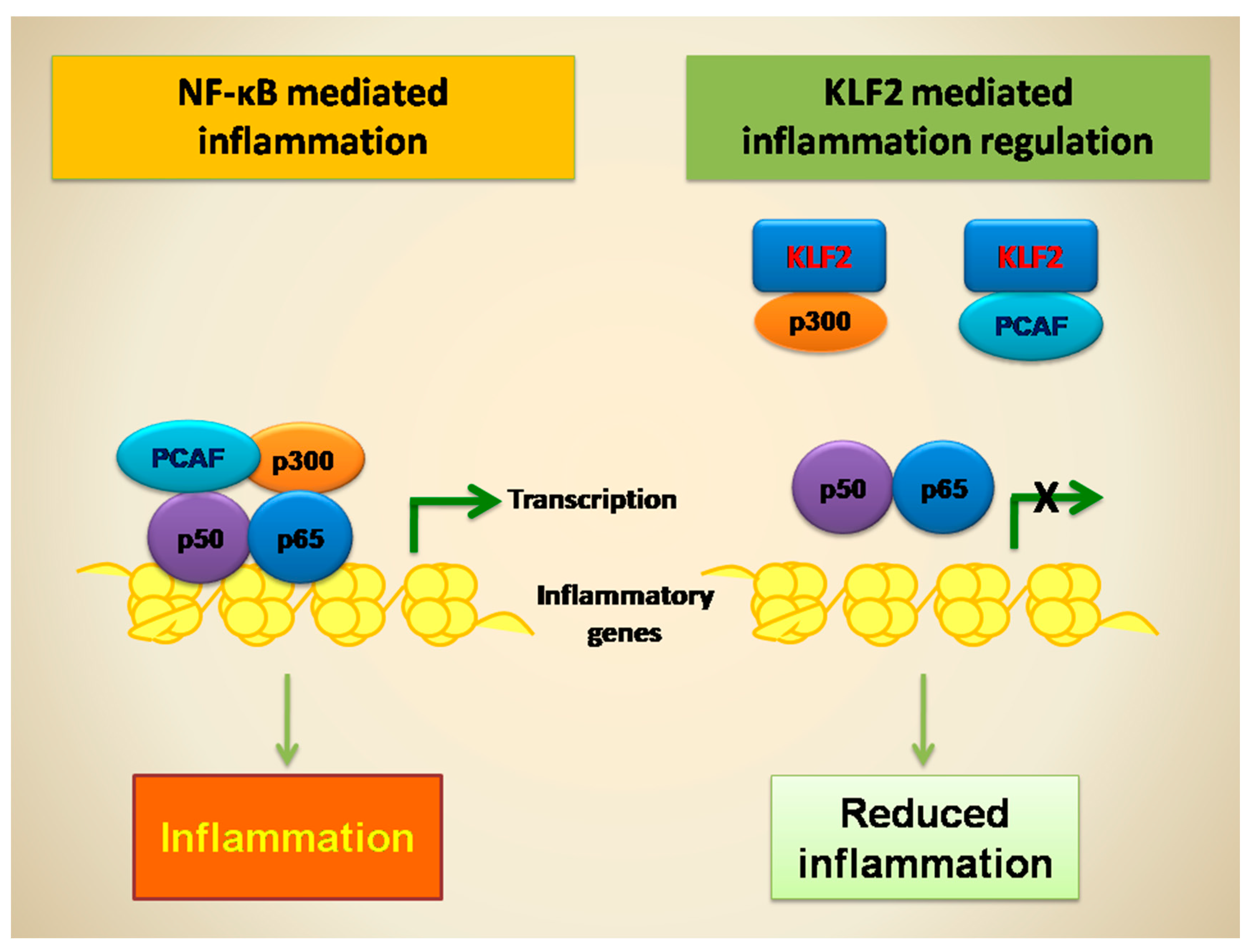
8. KLF2-Mediated Regulation of NF-κB Transcriptional Activity and Function in Monocytes
9. KLF2-Mediated Regulation of NF-κB Transcriptional Activity and Function in Endothelial Cells
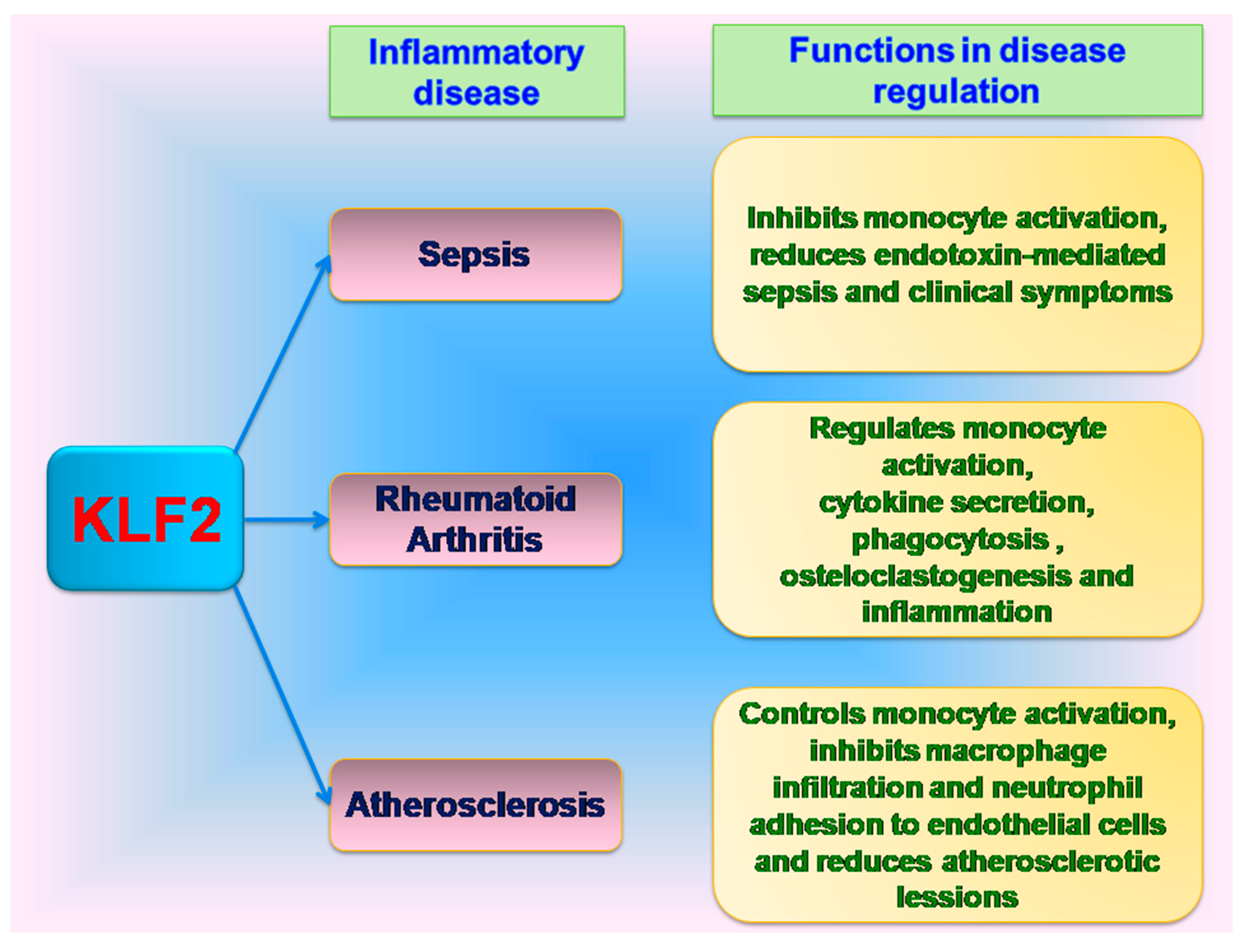
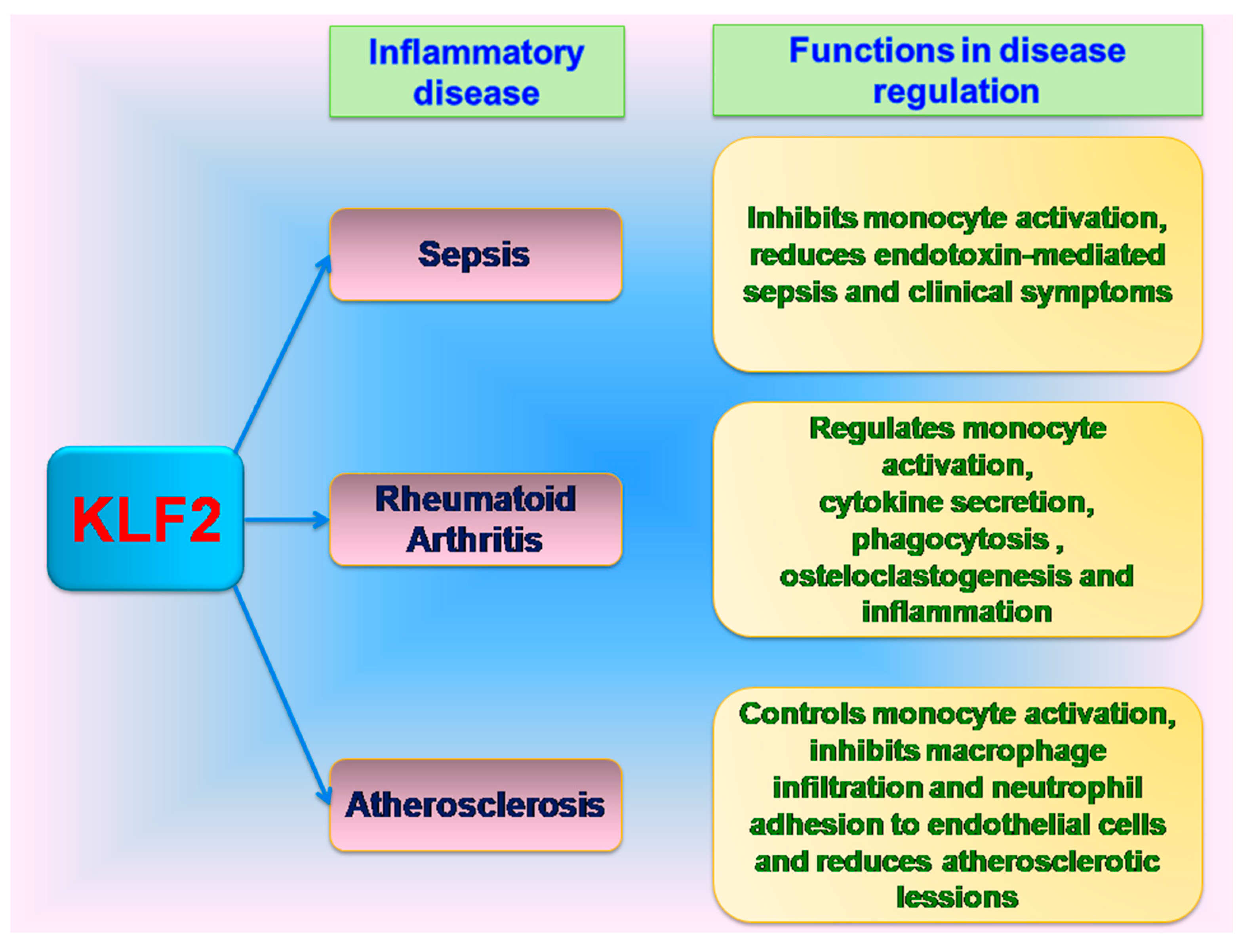
10. KLF2 in Inflammatory Diseases
11. KLF2 in Sepsis
12. KLF2 in Rheumatoid Arthritis
13. KLF2 in Atherosclerosis
14. Conclusions
15. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| KLF2 | Kruppel-like factor 2 |
| NF-κB | Nuclear factor-κB |
| PCAF | p300/cyclic adenosine monophosphate response element binding protein (CBP)-associated factor |
| MEF2 | MADS box transcription enhancer factor 2 |
| HDAC | Histone deacetylases |
| EKLF | Erythroid Kruppel-like factor |
| CtBP | Carboxy-terminal binding protein |
| LKLF | Lung Kruppel-like factor |
| SMRT | Silencing mediator of retinoid and thyroid hormone receptors |
| LPS | Lipopolysaccharides |
| RAPGEF3 | Guanine nucleotide exchange factor 3 |
| RAC | Ras-related C3 botulinum toxin substrate |
| RAG | Recombination activating gene |
| Myc | Myelocytomatosis |
| Mad | Mitotic arrest deficient |
| Cip | Cyclin-dependent kinase interacting protein |
| MHC | Major histocompatibility complex |
| S1P1 | Sphingosine 1-phosphate receptor 1 |
| TCR | T cell receptor |
| MAPK | Mitogen-activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| IL | Interleukin |
| CXCR | C-X-C motif chemokine receptor |
| CXCL10 | C-X-C motif chemokine 10 |
| T-regs | Regulatory T cells |
| FoxP3 | Forkhead box P3 |
| i-T-regs | Induced T-regulatory cells |
| ChIP | Chromatin immunoprecipitation |
| H4Ac | Acetylated histone H4 |
| TGF-β | Transforming growth factor-beta |
| WWP1 | WW domain-containing protein 1 |
| SMURF1 | Smad ubiquitination regulatory factor 1 |
| FBW7 | F-box/WD repeat-containing protein 7 |
| FO | Follicular |
| MZ | Marginal zone |
| BCR | B cell receptor |
| VCAM | Vascular cell adhesion molecule |
| eNOS | Endothelial nitric oxide synthase |
| GTP | Guanosine triphosphate |
| RhoA | Ras homolog gene family member A |
| Cdc42 | Cell division cycle 42 |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-3 |
| ICAM | Intercellular adhesion molecule |
| Rel | Reticuloendotheliosis |
| IKK | Inhibitor of κB kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| COX-2 | Cyclooxygenase-2 |
| GSK | Glycogen synthase kinase |
| HIF-1α | Hypoxia induced factor-1α |
| mBSA | Methylated bovine serum albumin |
| PAI-1 | Plasminogen activator inhibitor-1 |
| TRAP | Triiodothyronine receptor auxiliary protein |
| MMP | Matrix metalloproteinases |
| TNF-α | Tumor necrosis factor-α |
| MIP | Macrophage inflammatory proteins |
| RHD | Rel homology domain |
| RANK | Receptor activator of NF-κB |
| CBP | Cyclic AMP response element binding protein (CREB)-binding protein |
| PKA | Protein kinase A |
| MIF | Macrophage migration inhibitory factor |
| H3K9 | Histone 3 lysine 9 |
References
- Feinberg, M.W.; Lin, Z.; Fisch, S.; Jain, M.K. An emerging role for Kruppel-like factors in vascular biology. Trends Cardiovasc. Med. 2004, 14, 241–246. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.B.; Yang, V.W. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Crossley, M. Mammalian Kruppel-like transcription factors: More than just a pretty finger. Trends Biochem. Sci. 1999, 24, 236–240. [Google Scholar] [CrossRef]
- Bieker, J.J. Kruppel-like factors: Three fingers in many pies. J. Biol. Chem. 2001, 276, 34355–34358. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.T.; Veselits, M.L.; Leiden, J.M. LKLF: A transcriptional regulator of single-positive T cell quiescence and survival. Science 1997, 277, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Das, H.; Kumar, A.; Lin, Z.; Patino, W.D.; Hwang, P.M.; Feinberg, M.W.; Majumder, P.K.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 6653–6658. [Google Scholar] [CrossRef] [PubMed]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Lu, J.; Joseph, M.; Aggarwal, R.; Kanji, S.; McMichael, B.; Lee, B.S.; Agarwal, S.; Ray-Chaudhury, A.; Iwenofu, O.H.; et al. Kruppel-like factor 2 (KLF2) regulates monocyte differentiation and functions in mBSA and IL-1β-induced arthritis. Curr. Mol. Med. 2012, 12, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Novodvorsky, P.; Chico, T.J. The role of the transcription factor KLF2 in vascular development and disease. Prog. Mol. Biol. Transl. Sci. 2014, 124, 155–188. [Google Scholar] [PubMed]
- Mahabeleshwar, G.H.; Qureshi, M.A.; Takami, Y.; Sharma, N.; Lingrel, J.B.; Jain, M.K. A Myeloid Hypoxia-inducible Factor 1α-Kruppel-like Factor 2 Pathway Regulates Gram-positive Endotoxin-mediated Sepsis. J. Biol. Chem. 2012, 287, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.J.; Bieker, J.J. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol. Cell. Biol. 1993, 13, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.C.; Sharpe, A.H.; Orkin, S.H. Lethal β-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature 1995, 375, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.A.; Conkright, M.D.; Jeffries, S.; Hughes, M.J.; Lingrel, J.B. cDNA isolation, genomic structure, regulation and chromosomal localization of human lung Kruppel-like factor. Genomics 1999, 60, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.P.; Kern, C.B.; Crable, S.C.; Lingrel, J.B. Isolation of a gene encoding a functional zinc finger protein homologous to erythroid Kruppel-like factor: Identification of a new multigene family. Mol. Cell. Biol. 1995, 15, 5957–5965. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Leblanc, M.; Barish, G.; Atkins, A.; Nofsinger, R.; Whyte, J.; Gold, D.; He, M.; Kawamura, K.; Li, H.R.; et al. Thyroid hormone receptor repression is linked to type I pneumocyte-associated respiratory distress syndrome. Nat. Med. 2011, 17, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.T.; Wu, D.; Meliton, A.; Oh, M.J.; Krause, M.; Lloyd, J.A.; Nigdelioglu, R.; Hamanaka, R.B.; Jain, M.K.; Birukova, A.; et al. Experimental Lung Injury Reduces Kruppel-like Factor 2 to Increase Endothelial Permeability via Regulation of RAPGEF3-Rac1 Signaling. Am. J. Respir. Crit. Care Med. 2017, 195, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.F.; Kuo, C.T.; Leiden, J.M. Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc—Dependent pathway. Nat. Immunol. 2001, 2, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Alberts-Grill, N.; Engelbertsen, D.; Bu, D.; Foks, A.; Grabie, N.; Herter, J.M.; Kuperwaser, F.; Chen, T.; Destefano, G.; Jarolim, P.; et al. Dendritic Cell KLF2 Expression Regulates T Cell Activation and Proatherogenic Immune Responses. J. Immunol. 2016, 197, 4651–4662. [Google Scholar] [CrossRef] [PubMed]
- Bai, A.; Hu, H.; Yeung, M.; Chen, J. Kruppel-like factor 2 controls T cell trafficking by activating L-selectin (CD62L) and sphingosine-1-phosphate receptor 1 transcription. J. Immunol. 2007, 178, 7632–7639. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.C.; Feijoo-Carnero, C.; Schurch, N.; Cowling, V.H.; Cantrell, D.A. The impact of KLF2 modulation on the transcriptional program and function of CD8 T cells. PLoS ONE 2013, 8, e77537. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Wang, X.; Hart, G.T.; Odumade, O.A.; Weinreich, M.A.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 is required for trafficking but not quiescence in postactivated T cells. J. Immunol. 2011, 186, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Pabbisetty, S.K.; Rabacal, W.; Maseda, D.; Cendron, D.; Collins, P.L.; Hoek, K.L.; Parekh, V.V.; Aune, T.M.; Sebzda, E. KLF2 is a rate-limiting transcription factor that can be targeted to enhance regulatory T-cell production. Proc. Natl. Acad. Sci. USA 2014, 111, 9579–9584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Srinivasan, S.V.; Lingrel, J.B. WWP1-dependent ubiquitination and degradation of the lung Kruppel-like factor, KLF2. Biochem. Biophys. Res. Commun. 2004, 316, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Tang, Y.; Shen, S.; Wang, Y.; Xing, G.; Yin, Y.; He, F.; Zhang, L. Smurf1 ubiquitin ligase targets Kruppel-like factor KLF2 for ubiquitination and degradation in human lung cancer H1299 cells. Biochem. Biophys. Res. Commun. 2011, 407, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Y.; Liu, N.; Ren, C.; Jiang, C.; Zhang, K.; Yu, S.; Chen, Y.; Tang, H.; Deng, Q.; et al. FBW7 regulates endothelial functions by targeting KLF2 for ubiquitination and degradation. Cell. Res. 2013, 23, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Schuh, W.; Meister, S.; Herrmann, K.; Bradl, H.; Jack, H.M. Transcriptome analysis in primary B lymphoid precursors following induction of the pre-B cell receptor. Mol. Immunol. 2008, 45, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Glynne, R.; Ghandour, G.; Rayner, J.; Mack, D.H.; Goodnow, C.C. B-lymphocyte quiescence, tolerance and activation as viewed by global gene expression profiling on microarrays. Immunol. Rev. 2000, 176, 216–246. [Google Scholar] [PubMed]
- Allman, D.; Pillai, S. Peripheral B cell subsets. Curr. Opin. Immunol. 2008, 20, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.T.; Wang, X.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation and trafficking molecule expression. Proc. Natl. Acad. Sci. USA 2011, 108, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.L.; Gordy, L.E.; Collins, P.L.; Parekh, V.V.; Aune, T.M.; Joyce, S.; Thomas, J.W.; Van Kaer, L.; Sebzda, E. Follicular B cell trafficking within the spleen actively restricts humoral immune responses. Immunity 2010, 33, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, R.; Sandrock, L.; Porstner, M.; Roth, E.; Mathews, M.; Hobeika, E.; Reth, M.; Kahn, M.L.; Schuh, W.; Jack, H.M. B cell homeostasis and plasma cell homing controlled by Kruppel-like factor 2. Proc. Natl. Acad. Sci. USA 2011, 108, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Dekker, R.J.; van Thienen, J.V.; Rohlena, J.; de Jager, S.C.; Elderkamp, Y.W.; Seppen, J.; de Vries, C.J.; Biessen, E.A.; van Berkel, T.J.; Pannekoek, H.; et al. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am. J. Pathol. 2005, 167, 609–618. [Google Scholar] [CrossRef]
- Hergenreider, E.; Heydt, S.; Treguer, K.; Boettger, T.; Horrevoets, A.J.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell. Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef] [PubMed]
- DiChiara, M.R.; Kiely, J.M.; Gimbrone, M.A., Jr.; Lee, M.E.; Perrella, M.A.; Topper, J.N. Inhibition of E-selectin gene expression by transforming growth factor β in endothelial cells involves coactivator integration of Smad and nuclear factor κB-mediated signals. J. Exp. Med. 2000, 192, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.S.; Truskey, G.A. Direct-contact co-culture between smooth muscle and endothelial cells inhibits TNF-α-mediated endothelial cell activation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H338–H346. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hoffman, T.A.; Dericco, J.; Naqvi, A.; Jain, M.K.; Irani, K. Transcriptional repression of Kruppel like factor-2 by the adaptor protein p66shc. FASEB J. 2009, 23, 4344–4352. [Google Scholar] [CrossRef] [PubMed]
- Komano, Y.; Nanki, T.; Hayashida, K.; Taniguchi, K.; Miyasaka, N. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Res. Ther. 2006, 8, R152. [Google Scholar] [CrossRef] [PubMed]
- Mahabeleshwar, G.H.; Kawanami, D.; Sharma, N.; Takami, Y.; Zhou, G.; Shi, H.; Nayak, L.; Jeyaraj, D.; Grealy, R.; White, M.; et al. The myeloid transcription factor KLF2 regulates the host response to polymicrobial infection and endotoxic shock. Immunity 2011, 34, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B.; Pilcher-Roberts, R.; Basford, J.E.; Manoharan, P.; Neumann, J.; Konaniah, E.S.; Srinivasan, R.; Bogdanov, V.Y.; Hui, D.Y. Myeloid-specific Kruppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ. Res. 2012, 110, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Goduni, L.; Takami, Y.; Sharma, N.; Kapil, P.; Jain, M.K.; Mahabeleshwar, G.H. Kruppel-like factor 2 is a transcriptional regulator of chronic and acute inflammation. Am. J. Pathol. 2013, 182, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Foxwell, B.; Browne, K.; Bondeson, J.; Clarke, C.; de Martin, R.; Brennan, F.; Feldmann, M. Efficient adenoviral infection with IκB α reveals that macrophage tumor necrosis factor α production in rheumatoid arthritis is NF-κB dependent. Proc. Natl. Acad. Sci. USA 1998, 95, 8211–8215. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. The NF-κ B and I κB proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.J.; Gerondakis, S. Alternative splicing of RNA transcripts encoded by the murine p105 NF-κ B gene generates I κB γ isoforms with different inhibitory activities. Proc. Natl. Acad. Sci. USA 1994, 91, 4367–4371. [Google Scholar] [CrossRef] [PubMed]
- Crisostomo, P.R.; Wang, Y.; Markel, T.A.; Wang, M.; Lahm, T.; Meldrum, D.R. Human mesenchymal stem cells stimulated by TNF-α, LPS, or hypoxia produce growth factors by an NF κB- but not JNK-dependent mechanism. Am. J. Physiol. Cell Physiol. 2008, 294, C675–C682. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D. NF-κB is 25. Nat. Immunol. 2011, 12, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.P.; Behar, M.; Birnbaum, H.A.; Hoffmann, A.; Wright, P.E.; Ghosh, G. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol. 2013, 11, e1001647. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wan, D.; Li, J.; Chen, H.; Huang, K.; Zheng, L. Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics 2015, 10, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, D.; Ichikawa, K. Regulation of NF-κB Oscillation by Nuclear Transport: Mechanisms Determining the Persistency and Frequency of Oscillation. PLoS ONE 2015, 10, e0127633. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Lin, Z.; Senbanerjee, S.; Jain, M.K. Tumor Necrosis Factor α-Mediated Reduction of KLF2 Is Due to Inhibition of MEF2 by NF-κB and Histone Deacetylases. Mol. Cell. Biol. 2005, 25, 5893–5903. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.S.; Wang, W.; Xu, S.; Jin, Z.G. Histone deacetylase 5 interacts with Kruppel-like factor 2 and inhibits its transcriptional activity in endothelium. Cardiovasc. Res. 2014, 104, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Cernotta, N.; Clocchiatti, A.; Florean, C.; Brancolini, C. Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol. Biol. Cell. 2011, 22, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Abe, J.J.; Min, W.; Suprapisitchat, J.; Yan, C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann. N. Y. Acad. Sci. 2001, 947, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; Topper, J.N.; Nagel, T.; Anderson, K.R.; Garcia-Cardena, G. Endothelial dysfunction, hemodynamic forces and atherogenesis. Ann. N. Y. Acad. Sci. 2000, 902, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Andreakos, E.T.; Foxwell, B.M.; Brennan, F.M.; Maini, R.N.; Feldmann, M. Cytokines and anti-cytokine biologicals in autoimmunity: Present and future. Cytokine Growth Factor Rev. 2002, 13, 299–313. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Welsch, D.J.; Pelletier, J.P. Metalloproteases and inhibitors in arthritic diseases. Best Pract. Res. Clin. Rheumatol. 2001, 15, 805–829. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Therneau, T.M.; Gabriel, S.E. Is the incidence of rheumatoid arthritis rising? Results from Olmsted County, Minnesota, 1955–2007. Arthritis Rheumatol. 2010, 62, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Bennink, R.J.; Thurlings, R.M.; van Hemert, F.J.; Voermans, C.; Dohmen, S.E.; van Eck-Smit, B.L.; Tak, P.P.; Busemann-Sokole, E. Biodistribution and radiation dosimetry of 99mTc-HMPAO-labeled monocytes in patients with rheumatoid arthritis. J. Nucl. Med. 2008, 49, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.B.; Wang, Y.; Mahabeleshwar, G.H.; Shi, H.; Gao, H.; Kawanami, D.; Natesan, V.; Lin, Z.; Simon, D.I.; Jain, M.K. Hemizygous deficiency of Kruppel-like factor 2 augments experimental atherosclerosis. Circ. Res. 2008, 103, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.P.; Liu, X.X.; Xia, R.; Yin, L.; Kong, R.; Chen, W.M.; Huang, M.D.; Shu, Y.Q. SP1-induced upregulation of the long noncoding RNA TINCR regulates cell proliferation and apoptosis by affecting KLF2 mRNA stability in gastric cancer. Oncogene 2015, 34, 5648–5661. [Google Scholar] [CrossRef] [PubMed]
- Ohguchi, H.; Hideshima, T.; Bhasin, M.K.; Gorgun, G.T.; Santo, L.; Cea, M.; Samur, M.K.; Mimura, N.; Suzuki, R.; Tai, Y.T.; et al. The KDM3A-KLF2-IRF4 axis maintains myeloma cell survival. Nat. Commun. 2016, 7, 10258. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Guo, G.; Wray, J.; Eyres, I.; Nichols, J.; Grotewold, L.; Morfopoulou, S.; Humphreys, P.; Mansfield, W.; Walker, R.; et al. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 2009, 5, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.C.; Jiang, J.; Tan, Z.Y.; Yim, G.R.; Ng, J.H.; Goke, J.; Kraus, P.; Liang, H.; Gonzales, K.A.; Chong, H.C.; et al. Klf2 is an essential factor that sustains ground state pluripotency. Cell Stem Cell 2014, 14, 864–872. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| KLF Group | Characteristics | KLF Family Members | Function |
|---|---|---|---|
| Group 1 | Presence of CtBP- binding sites | KLFs 3, 8 and 12 | Transcriptional repressors through their interaction with the carboxy-terminal binding protein (CtBP) |
| Group 2 | Ability to bind deacetylases | KLFs 1, 2, 4, 5, 6 and 7 | Transcriptional activators |
| Group 3 | Presence of a Sin3A-binding sites | KLFs 9, 10, 11, 13, 14 and 16 | Repressor activity through their interaction with the common transcriptional co-repressor Sin3A |
| Subfamily | Protein | Precursors | Gene |
|---|---|---|---|
| NF-κB | NF-κB1 | p105→p50 | NFKB1 |
| NF-κB2 | p100→p52 | NFKB2 | |
| Rel | RelA | p65 | RELA |
| RelB | RELB | ||
| c-Rel | REL |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jha, P.; Das, H. KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation. Int. J. Mol. Sci. 2017, 18, 2383. https://doi.org/10.3390/ijms18112383
Jha P, Das H. KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation. International Journal of Molecular Sciences. 2017; 18(11):2383. https://doi.org/10.3390/ijms18112383
Chicago/Turabian StyleJha, Prerana, and Hiranmoy Das. 2017. "KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation" International Journal of Molecular Sciences 18, no. 11: 2383. https://doi.org/10.3390/ijms18112383
APA StyleJha, P., & Das, H. (2017). KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation. International Journal of Molecular Sciences, 18(11), 2383. https://doi.org/10.3390/ijms18112383