Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update


Abstract
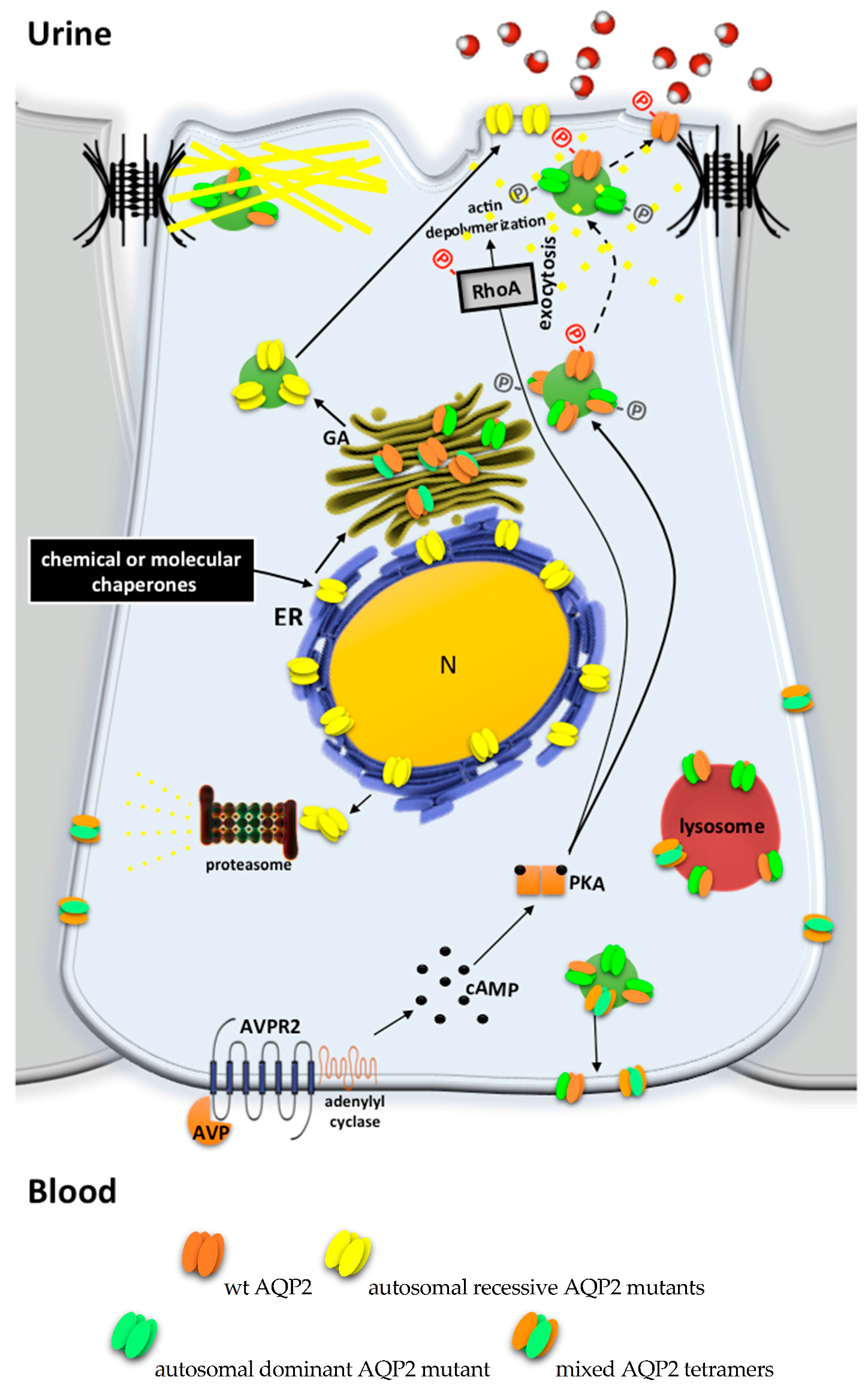
:1. Introduction
2. Pathophysiology of Congenital Nephrogenic Diabetes Insipidus
2.1. AVPR2 Mutations Leading to X-Linked NDI
2.2. AQP2 Mutations Leading to Autosomal Recessive/Dominant NDI
2.3. Partial NDI
3. Animal Models to Study NDI
3.1. Models of Autosomal Recessive NDI
3.2. Models of Autosomal Dominant NDI
3.3. Models of X-Linked NDI
4. Current Conventional Treatment of Congenital NDI
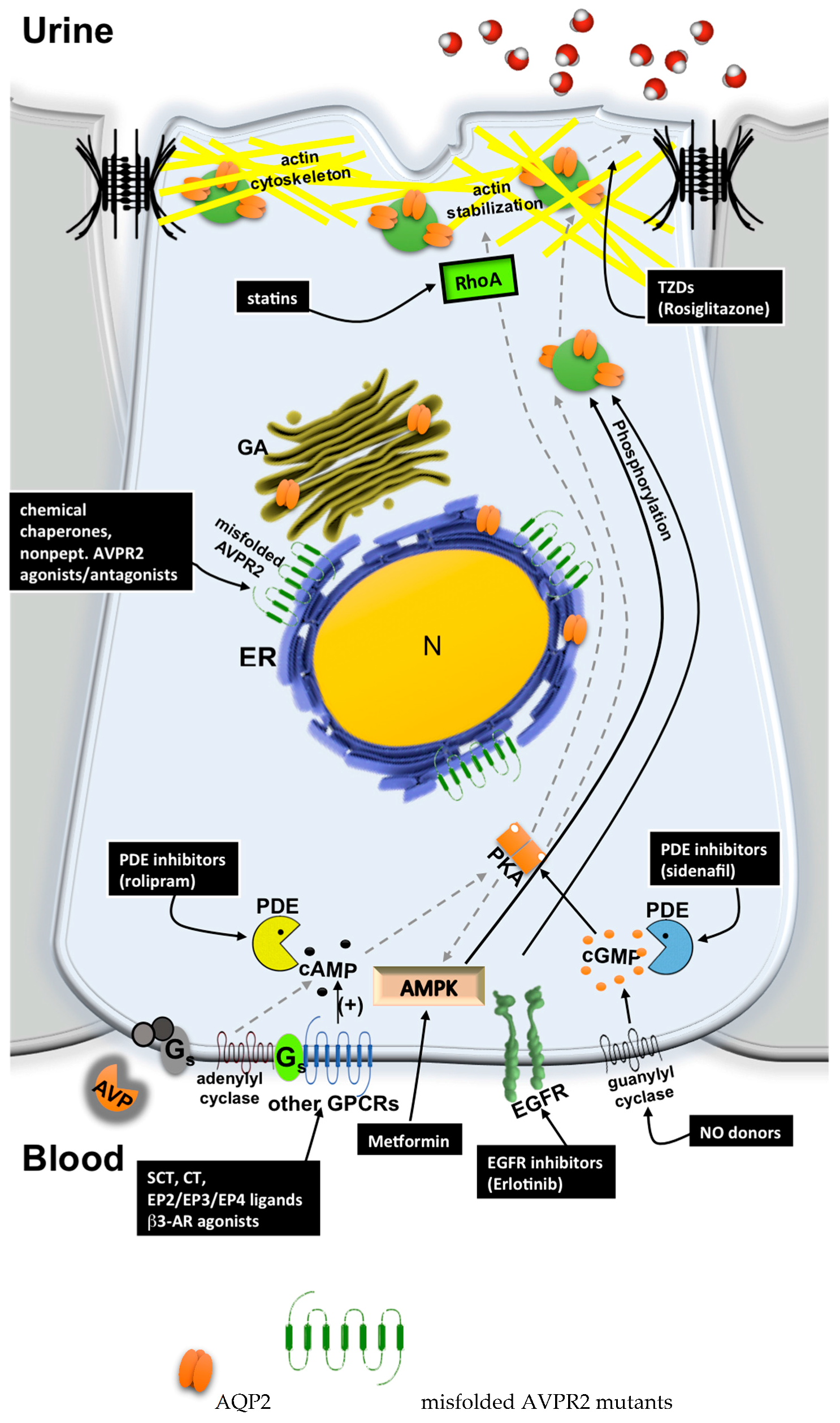
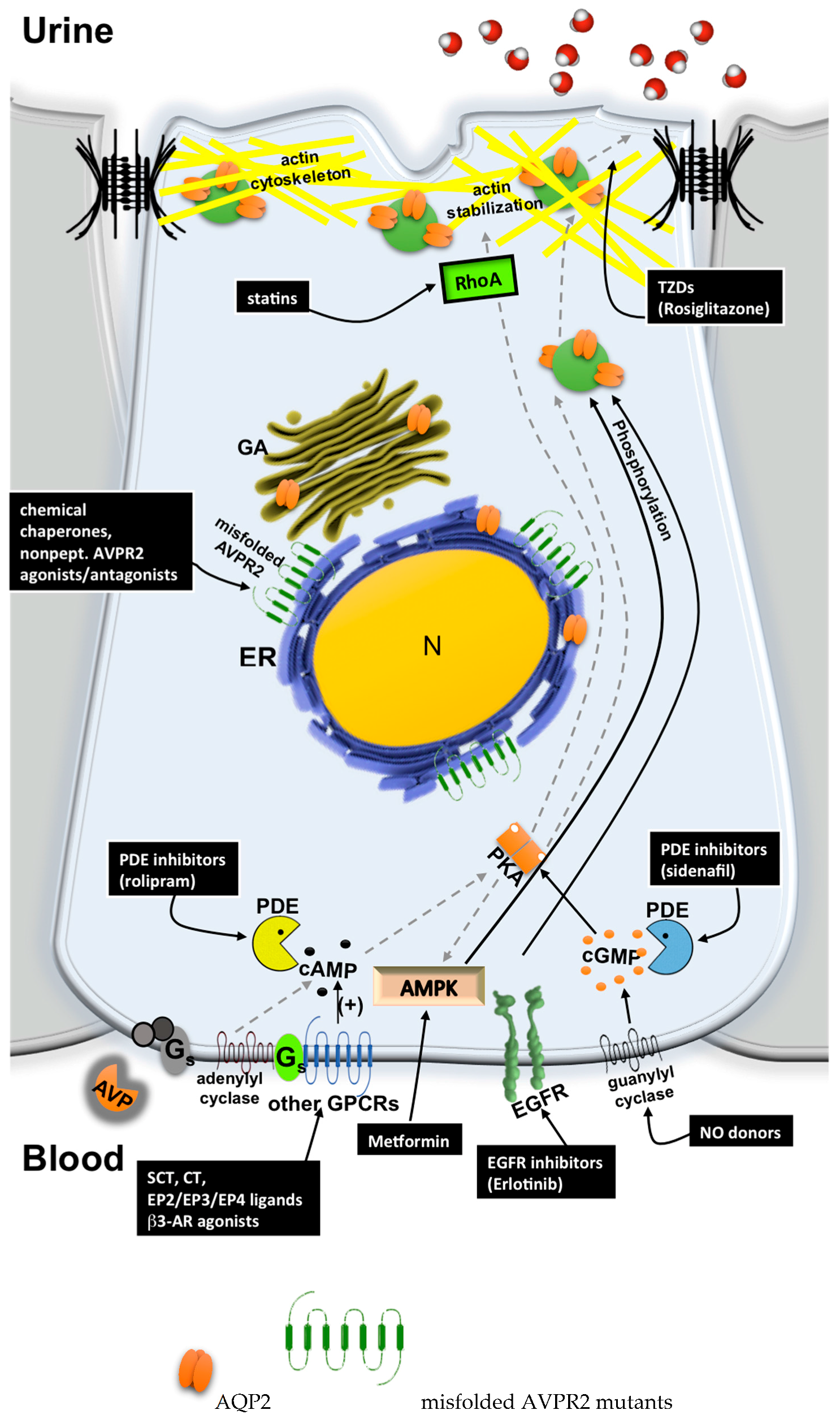
5. Possible Therapeutic Strategies to Cure Congenital NDI
5.1. Chemical Chaperones
5.2. Nonpeptide AVPR2 Antagonists: Pharmacological Chaperones
5.3. Nonpeptide AVPR2 Agonists
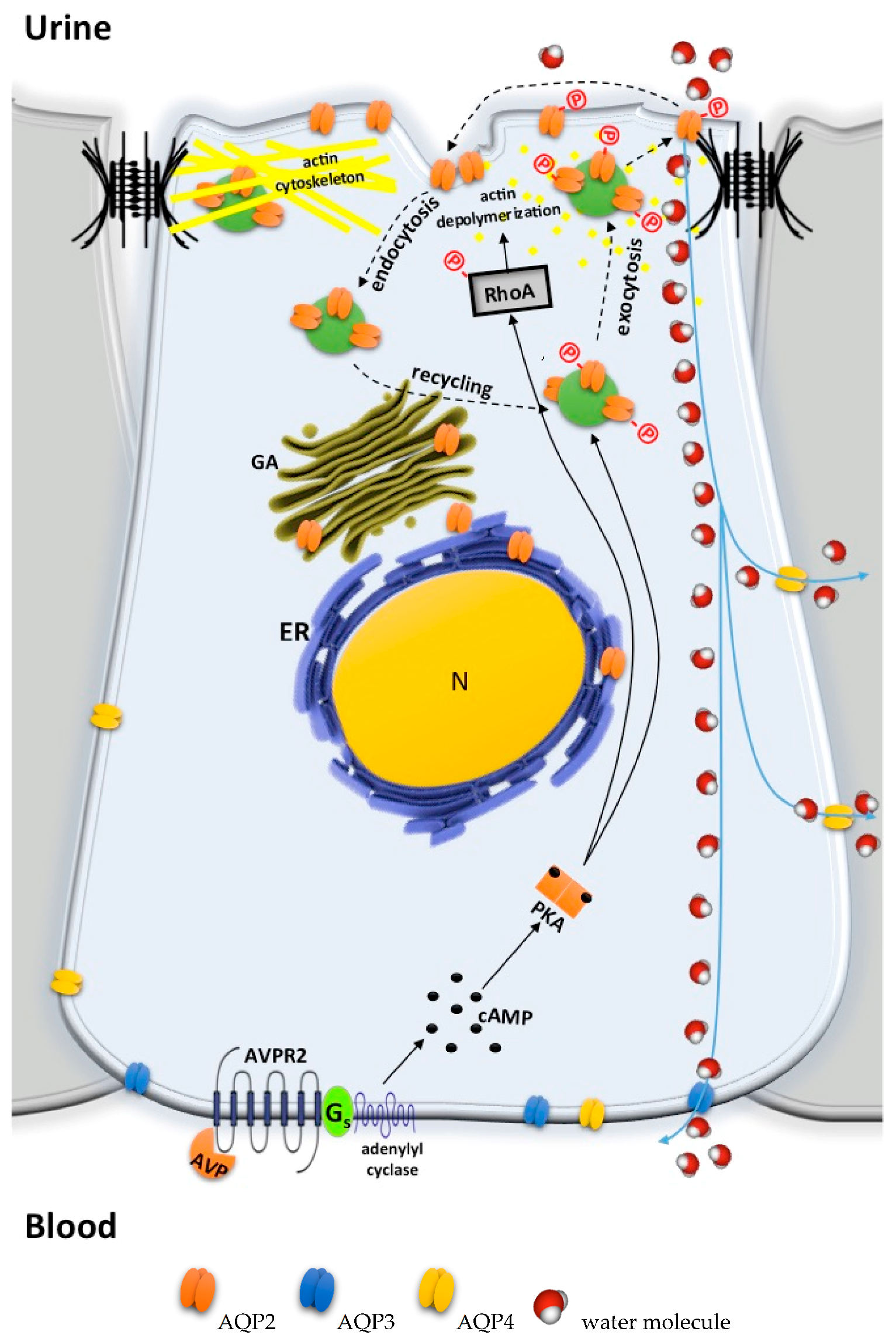
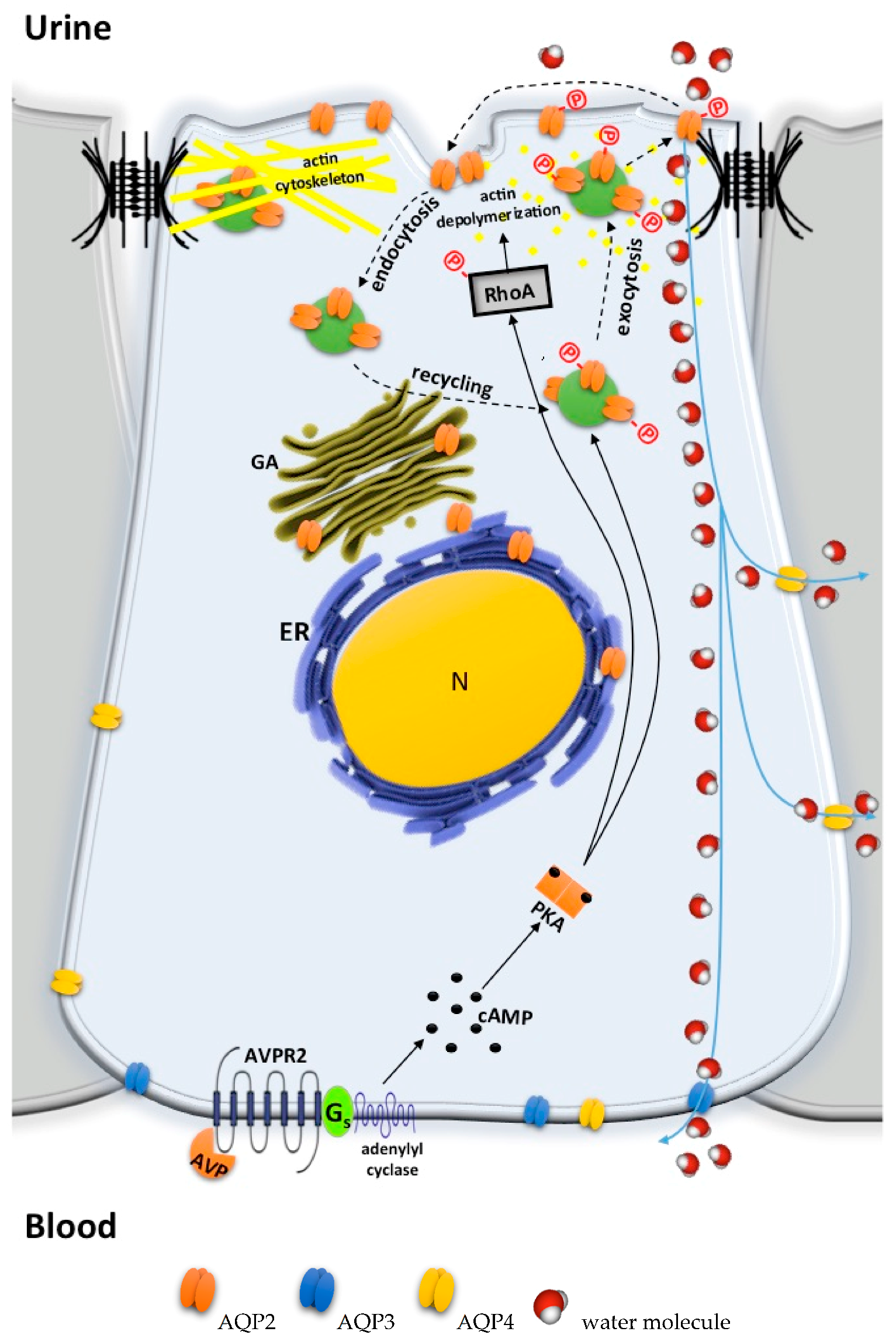
5.4. Bypassing AVPR2 Signaling
- Cytosolic cAMP elevation: activation of other G protein-coupled receptors (GPCRs) coupled to Gs/adenylyl cyclase expressed in the collecting duct (CD) principal cells; the inhibition of phosphodiesterases (PDE).
- Activation of cAMP-independent pathways.
5.4.1. Cytosolic cAMP Elevation
5.4.2. Activation of cAMP-Independent Signaling Cascades
Acknowledgments
Conflicts of Interest
References
- Du, B.; Jiang, X.; Das, A.; Zhou, Q.; Yu, M.; Jin, R.; Zheng, J. Glomerular barrier behaves as an atomically precise bandpass filter in a sub-nanometre regime. Nat. Nanotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kortenoeven, M.L.; Fenton, R.A. Renal aquaporins and water balance disorders. Biochim. Biophys. Acta 2014, 1840, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Thrasher, T.N. Baroreceptor regulation of vasopressin and renin secretion: Low-pressure versus high-pressure receptors. Front. Neuroendocrinol. 1994, 15, 157–196. [Google Scholar] [CrossRef] [PubMed]
- Bourque, C.W. Central mechanisms of osmosensation and systemic osmoregulation. Nat. Rev. Neurosci. 2008, 9, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.L. Physiology of adh secretion. Kidney Int. Suppl. 1987, 21, S20–S26. [Google Scholar] [PubMed]
- Lolait, S.J.; O’Carroll, A.M.; McBride, O.W.; Konig, M.; Morel, A.; Brownstein, M.J. Cloning and characterization of a vasopressin v2 receptor and possible link to nephrogenic diabetes insipidus. Nature 1992, 357, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Tamma, G.; Klussmann, E.; Procino, G.; Svelto, M.; Rosenthal, W.; Valenti, G. Camp-induced aqp2 translocation is associated with rhoa inhibition through rhoa phosphorylation and interaction with rhogdi. J. Cell Sci. 2003, 116, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, K.; Sasaki, S.; Marumo, F. Phosphorylation of serine 256 is required for camp-dependent regulatory exocytosis of the aquaporin-2 water channel. J. Biol. Chem. 1997, 272, 14800–14804. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Chou, C.L.; Marples, D.; Christensen, E.I.; Kishore, B.K.; Knepper, M.A. Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-cd water channels to plasma membrane. Proc. Natl. Acad. Sci. USA 1995, 92, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Sands, J.M.; Layton, H.E. The physiology of urinary concentration: An update. Semin. Nephrol. 2009, 29, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Katsura, T.; Ausiello, D.A.; Brown, D. Direct demonstration of aquaporin-2 water channel recycling in stably transfected llc-pk1 epithelial cells. Am. J. Physiol. 1996, 270, F548–F553. [Google Scholar] [PubMed]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Barak, L.S.; Caron, M.G. Association of beta-arrestin with g protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem. 1999, 274, 32248–32257. [Google Scholar] [CrossRef] [PubMed]
- Baertschi, A.J.; Pence, R.A. Gut-brain signaling of water absorption inhibits vasopressin in rats. Am. J. Physiol. 1995, 268, R236–R247. [Google Scholar] [PubMed]
- Huang, W.; Sved, A.F.; Stricker, E.M. Water ingestion provides an early signal inhibiting osmotically stimulated vasopressin secretion in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R756–R760. [Google Scholar] [PubMed]
- Stricker, E.M.; Hoffmann, M.L. Presystemic signals in the control of thirst, salt appetite, and vasopressin secretion. Physiol. Behav. 2007, 91, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Mandelblat-Cerf, Y.; Kim, A.; Burgess, C.R.; Subramanian, S.; Tannous, B.A.; Lowell, B.B.; Andermann, M.L. Bidirectional anticipation of future osmotic challenges by vasopressin neurons. Neuron 2017, 93, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, C.A.; Lin, Y.C.; Leib, D.E.; Guo, L.; Huey, E.L.; Daly, G.E.; Chen, Y.; Knight, Z.A. Thirst neurons anticipate the homeostatic consequences of eating and drinking. Nature 2016, 537, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, C.A.; Leib, D.E.; Knight, Z.A. Neural circuits underlying thirst and fluid homeostasis. Nat. Rev. Neurosci. 2017, 18, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, D.; Wennemuth, G.; Oka, Y. The cellular mechanism for water detection in the mammalian taste system. Nat. Neurosci. 2017, 20, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Bedford, J.J.; Weggery, S.; Ellis, G.; McDonald, F.J.; Joyce, P.R.; Leader, J.P.; Walker, R.J. Lithium-induced nephrogenic diabetes insipidus: Renal effects of amiloride. Clin. J. Am. Soc. Nephrol. 2008, 3, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Garofeanu, C.G.; Weir, M.; Rosas-Arellano, M.P.; Henson, G.; Garg, A.X.; Clark, W.F. Causes of reversible nephrogenic diabetes insipidus: A systematic review. Am. J. Kidney Dis. 2005, 45, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Castell, D.O.; Sparks, H.A. Nephrogenic diabetes insipidus due to demethylchlortetracycline hydrochloride. JAMA 1965, 193, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Metzger, N.L.; Varney Gill, K.L. Nephrogenic diabetes insipidus induced by two amphotericin b liposomal formulations. Pharmacotherapy 2009, 29, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Quereda, C.; Quereda, C.; Gallego, N.; Antela, A.; Mora, C.; Ortuno, J. Nephrogenic diabetes insipidus and renal tubular acidosis secondary to foscarnet therapy. Am. J. Kidney Dis. 1996, 27, 431–434. [Google Scholar] [CrossRef]
- Roth, H.; Becker, K.L.; Shalhoub, R.J.; Katz, S. Nephrotoxicity of demethylchlortetracycline hydrochloride. A prospective study. Arch. Intern. Med. 1967, 120, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Singer, I.; Rotenberg, D. Demeclocycline-induced nephrogenic diabetes insipidus. In-vivo and in-vitro studies. Ann. Intern. Med. 1973, 79, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Torin, D.E. Nephrogenic diabetes insipidus induced by demethylchlortetracycline (declomycin). Calif. Med. 1967, 107, 420–422. [Google Scholar] [PubMed]
- Marples, D.; Frokiaer, J.; Dorup, J.; Knepper, M.A.; Nielsen, S. Hypokalemia-induced downregulation of aquaporin-2 water channel expression in rat kidney medulla and cortex. J. Clin. Investig. 1996, 97, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, C.; Kwon, T.H.; Knepper, M.A.; Frokiaer, J.; Nielsen, S. Aqp3, p-aqp2, and aqp2 expression is reduced in polyuric rats with hypercalcemia: Prevention by camp-pde inhibitors. Am. J. Physiol. Ren. Physiol. 2002, 283, F1313–F1325. [Google Scholar] [CrossRef] [PubMed]
- Sands, J.M.; Flores, F.X.; Kato, A.; Baum, M.A.; Brown, E.M.; Ward, D.T.; Hebert, S.C.; Harris, H.W. Vasopressin-elicited water and urea permeabilities are altered in imcd in hypercalcemic rats. Am. J. Physiol. 1998, 274, F978–F985. [Google Scholar] [PubMed]
- Bustamante, M.; Hasler, U.; Leroy, V.; de Seigneux, S.; Dimitrov, M.; Mordasini, D.; Rousselot, M.; Martin, P.Y.; Feraille, E. Calcium-sensing receptor attenuates avp-induced aquaporin-2 expression via a calmodulin-dependent mechanism. J. Am. Soc. Nephrol. 2008, 19, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Mastrofrancesco, L.; Tamma, G.; Lasorsa, D.R.; Ranieri, M.; Stringini, G.; Emma, F.; Svelto, M.; Valenti, G. Calcium-sensing receptor and aquaporin 2 interplay in hypercalciuria-associated renal concentrating defect in humans. An in vivo and in vitro study. PLoS ONE 2012, 7, e33145. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Llama, P.; Andrews, P.; Turner, R.; Saggi, S.; Dimari, J.; Kwon, T.H.; Nielsen, S.; Safirstein, R.; Knepper, M.A. Decreased abundance of collecting duct aquaporins in post-ischemic renal failure in rats. J. Am. Soc. Nephrol. 1999, 10, 1658–1668. [Google Scholar] [PubMed]
- Gong, H.; Wang, W.; Kwon, T.H.; Jonassen, T.; Frokiaer, J.; Nielsen, S. Reduced renal expression of aqp2, p-aqp2 and aqp3 in haemorrhagic shock-induced acute renal failure. Nephrol. Dial. Transpl. 2003, 18, 2551–2559. [Google Scholar] [CrossRef]
- Kwon, T.H.; Frokiaer, J.; Fernandez-Llama, P.; Knepper, M.A.; Nielsen, S. Reduced abundance of aquaporins in rats with bilateral ischemia-induced acute renal failure: Prevention by alpha-msh. Am. J. Physiol. 1999, 277, F413–F427. [Google Scholar] [PubMed]
- Kwon, T.H.; Frokiaer, J.; Knepper, M.A.; Nielsen, S. Reduced aqp1, -2, and -3 levels in kidneys of rats with crf induced by surgical reduction in renal mass. Am. J. Physiol. 1998, 275, F724–F741. [Google Scholar] [PubMed]
- Tannen, R.L.; Regal, E.M.; Dunn, M.J.; Schrier, R.W. Vasopressin-resistant hyposthenuria in advanced chronic renal disease. N. Engl. J. Med. 1969, 280, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, I.; McGuinness, S. Vasopressin resistance in chronic renal failure. Evidence for the role of decreased v2 receptor mrna. J. Clin. Investig. 1995, 96, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Wesche, D.; Deen, P.M.; Knoers, N.V. Congenital nephrogenic diabetes insipidus: The current state of affairs. Pediatr. Nephrol. 2012, 27, 2183–2204. [Google Scholar] [CrossRef] [PubMed]
- Van Lieburg, A.F.; Knoers, N.V.; Monnens, L.A. Clinical presentation and follow-up of 30 patients with congenital nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 1999, 10, 1958–1964. [Google Scholar] [PubMed]
- Bichet, D.G. Nephrogenic diabetes insipidus. Adv. Chronic Kidney Dis. 2006, 13, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G. Hereditary polyuric disorders: New concepts and differential diagnosis. Semin. Nephrol. 2006, 26, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, J.A.; van Lieburg, A.F.; Monnens, L.A.; Hulstijn-Dirkmaat, G.M.; Knoers, V.V. Cognitive and psychosocial functioning of patients with congenital nephrogenic diabetes insipidus. Am. J. Med. Genet. 1996, 61, 81–88. [Google Scholar] [CrossRef]
- Rosenthal, W.; Seibold, A.; Antaramian, A.; Lonergan, M.; Arthus, M.F.; Hendy, G.N.; Birnbaumer, M.; Bichet, D.G. Molecular identification of the gene responsible for congenital nephrogenic diabetes insipidus. Nature 1992, 359, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Seibold, A.; Brabet, P.; Rosenthal, W.; Birnbaumer, M. Structure and chromosomal localization of the human antidiuretic hormone receptor gene. Am. J. Hum. Genet. 1992, 51, 1078–1083. [Google Scholar] [PubMed]
- Morello, J.P.; Bichet, D.G. Nephrogenic diabetes insipidus. Annu. Rev. Physiol. 2001, 63, 607–630. [Google Scholar] [CrossRef] [PubMed]
- Birnbaumer, M.; Seibold, A.; Gilbert, S.; Ishido, M.; Barberis, C.; Antaramian, A.; Brabet, P.; Rosenthal, W. Molecular cloning of the receptor for human antidiuretic hormone. Nature 1992, 357, 333–335. [Google Scholar] [CrossRef] [PubMed]
- Knoers, N.V.; Deen, P.M. Molecular and cellular defects in nephrogenic diabetes insipidus. Pediatr. Nephrol. 2001, 16, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Arthus, M.F.; Lonergan, M.; Crumley, M.J.; Naumova, A.K.; Morin, D.; De Marco, L.A.; Kaplan, B.S.; Robertson, G.L.; Sasaki, S.; Morgan, K.; et al. Report of 33 novel avpr2 mutations and analysis of 117 families with x-linked nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2000, 11, 1044–1054. [Google Scholar] [PubMed]
- Faerch, M.; Corydon, T.J.; Rittig, S.; Christensen, J.H.; Hertz, J.M.; Jendle, J. Skewed x-chromosome inactivation causing diagnostic misinterpretation in congenital nephrogenic diabetes insipidus. Scand. J. Urol. Nephrol. 2010, 44, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Ogikubo, S.; Yoshizawa-Ogasawara, A. Correlation between clinical phenotypes and x-inactivation patterns in six female carriers with heterozygote vasopressin type 2 receptor gene mutations. Endocr. J. 2008, 55, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.B.; Rittig, S.; Fenton, R.A. Nephrogenic diabetes insipidus: Essential insights into the molecular background and potential therapies for treatment. Endocr. Rev. 2013, 34, 278–301. [Google Scholar] [CrossRef] [PubMed]
- Robben, J.H.; Knoers, N.V.; Deen, P.M. Characterization of vasopressin v2 receptor mutants in nephrogenic diabetes insipidus in a polarized cell model. Am. J. Physiol. Ren. Physiol. 2005, 289, F265–F272. [Google Scholar] [CrossRef] [PubMed]
- Robben, J.H.; Knoers, N.V.; Deen, P.M. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am. J. Physiol. Ren. Physiol. 2006, 291, F257–F270. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. ER quality control: Towards an understanding at the molecular level. Curr. Opin. Cell Biol. 2001, 13, 431–437. [Google Scholar] [CrossRef]
- Barak, L.S.; Oakley, R.H.; Laporte, S.A.; Caron, M.G. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. USA 2001, 98, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Bernier, V.; Lagace, M.; Lonergan, M.; Arthus, M.F.; Bichet, D.G.; Bouvier, M. Functional rescue of the constitutively internalized v2 vasopressin receptor mutant r137h by the pharmacological chaperone action of sr49059. Mol. Endocrinol. 2004, 18, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Nossent, A.Y.; Robben, J.H.; Deen, P.M.; Vos, H.L.; Rosendaal, F.R.; Doggen, C.J.; Hansen, J.L.; Sheikh, S.P.; Bertina, R.M.; Eikenboom, J.C. Functional variation in the arginine vasopressin 2 receptor as a modifier of human plasma von willebrand factor levels. J. Thromb. Haemost. 2010, 8, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Rosenthal, S.M.; Vargas, G.A.; Fenwick, R.G.; Huang, E.A.; Matsuda-Abedini, M.; Lustig, R.H.; Mathias, R.S.; Portale, A.A.; Miller, W.L.; et al. Nephrogenic syndrome of inappropriate antidiuresis. N. Engl. J. Med. 2005, 352, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, J.; Ayoub, M.A.; Perkovska, S.; Adra-Delenne, A.L.; Mendre, C.; Ranchin, B.; Bricca, G.; Geelen, G.; Mouillac, B.; Durroux, T.; et al. The constitutively active v2 receptor mutants conferring nsiad are weakly sensitive to agonist and antagonist regulation. PLoS ONE 2009, 4, e8383. [Google Scholar] [CrossRef] [PubMed]
- Rochdi, M.D.; Vargas, G.A.; Carpentier, E.; Oligny-Longpre, G.; Chen, S.; Kovoor, A.; Gitelman, S.E.; Rosenthal, S.M.; von Zastrow, M.; Bouvier, M. Functional characterization of vasopressin type 2 receptor substitutions (r137h/c/l) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: Implications for treatments. Mol. Pharmacol. 2010, 77, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Frattini, A.; Zucchi, I.; Villa, A.; Patrosso, C.; Repetto, M.; Susani, L.; Strina, D.; Redolfi, E.; Vezzoni, P.; Romano, G.; et al. Type 2 vasopressin receptor gene, the gene responsible nephrogenic diabetes insipidus, maps to xq28 close to the licam gene. Biochem. Biophys. Res. Commun. 1993, 193, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Knops, N.B.; Bos, K.K.; Kerstjens, M.; van Dael, K.; Vos, Y.J. Nephrogenic diabetes insipidus in a patient with l1 syndrome: A new report of a contiguous gene deletion syndrome including l1cam and avpr2. Am. J. Med. Genet. Part A 2008, 146A, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Tegay, D.H.; Lane, A.H.; Roohi, J.; Hatchwell, E. Contiguous gene deletion involving l1cam and avpr2 causes x-linked hydrocephalus with nephrogenic diabetes insipidus. Am. J. Med. Genet. Part A 2007, 143, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Broides, A.; Ault, B.H.; Arthus, M.F.; Bichet, D.G.; Conley, M.E. Severe combined immunodeficiency associated with nephrogenic diabetes insipidus and a deletion in the xq28 region. Clin. Immunol. 2006, 120, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.; Gartner, J. Genetic and clinical aspects of x-linked hydrocephalus (l1 disease): Mutations in the l1cam gene. Hum. Mutat. 2001, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Izzi, C.; Dallera, N.; Manenti, C.; Caridi, G.; Ghiggeri, G.; Rampoldi, L.; Scolari, F. The case | cystic renal disease, nephrogenic diabetes insipidus, and polycytemia. Kidney Int. 2014, 86, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Fushimi, K.; Sasaki, S.; Marumo, F. Structure of aquaporin-2 vasopressin water channel. J. Biol. Chem. 1996, 271, 5171–5176. [Google Scholar] [PubMed]
- Hoffert, J.D.; Fenton, R.A.; Moeller, H.B.; Simons, B.; Tchapyjnikov, D.; McDill, B.W.; Yu, M.J.; Pisitkun, T.; Chen, F.; Knepper, M.A. Vasopressin-stimulated increase in phosphorylation at ser269 potentiates plasma membrane retention of aquaporin-2. J. Biol. Chem. 2008, 283, 24617–24627. [Google Scholar] [CrossRef] [PubMed]
- Nejsum, L.N.; Zelenina, M.; Aperia, A.; Frokiaer, J.; Nielsen, S. Bidirectional regulation of aqp2 trafficking and recycling: Involvement of aqp2-s256 phosphorylation. Am. J. Physiol. Ren. Physiol. 2005, 288, F930–F938. [Google Scholar] [CrossRef] [PubMed]
- Van Balkom, B.W.; Savelkoul, P.J.; Markovich, D.; Hofman, E.; Nielsen, S.; van der Sluijs, P.; Deen, P.M. The role of putative phosphorylation sites in the targeting and shuttling of the aquaporin-2 water channel. J. Biol. Chem. 2002, 277, 41473–41479. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.B.; Praetorius, J.; Rutzler, M.R.; Fenton, R.A. Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc. Natl. Acad. Sci. USA 2010, 107, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.J.; Matsuzaki, T.; Bouley, R.; Hasler, U.; Qin, Q.H.; Brown, D. The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of aqp2 trafficking in renal epithelial cells. Am. J. Physiol. Ren. Physiol. 2008, 295, F290–F294. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Bichet, D.G.; Sasaki, S.; Kuwahara, M.; Arthus, M.F.; Lonergan, M.; Lin, Y.F. Two novel aquaporin-2 mutations responsible for congenital nephrogenic diabetes insipidus in chinese families. J. Clin. Endocrinol. Metab. 2002, 87, 2694–2700. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.J.; Hall, F.W.; Tarantino, L.M.; Gekakis, N. Diabetes insipidus in mice with a mutation in aquaporin-2. PLoS Genet. 2005, 1, e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamarappoo, B.K.; Verkman, A.S. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J. Clin. Investig. 1998, 101, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Bichet, D.G.; Hoefs, S.; Savelkoul, P.J.; Konings, I.B.; De Mattia, F.; Graat, M.P.; Arthus, M.F.; Lonergan, M.; Fujiwara, T.M.; et al. Cell-biologic and functional analyses of five new aquaporin-2 missense mutations that cause recessive nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2002, 13, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Aglio, V.; Tamma, G.; D’Apolito, M.; Addabbo, F.; Procino, G.; Simonetti, M.C.; Montini, G.; Gesualdo, L.; Debler, E.W.; et al. Characterization of two novel missense mutations in the aqp2 gene causing nephrogenic diabetes insipidus. Nephron. Physiol. 2007, 105, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Nadeau, A.; Lussier, Y.; Arthus, M.F.; Lonergan, M.; Martinez-Aguayo, A.; Riveira-Munoz, E.; Devuyst, O.; Bissonnette, P.; Bichet, D.G. New autosomal recessive mutations in aquaporin-2 causing nephrogenic diabetes insipidus through deficient targeting display normal expression in xenopus oocytes. J. Physiol. 2010, 588, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Iwai, K.; Ooeda, T.; Igarashi, T.; Ogawa, E.; Katsushima, Y.; Shinbo, I.; Uchida, S.; Terada, Y.; Arthus, M.F.; et al. Three families with autosomal dominant nephrogenic diabetes insipidus caused by aquaporin-2 mutations in the c-terminus. Am. J. Hum. Genet. 2001, 69, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Sohara, E.; Rai, T.; Yang, S.S.; Uchida, K.; Nitta, K.; Horita, S.; Ohno, M.; Harada, A.; Sasaki, S.; Uchida, S. Pathogenesis and treatment of autosomal-dominant nephrogenic diabetes insipidus caused by an aquaporin 2 mutation. Proc. Natl. Acad. Sci. USA 2006, 103, 14217–14222. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Kuwahara, M.; Kurihara, H.; Sakai, T.; Terada, Y.; Marumo, F.; Sasaki, S. Pathogenesis of nephrogenic diabetes insipidus by aquaporin-2 c-terminus mutations. Kidney Int. 2003, 64, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Mulders, S.M.; Bichet, D.G.; Rijss, J.P.; Kamsteeg, E.J.; Arthus, M.F.; Lonergan, M.; Fujiwara, M.; Morgan, K.; Leijendekker, R.; van der Sluijs, P.; et al. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the golgi complex. J. Clin. Investig. 1998, 102, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Carmosino, M.; Marin, O.; Brunati, A.M.; Contri, A.; Pinna, L.A.; Mannucci, R.; Nielsen, S.; Kwon, T.H.; Svelto, M.; et al. Ser-256 phosphorylation dynamics of aquaporin 2 during maturation from the er to the vesicular compartment in renal cells. FASEB J. 2003, 17, 1886–1888. [Google Scholar] [PubMed]
- Kamsteeg, E.J.; Bichet, D.G.; Konings, I.B.; Nivet, H.; Lonergan, M.; Arthus, M.F.; van Os, C.H.; Deen, P.M. Reversed polarized delivery of an aquaporin-2 mutant causes dominant nephrogenic diabetes insipidus. J. Cell Biol. 2003, 163, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, W.; Antaramian, A.; Gilbert, S.; Birnbaumer, M. Nephrogenic diabetes insipidus. A v2 vasopressin receptor unable to stimulate adenylyl cyclase. J. Biol. Chem. 1993, 268, 13030–13033. [Google Scholar] [PubMed]
- Neocleous, V.; Skordis, N.; Shammas, C.; Efstathiou, E.; Mastroyiannopoulos, N.P.; Phylactou, L.A. Identification and characterization of a novel x-linked avpr2 mutation causing partial nephrogenic diabetes insipidus: A case report and review of the literature. Metab. Clin. Exp. 2012, 61, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Sato, T.; Yajima-Shoji, Y.; Sato, J.; Manaka, K.; Eda-Hashimoto, M.; Ootaki, M.; Matsumoto, N.; Nangaku, M.; Iiri, T. Analysis of the v2 vasopressin receptor (v2r) mutations causing partial nephrogenic diabetes insipidus highlights a sustainable signaling by a non-peptide v2r agonist. J. Biol. Chem. 2016, 291, 22460–22471. [Google Scholar] [CrossRef] [PubMed]
- Shinbo, I.; Fushimi, K.; Kasahara, M.; Yamauchi, K.; Sasaki, S.; Marumo, F. Functional analysis of aquaporin-2 mutants associated with nephrogenic diabetes insipidus by yeast expression. Am. J. Physiol. 1999, 277, F734–F741. [Google Scholar] [PubMed]
- Kamsteeg, E.J.; Wormhoudt, T.A.; Rijss, J.P.; van Os, C.H.; Deen, P.M. An impaired routing of wild-type aquaporin-2 after tetramerization with an aquaporin-2 mutant explains dominant nephrogenic diabetes insipidus. EMBO J. 1999, 18, 2394–2400. [Google Scholar] [CrossRef] [PubMed]
- Preisser, L.; Teillet, L.; Aliotti, S.; Gobin, R.; Berthonaud, V.; Chevalier, J.; Corman, B.; Verbavatz, J.M. Downregulation of aquaporin-2 and -3 in aging kidney is independent of v(2) vasopressin receptor. Am. J. Physiol. Ren. Physiol. 2000, 279, F144–F152. [Google Scholar]
- Trinh-Trang-Tan, M.M.; Geelen, G.; Teillet, L.; Corman, B. Urea transporter expression in aging kidney and brain during dehydration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R1355–R1365. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Yang, B.; Gillespie, A.; Carlson, E.J.; Epstein, C.J.; Verkman, A.S. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J. Biol. Chem. 1998, 273, 4296–4299. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Song, Y.; Yang, B.; Gillespie, A.; Carlson, E.J.; Epstein, C.J.; Verkman, A.S. Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc. Natl. Acad. Sci. USA 2000, 97, 4386–4391. [Google Scholar] [CrossRef] [PubMed]
- Fenton, R.A.; Knepper, M.A. Mouse models and the urinary concentrating mechanism in the new millennium. Physiol. Rev. 2007, 87, 1083–1112. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Gillespie, A.; Carlson, E.J.; Epstein, C.J.; Verkman, A.S. Neonatal mortality in an aquaporin-2 knock-in mouse model of recessive nephrogenic diabetes insipidus. J. Biol. Chem. 2001, 276, 2775–2779. [Google Scholar] [CrossRef] [PubMed]
- Rojek, A.; Fuchtbauer, E.M.; Kwon, T.H.; Frokiaer, J.; Nielsen, S. Severe urinary concentrating defect in renal collecting duct-selective aqp2 conditional-knockout mice. Proc. Natl. Acad. Sci. USA 2006, 103, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- McDill, B.W.; Li, S.Z.; Kovach, P.A.; Ding, L.; Chen, F. Congenital progressive hydronephrosis (cph) is caused by an s256l mutation in aquaporin-2 that affects its phosphorylation and apical membrane accumulation. Proc. Natl. Acad. Sci. USA 2006, 103, 6952–6957. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.P.; Cao, X.R.; Qu, J.; Volk, K.A.; Kirby, P.; Williamson, R.A.; Stokes, J.B.; Yang, B. Nephrogenic diabetes insipidus in mice caused by deleting cooh-terminal tail of aquaporin-2. Am. J. Physiol. Ren. Physiol. 2007, 292, F1334–F1344. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhao, D.; Verkman, A.S. Hsp90 inhibitor partially corrects nephrogenic diabetes insipidus in a conditional knock-in mouse model of aquaporin-2 mutation. FASEB J. 2009, 23, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Schoneberg, T.; Liu, J.; Schulz, A.; Ecelbarger, C.A.; Promeneur, D.; Nielsen, S.; Sheng, H.; Grinberg, A.; Deng, C.; et al. Generation and phenotype of mice harboring a nonsense mutation in the v2 vasopressin receptor gene. J. Clin. Investig. 2000, 106, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Schliebe, N.; Strotmann, R.; Busse, K.; Mitschke, D.; Biebermann, H.; Schomburg, L.; Kohrle, J.; Bar, J.; Rompler, H.; Wess, J.; et al. V2 vasopressin receptor deficiency causes changes in expression and function of renal and hypothalamic components involved in electrolyte and water homeostasis. Am. J. Physiol. Ren. Physiol. 2008, 295, F1177–F1190. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Chou, C.L.; Li, B.; Gavrilova, O.; Eisner, C.; Schnermann, J.; Anderson, S.A.; Deng, C.X.; Knepper, M.A.; Wess, J. A selective ep4 pge2 receptor agonist alleviates disease in a new mouse model of x-linked nephrogenic diabetes insipidus. J. Clin. Investig. 2009, 119, 3115–3126. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.A.; Lee, J.W.; Chou, C.L.; Knepper, M.A. Tolvaptan as a tool in renal physiology. Am. J. Physiol. Ren. Physiol. 2014, 306, F359–F366. [Google Scholar] [CrossRef] [PubMed]
- Earley, L.E.; Orloff, J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J. Clin. Investig. 1962, 41, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Knoers, N.; Monnens, L.A. Amiloride-hydrochlorothiazide versus indomethacin-hydrochlorothiazide in the treatment of nephrogenic diabetes insipidus. J. Pediatr. 1990, 117, 499–502. [Google Scholar] [CrossRef]
- Libber, S.; Harrison, H.; Spector, D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J. Pediatr. 1986, 108, 305–311. [Google Scholar] [CrossRef]
- Monnens, L.; Jonkman, A.; Thomas, C. Response to indomethacin and hydrochlorothiazide in nephrogenic diabetes insipidus. Clin. Sci. 1984, 66, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Dayal, D.; Verma Attri, S.; Kumar Bhalla, A.; Kumar, R. Response to low dose indomethacin in two children with nephrogenic diabetes insipidus. Pediatr. Endocrinol. Diabetes Metab. 2015, 20, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D.; Reardon, S. Embryo editing sparks epic debate. Nature 2015, 520, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Robben, J.H.; Sze, M.; Knoers, N.V.; Deen, P.M. Functional rescue of vasopressin v2 receptor mutants in mdck cells by pharmacochaperones: Relevance to therapy of nephrogenic diabetes insipidus. Am. J. Physiol. Ren. Physiol. 2007, 292, F253–F260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, G.S.; Tang, W.H. Vasopressin receptor antagonists: Will the ‘vaptans’ fulfill their promise? JAMA 2004, 291, 2017–2018. [Google Scholar] [CrossRef] [PubMed]
- Mouillac, B.; Chini, B.; Balestre, M.N.; Elands, J.; Trumpp-Kallmeyer, S.; Hoflack, J.; Hibert, M.; Jard, S.; Barberis, C. The binding site of neuropeptide vasopressin v1a receptor. Evidence for a major localization within transmembrane regions. J. Biol. Chem. 1995, 270, 25771–25777. [Google Scholar] [CrossRef] [PubMed]
- Los, E.L.; Deen, P.M.; Robben, J.H. Potential of nonpeptide (ant)agonists to rescue vasopressin v2 receptor mutants for the treatment of x-linked nephrogenic diabetes insipidus. J. Neuroendocrinol. 2010, 22, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Morello, J.P.; Salahpour, A.; Laperriere, A.; Bernier, V.; Arthus, M.F.; Lonergan, M.; Petaja-Repo, U.; Angers, S.; Morin, D.; Bichet, D.G.; et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded v2 vasopressin receptor mutants. J. Clin. Investig. 2000, 105, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Wuller, S.; Wiesner, B.; Loffler, A.; Furkert, J.; Krause, G.; Hermosilla, R.; Schaefer, M.; Schulein, R.; Rosenthal, W.; Oksche, A. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin v2 receptors. J. Biol. Chem. 2004, 279, 47254–47263. [Google Scholar] [CrossRef] [PubMed]
- Bernier, V.; Morello, J.P.; Zarruk, A.; Debrand, N.; Salahpour, A.; Lonergan, M.; Arthus, M.F.; Laperriere, A.; Brouard, R.; Bouvier, M.; et al. Pharmacologic chaperones as a potential treatment for x-linked nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2006, 17, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Janovick, J.A.; Bannister, T.D.; Shumate, J.; Scampavia, L.; Conn, P.M.; Spicer, T.P. Identification of potential pharmacoperones capable of rescuing the functionality of misfolded vasopressin 2 receptor involved in nephrogenic diabetes insipidus. J. Biomol. Screen. 2016, 21, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Robben, J.H.; Knoers, N.V.; Deen, P.M. Regulation of the vasopressin v2 receptor by vasopressin in polarized renal collecting duct cells. Mol. Biol. Cell 2004, 15, 5693–5699. [Google Scholar] [CrossRef] [PubMed]
- Robben, J.H.; Kortenoeven, M.L.; Sze, M.; Yae, C.; Milligan, G.; Oorschot, V.M.; Klumperman, J.; Knoers, N.V.; Deen, P.M. Intracellular activation of vasopressin v2 receptor mutants in nephrogenic diabetes insipidus by nonpeptide agonists. Proc. Natl. Acad. Sci. USA 2009, 106, 12195–12200. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Hirano, T.; Tsujimae, K.; Aoyama, M.; Kondo, K.; Yamamura, Y.; Mori, T.; Tominaga, M. Antidiuretic effects of a nonpeptide vasopressin v(2)-receptor agonist, opc-51803, administered orally to rats. J. Pharmacol. Exp. Ther. 2000, 295, 1005–1011. [Google Scholar] [PubMed]
- Jean-Alphonse, F.; Perkovska, S.; Frantz, M.C.; Durroux, T.; Mejean, C.; Morin, D.; Loison, S.; Bonnet, D.; Hibert, M.; Mouillac, B.; et al. Biased agonist pharmacochaperones of the avp v2 receptor may treat congenital nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2009, 20, 2190–2203. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Carmosino, M.; Milano, S.; Dal Monte, M.; Schena, G.; Mastrodonato, M.; Gerbino, A.; Bagnoli, P.; Svelto, M. Beta3 adrenergic receptor in the kidney may be a new player in sympathetic regulation of renal function. Kidney Int. 2016, 90, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.Y.; Chung, S.C.; Lam, A.K.; Tam, S.; Chung, S.K.; Chow, B.K. Phenotypes developed in secretin receptor-null mice indicated a role for secretin in regulating renal water reabsorption. Mol. Cell. Biol. 2007, 27, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Charlton, C.G.; Quirion, R.; Handelmann, G.E.; Miller, R.L.; Jensen, R.T.; Finkel, M.S.; O’Donohue, T.L. Secretin receptors in the rat kidney: Adenylate cyclase activation and renal effects. Peptides 1986, 7, 865–871. [Google Scholar] [CrossRef]
- Procino, G.; Milano, S.; Carmosino, M.; Barbieri, C.; Nicoletti, M.C.; H Li, J.; Wess, J.; Svelto, M. Combination of secretin and fluvastatin ameliorates the polyuria associated with x-linked nephrogenic diabetes insipidus in mice. Kidney Int. 2014, 86, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Barbieri, C.; Carmosino, M.; Tamma, G.; Milano, S.; De Benedictis, L.; Mola, M.G.; Lazo-Fernandez, Y.; Valenti, G.; Svelto, M. Fluvastatin modulates renal water reabsorption in vivo through increased aqp2 availability at the apical plasma membrane of collecting duct cells. Pflug. Arch. Eur. J. Physiol. 2011, 462, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Carney, S.; Thompson, L. Acute effect of calcitonin on rat renal electrolyte transport. Am. J. Physiol. 1981, 240, F12–F16. [Google Scholar] [PubMed]
- De Rouffignac, C.; Elalouf, J.M. Effects of calcitonin on the renal concentrating mechanism. Am. J. Physiol. 1983, 245, F506–F511. [Google Scholar] [PubMed]
- Chai, S.Y.; Christopoulos, G.; Cooper, M.E.; Sexton, P.M. Characterization of binding sites for amylin, calcitonin, and cgrp in primate kidney. Am. J. Physiol. 1998, 274, F51–F62. [Google Scholar] [PubMed]
- Morel, F. Sites of hormone action in the mammalian nephron. Am. J. Physiol. 1981, 240, F159–F164. [Google Scholar] [PubMed]
- Chabardès, D.; Imbert-Teboul, M.; Montégut, M.; Clique, A.; Morel, F. Distribution of calcitonin-sensitive adenylate cyclase activity along the rabbit kidney tubule. Proc. Natl. Acad. Sci. USA 1976, 73, 3608–3612. [Google Scholar] [CrossRef] [PubMed]
- Morel, F.; Chabardès, D.; Imbert-Teboul, M.; Le Bouffant, F.; Hus-Citharel, A.; Montégut, M. Multiple hormonal control of adenylate cyclase in distal segments of the rat kidney. Kidney Int. Suppl. 1982, 11, S55–S62. [Google Scholar] [PubMed]
- Sexton, P.M.; Adam, W.R.; Moseley, J.M.; Martin, T.J.; Mendelsohn, F.A. Localization and characterization of renal calcitonin receptors by in vitro autoradiography. Kidney Int. 1987, 32, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Bouley, R.; Lu, H.A.; Nunes, P.; Da Silva, N.; McLaughlin, M.; Chen, Y.; Brown, D. Calcitonin has a vasopressin-like effect on aquaporin-2 trafficking and urinary concentration. J. Am. Soc. Nephrol. 2011, 22, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Uawithya, P.; Pisitkun, T.; Ruttenberg, B.E.; Knepper, M.A. Transcriptional profiling of native inner medullary collecting duct cells from rat kidney. Physiol. Genom. 2008, 32, 229–253. [Google Scholar] [CrossRef] [PubMed]
- Olesen, E.T.; Rützler, M.R.; Moeller, H.B.; Praetorius, H.A.; Fenton, R.A. Vasopressin-independent targeting of aquaporin-2 by selective e-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. USA 2011, 108, 12949–12954. [Google Scholar] [CrossRef] [PubMed]
- Hockel, G.M.; Cowley, A.W. Prostaglandin e2-induced hypertension in conscious dogs. Am. J. Physiol. 1979, 237, H449–H454. [Google Scholar] [PubMed]
- Breyer, M.D.; Breyer, R.M. G protein-coupled prostanoid receptors and the kidney. Annu. Rev. Physiol. 2001, 63, 579–605. [Google Scholar] [CrossRef] [PubMed]
- Bouley, R.; Hasler, U.; Lu, H.A.; Nunes, P.; Brown, D. Bypassing vasopressin receptor signaling pathways in nephrogenic diabetes insipidus. Semin. Nephrol. 2008, 28, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Takahashi, N.; Abe, T.; Abe, K. Two isoforms of the rat kidney ep3 receptor derived by alternative rna splicing: Intrarenal expression co-localization. Biochem. Biophys. Res. Commun. 1994, 199, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Dousa, T.P. Cyclic-3′,5′-nucleotide phosphodiesterase isozymes in cell biology and pathophysiology of the kidney. Kidney Int. 1999, 55, 29–62. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Ruel, N.; Arthus, M.F.; Lonergan, M. Rolipram, a phosphodiesterase inhibitor, in the treatment of two male patients with congenital nephrogenic diabetes insipidus. Nephron 1990, 56, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Bouley, R.; Breton, S.; Sun, T.; McLaughlin, M.; Nsumu, N.N.; Lin, H.Y.; Ausiello, D.A.; Brown, D. Nitric oxide and atrial natriuretic factor stimulate cgmp-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J. Clin. Investig. 2000, 106, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Bouley, R.; Pastor-Soler, N.; Cohen, O.; McLaughlin, M.; Breton, S.; Brown, D. Stimulation of aqp2 membrane insertion in renal epithelial cells in vitro and in vivo by the cgmp phosphodiesterase inhibitor sildenafil citrate (viagra). Am. J. Physiol. Ren. Physiol. 2005, 288, F1103–F1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, C.; Nejsum, L.N.; Li, H.; Kim, S.W.; Kwon, T.H.; Jonassen, T.E.; Knepper, M.A.; Thomsen, K.; Frøkiaer, J.; et al. Biphasic effects of anp infusion in conscious, euvolumic rats: Roles of aqp2 and enac trafficking. Am. J. Physiol. Ren. Physiol. 2006, 290, F530–F541. [Google Scholar] [CrossRef] [PubMed]
- Marples, D.; Christensen, B.M.; Frokiaer, J.; Knepper, M.A.; Nielsen, S. Dehydration reverses vasopressin antagonist-induced diuresis and aquaporin-2 downregulation in rats. Am. J. Physiol. 1998, 275, F400–F409. [Google Scholar] [PubMed]
- Marples, D.; Knepper, M.A.; Christensen, E.I.; Nielsen, S. Redistribution of aquaporin-2 water channels induced by vasopressin in rat kidney inner medullary collecting duct. Am. J. Physiol. 1995, 269, C655–C664. [Google Scholar] [PubMed]
- Matsumura, Y.; Uchida, S.; Rai, T.; Sasaki, S.; Marumo, F. Transcriptional regulation of aquaporin-2 water channel gene by camp. J. Am. Soc. Nephrol. 1997, 8, 861–867. [Google Scholar] [PubMed]
- Yasui, M.; Zelenin, S.M.; Celsi, G.; Aperia, A. Adenylate cyclase-coupled vasopressin receptor activates aqp2 promoter via a dual effect on cre and ap1 elements. Am. J. Physiol. 1997, 272, F443–F450. [Google Scholar] [PubMed]
- Boone, M.; Kortenoeven, M.; Robben, J.H.; Deen, P.M. Effect of the cgmp pathway on aqp2 expression and translocation: Potential implications for nephrogenic diabetes insipidus. Nephrol. Dial. Transpl. 2010, 25, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Sanches, T.R.; Volpini, R.A.; Massola Shimizu, M.H.; Bragança, A.C.; Oshiro-Monreal, F.; Seguro, A.C.; Andrade, L. Sildenafil reduces polyuria in rats with lithium-induced ndi. Am. J. Physiol. Ren. Physiol. 2012, 302, F216–F225. [Google Scholar] [CrossRef] [PubMed]
- Assadi, F.; Sharbaf, F.G. Sildenafil for the treatment of congenital nephrogenic diabetes insipidus. Am. J. Nephrol. 2015, 42, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Gow, C.B.; Phillips, P.A. Epidermal growth factor as a diuretic in sheep. J. Physiol. 1994, 477, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.W.; Nomura, N.; Nair, A.V.; Pathomthongtaweechai, N.; Ueberdiek, L.; Lu, H.A.; Brown, D.; Bouley, R. Egf receptor inhibition by erlotinib increases aquaporin 2-mediated renal water reabsorption. J. Am. Soc. Nephrol. 2016, 27, 3105–3116. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Nunes, P.; Bouley, R.; Nair, A.V.; Shaw, S.; Ueda, E.; Pathomthongtaweechai, N.; Lu, H.A.; Brown, D. High-throughput chemical screening identifies ag-490 as a stimulator of aquaporin 2 membrane expression and urine concentration. Am. J. Physiol. Cell Physiol. 2014, 307, C597–C605. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor creb. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of amp-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.D.; Wang, Y.; Blount, M.A.; Molina, P.A.; LaRocque, L.M.; Ruiz, J.A.; Sands, J.M. Metformin, an ampk activator, stimulates the phosphorylation of aquaporin 2 and urea transporter a1 in inner medullary collecting ducts. Am. J. Physiol. Ren. Physiol. 2016, 310, F1008–F1012. [Google Scholar] [CrossRef] [PubMed]
- Efe, O.; Klein, J.D.; LaRocque, L.M.; Ren, H.; Sands, J.M. Metformin improves urine concentration in rodents with nephrogenic diabetes insipidus. JCI Insight 2016, 1, e88409. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Barbieri, C.; Carmosino, M.; Rizzo, F.; Valenti, G.; Svelto, M. Lovastatin-induced cholesterol depletion affects both apical sorting and endocytosis of aquaporin-2 in renal cells. Am. J. Physiol. Ren. Physiol. 2010, 298, F266–F278. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; La Rovere, R.M.; Fulle, S.; Miscia, S.; Marchisio, M.; Pierdomenico, L.; Lanuti, P.; Procino, G.; Barbieri, C.; Svelto, M.; et al. Extracellular gtp is a potent water-transport regulator via aquaporin 5 plasma-membrane insertion in m1-ccd epithelial cortical collecting duct cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 33, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, Y.; Bouley, R.; Chen, Y.; Matsuzaki, T.; Nunes, P.; Hasler, U.; Brown, D.; Lu, H.A. Simvastatin enhances aquaporin-2 surface expression and urinary concentration in vasopressin-deficient brattleboro rats through modulation of rho gtpase. Am. J. Physiol. Ren. Physiol. 2011, 301, F309–F318. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Maiolo, D.; Barbieri, C.; Milano, S.; Squatrito, S.; Svelto, M.; Gullo, D. Fluvastatin increases aqp2 urine excretion in a dyslipidemic patient with nephrogenic diabetes insipidus: An in vivo and in vitro study. J. Diabetes Metab. 2014, 5, 408. [Google Scholar] [CrossRef]
- Bonfrate, L.; Procino, G.; Wang, D.Q.; Svelto, M.; Portincasa, P. A novel therapeutic effect of statins on nephrogenic diabetes insipidus. J. Cell. Mol. Med. 2015, 19, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Portincasa, P.; Mastrofrancesco, L.; Castorani, L.; Bonfrate, L.; Addabbo, F.; Carmosino, M.; Di Ciaula, A.; Svelto, M. Simvastatin increases aqp2 urinary excretion in hypercholesterolemic patients: A pleiotropic effect of interest for patients with impaired aqp2 trafficking. Clin. Pharmacol. Ther. 2016, 99, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Elie, D.; Segal, M.; Low, N.C.; Mucsi, I.; Holcroft, C.; Shulman, K.; Looper, K.J.; Rej, S. Statins in the prevention of lithium-associated diabetes insipidus: Preliminary findings. Kidney Int. 2015, 87, 862. [Google Scholar] [CrossRef] [PubMed]
- Beltowski, J.; Rachanczyk, J.; Wlodarczyk, M. Thiazolidinedione-induced fluid retention: Recent insights into the molecular mechanisms. PPAR Res. 2013, 2013, 628628. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Wyne, K.L.; McGuire, D.K. Thiazolidinediones, peripheral oedema and congestive heart failure: What is the evidence? Diabetes Vasc. Dis. Res. 2005, 2, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Knepper, M.A.; Hu, X.; Verbalis, J.G.; Ecelbarger, C.A. Rosiglitazone activates renal sodium- and water-reabsorptive pathways and lowers blood pressure in normal rats. J. Pharmacol. Exp. Ther. 2004, 308, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, A.; Kohan, D.E.; Nelson, R.D.; Gonzalez, F.J.; Yang, T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc. Natl. Acad. Sci. USA 2005, 102, 9406–9411. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Hummler, E.; Rieg, T.; Pochynyuk, O.; Bugaj, V.; Schroth, J.; Dechenes, G.; Rossier, B.; Cunard, R.; Stockand, J. Thiazolidinedione-induced fluid retention is independent of collecting duct alphaenac activity. J. Am. Soc. Nephrol. 2009, 20, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Gerbino, A.; Milano, S.; Nicoletti, M.C.; Mastrofrancesco, L.; Carmosino, M.; Svelto, M. Rosiglitazone promotes aqp2 plasma membrane expression in renal cells via a ca-dependent/camp-independent mechanism. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 35, 1070–1085. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene/Mutation Type | Location | Disease | Number of Mutations |
|---|---|---|---|
| AQP2 | |||
| Missense/nonsense | 12q12–q13 | Autosomal recessive NDI | 46 |
| 12q12–q13 | Autosomal dominant NDI | 4 | |
| Splicing | 12q12–q13 | Autosomal recessive NDI | 4 |
| Small deletions | 12q12–q13 | Autosomal recessive NDI | 3 |
| 12q12–q13 | Autosomal dominant NDI | 6 | |
| Small insertions | 12q12–q13 | Autosomal dominant NDI | 1 |
| 12q12–q13 | Autosomal recessive NDI | 1 | |
| TOTAL | 65 | ||
| AVPR2 | |||
| Missense/nonsense | Xq28 | X-linked NDI | 166 (1 partial) |
| Splicing | Xq28 | X-linked NDI | 4 |
| Small deletions | Xq28 | X-linked NDI | 52 |
| Small insertions | Xq28 | X-linked NDI | 19 |
| Small indels | Xq28 | X-linked NDI | 5 |
| Gross deletions | Xq28 | X-linked NDI | 23 |
| Gross insertions | Xq28 | X-linked NDI | 1 |
| Complex rearrangements | Xq28 | X-linked NDI | 4 |
| TOTAL | 274 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milano, S.; Carmosino, M.; Gerbino, A.; Svelto, M.; Procino, G. Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update. Int. J. Mol. Sci. 2017, 18, 2385. https://doi.org/10.3390/ijms18112385
Milano S, Carmosino M, Gerbino A, Svelto M, Procino G. Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update. International Journal of Molecular Sciences. 2017; 18(11):2385. https://doi.org/10.3390/ijms18112385
Chicago/Turabian StyleMilano, Serena, Monica Carmosino, Andrea Gerbino, Maria Svelto, and Giuseppe Procino. 2017. "Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update" International Journal of Molecular Sciences 18, no. 11: 2385. https://doi.org/10.3390/ijms18112385