The MEK Inhibitors Trametinib and Cobimetinib Induce a Type I Interferon Response in Human Keratinocytes

Abstract
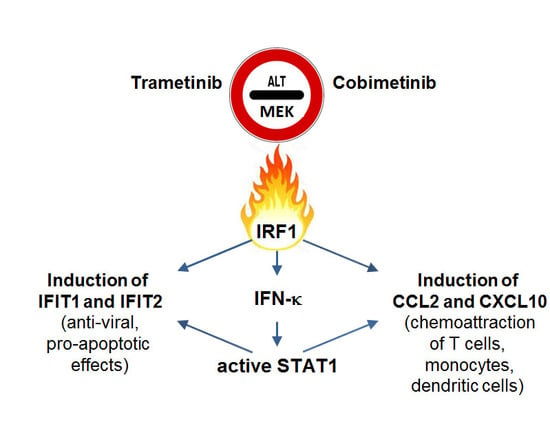
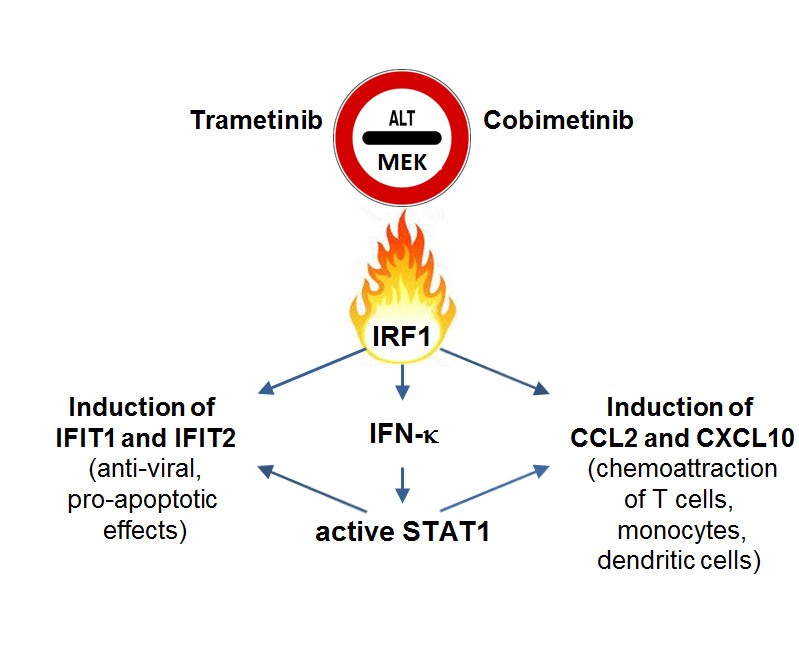
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
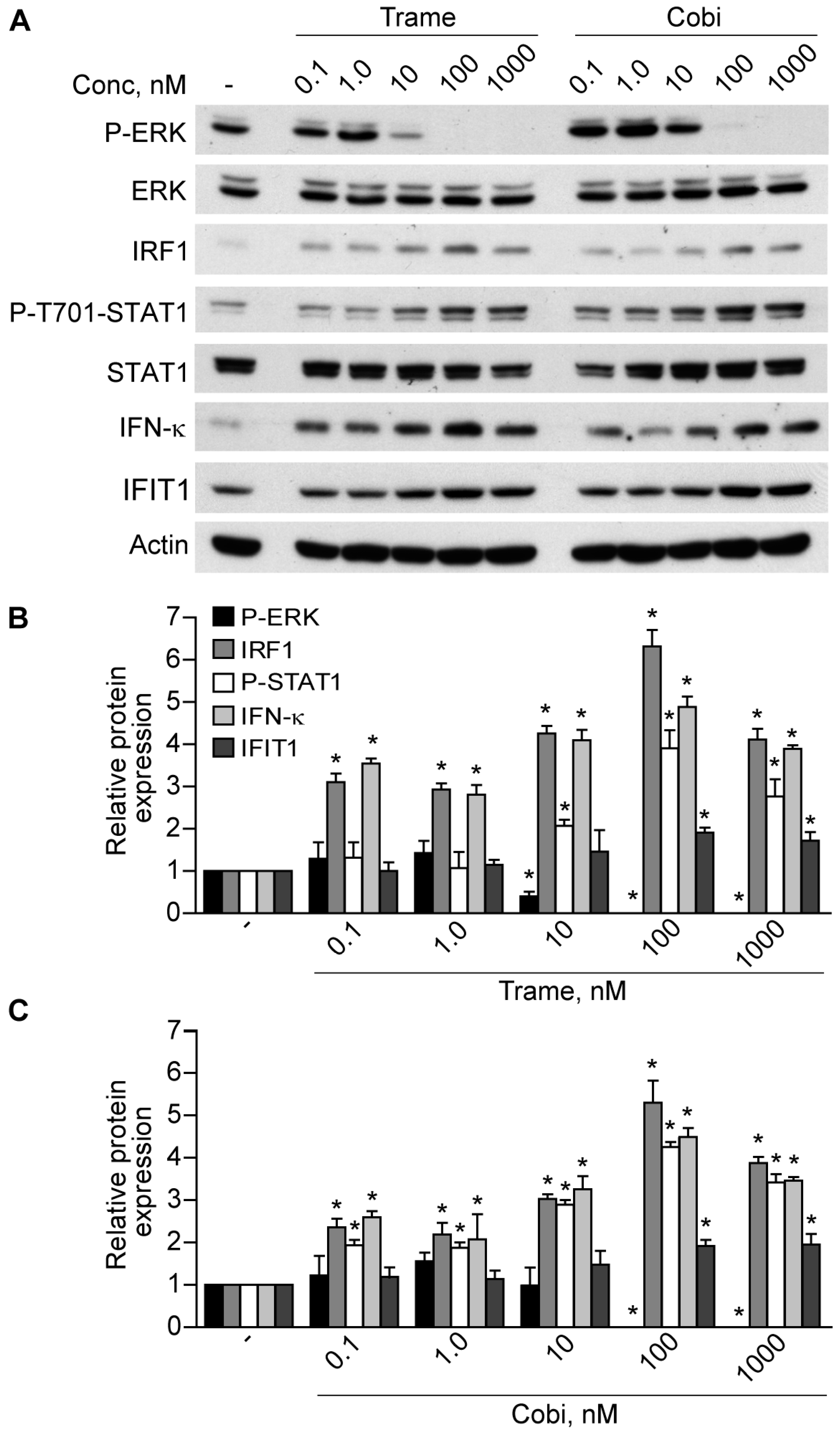
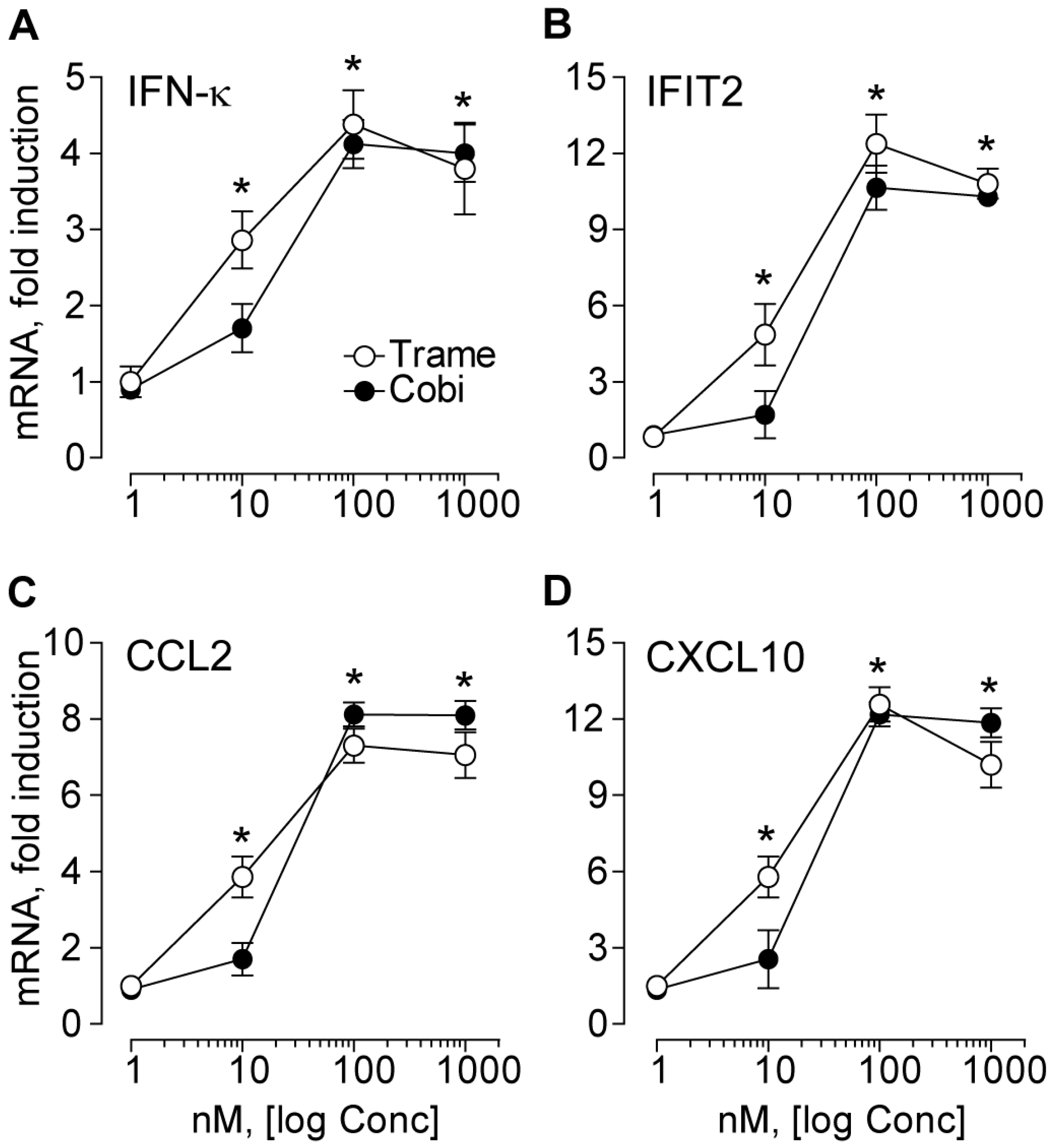
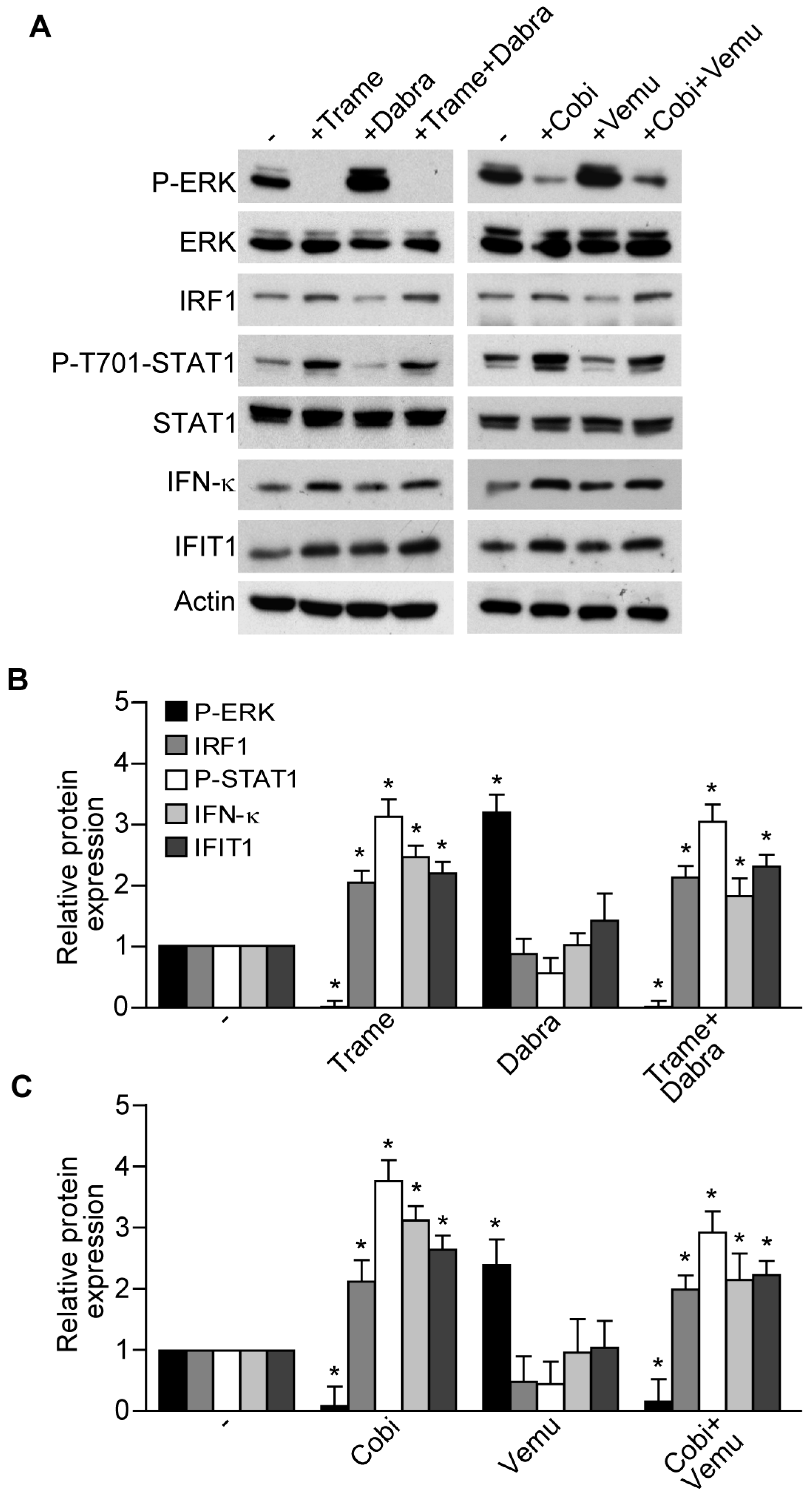
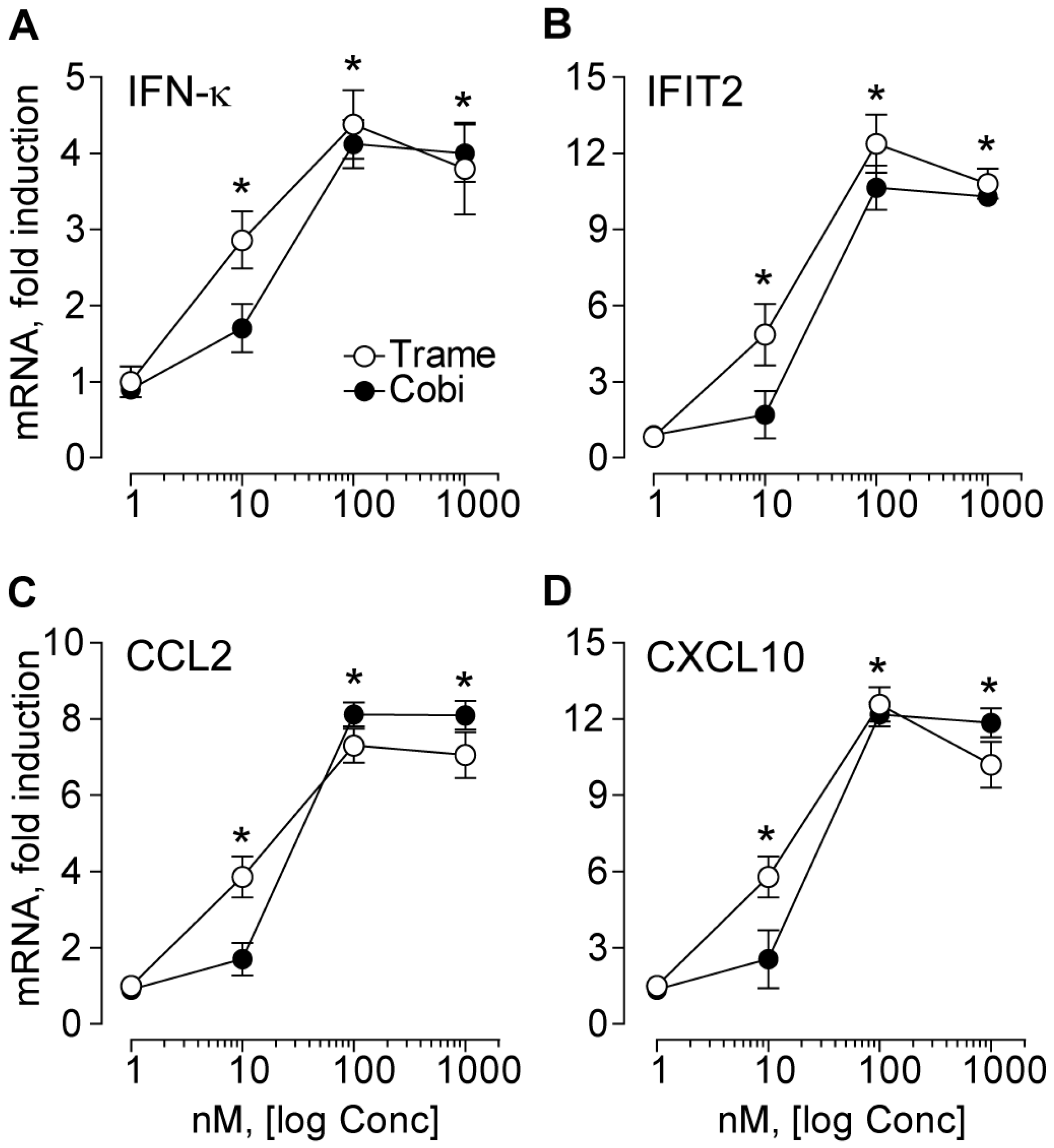
2.1. Trametinib and Cobimetinib Induce a Type I Interferon Response in Keratinocytes
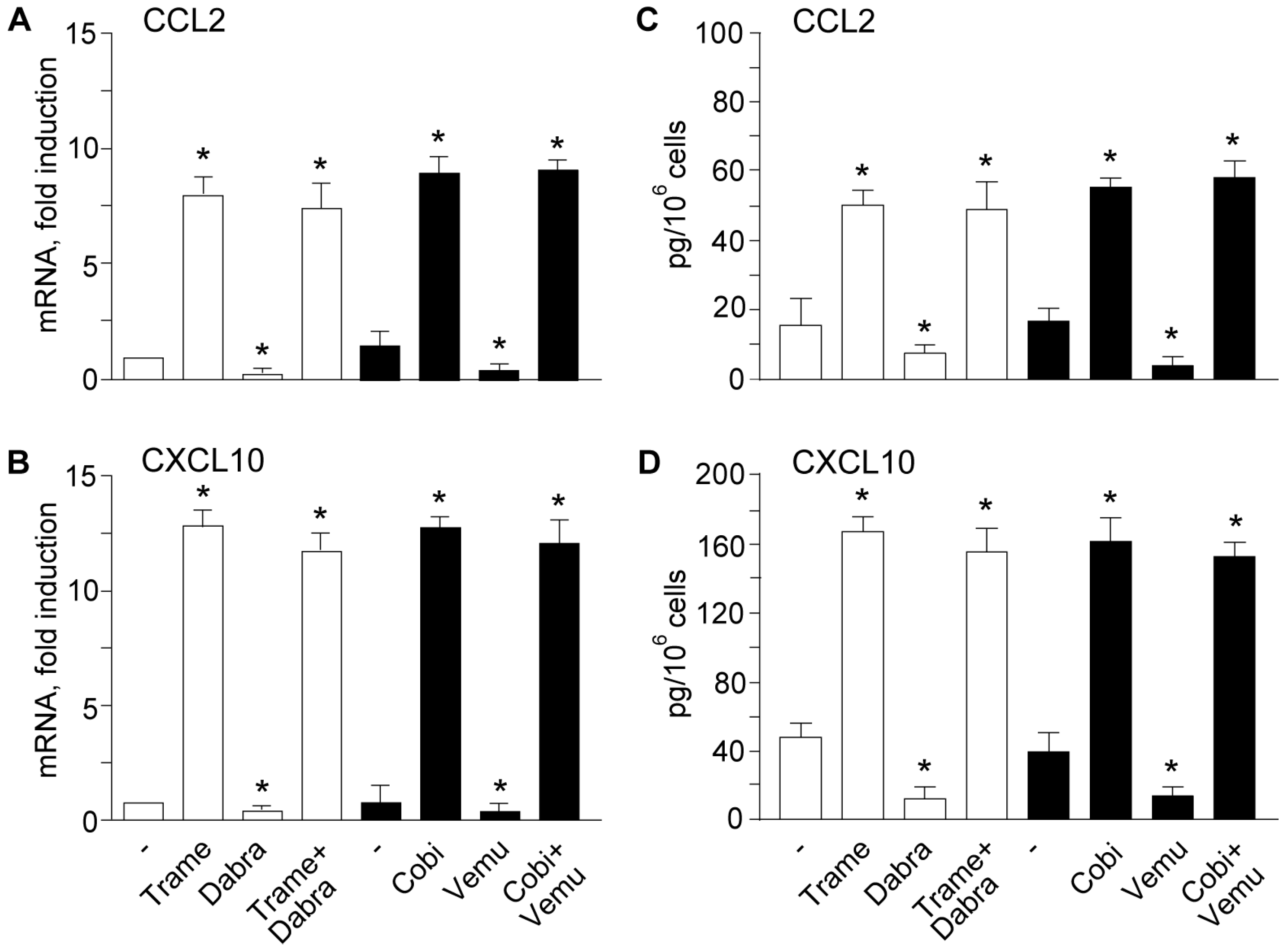
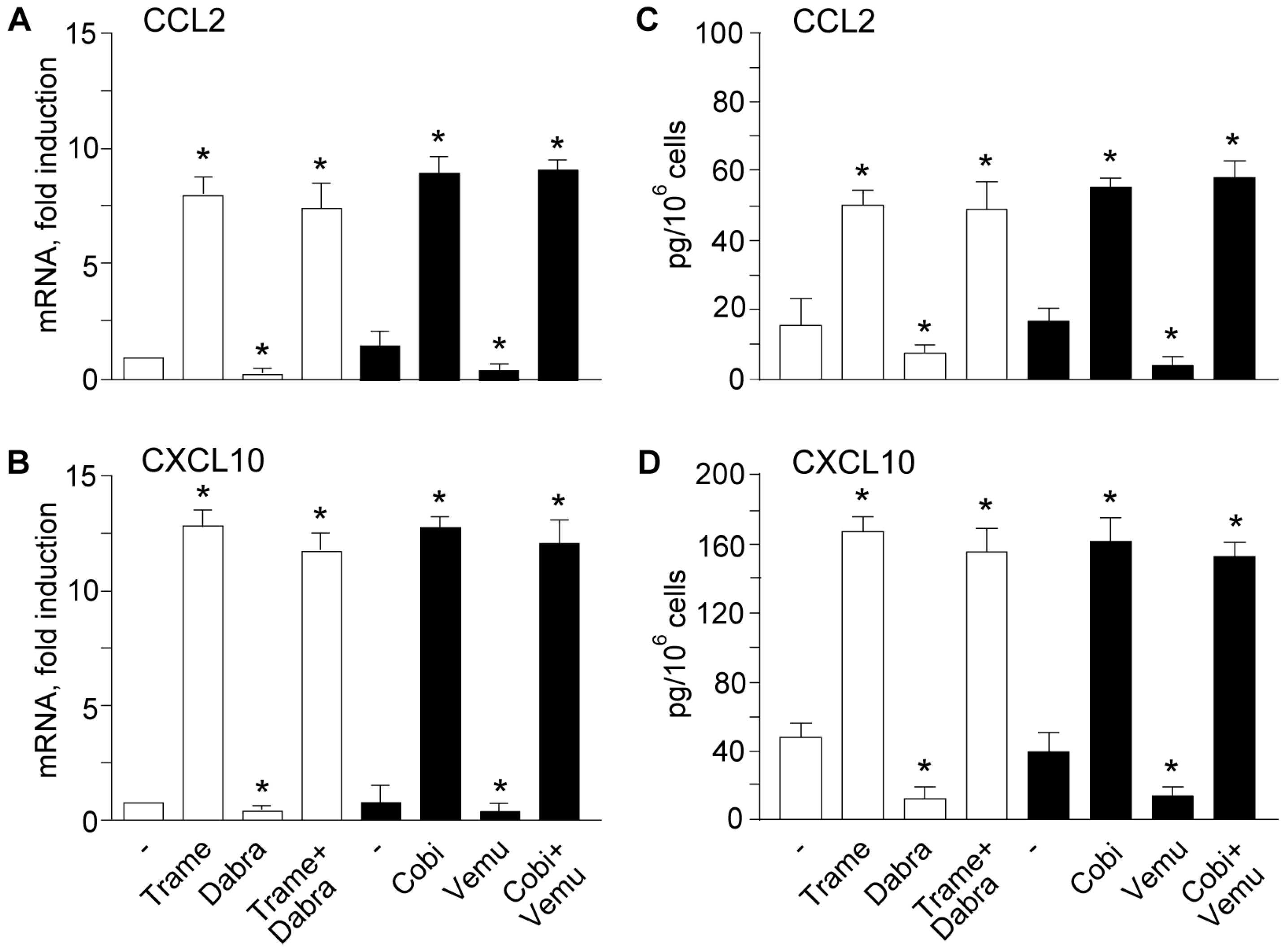
2.2. Activation of Type I Interferon Signature is Maintained When Mitogen Activated Kinase Kinase (MEK) Inhibitors Are Associated to v-Raf Murine Sarcoma Viral Oncogene Homolog B (BRAF) Inhibitors
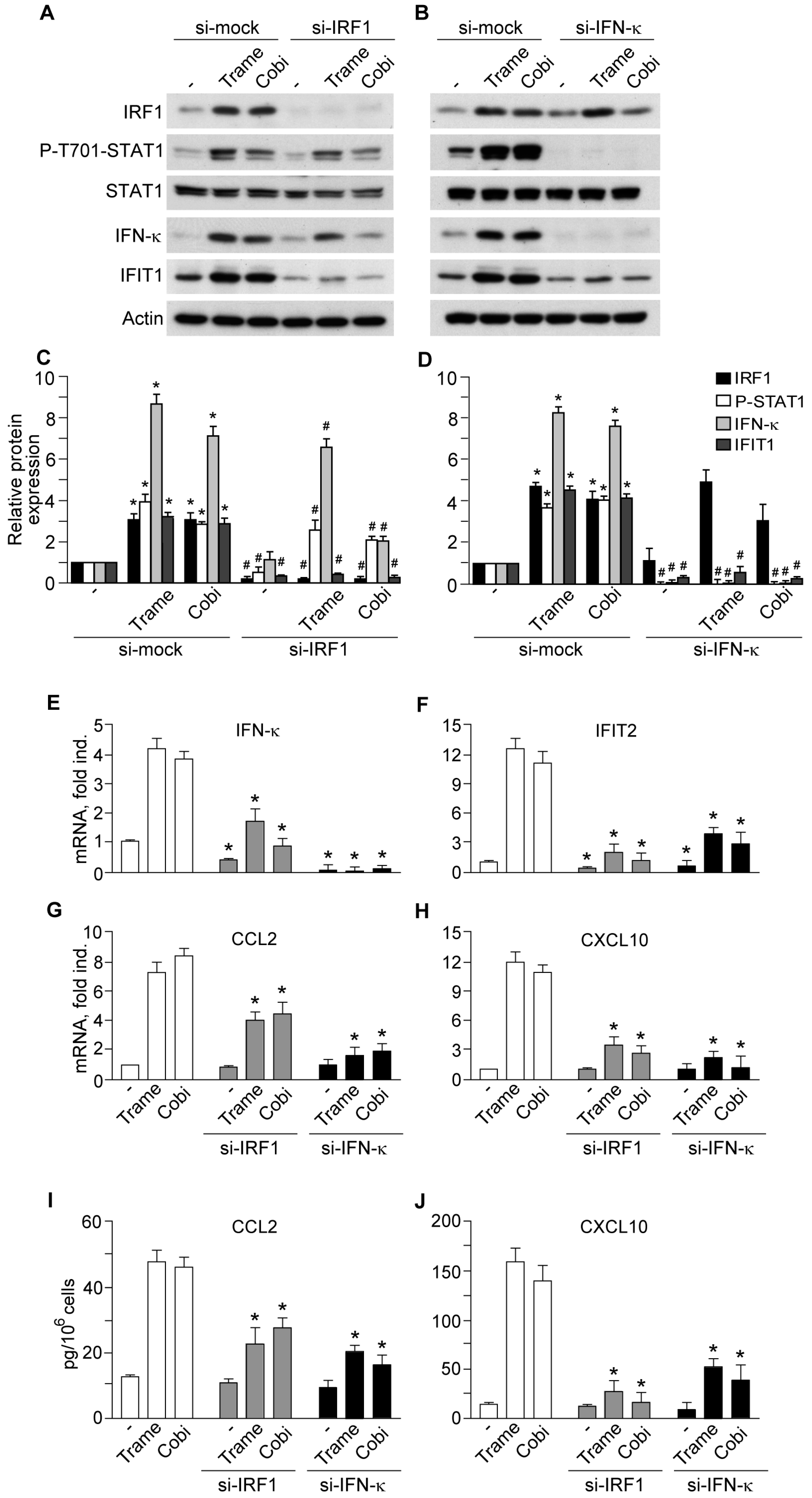
2.3. Impact of Interferon Regulatory Factor 1 (IRF1) and Activated Signal Transducer and Activator of Transcription 1 (STAT1) in MEK Inhibitor-Induced Gene Expression
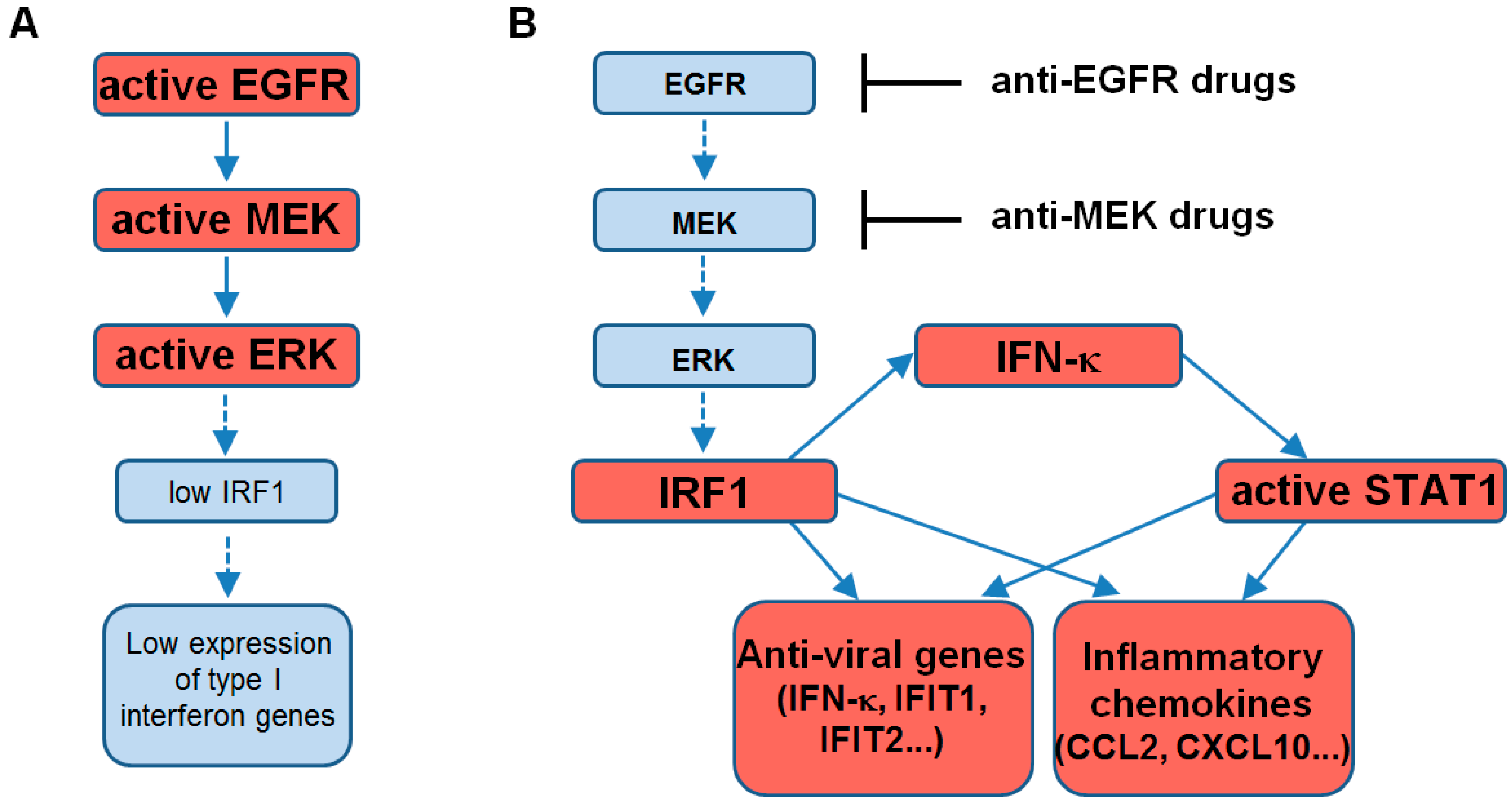
3. Discussion
4. Materials and Methods
4.1. Keratinocyte Source and Culture
4.2. Keratinocyte Lysis
4.3. Kinase Inhibitors
4.4. Western Blot Analysis
4.5. Quantitative Real-Time RT-PCR Analysis
4.6. Enzyme-linked Immunosorbent Assay (ELISA)
4.7. Transfection with Specific Small Interference (si) RNA
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK Signalling in Cancer: Promises and Challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF Inhibitors Prime Wild-Type RAF to Activate the MAPK Pathway and Enhance Growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF Inhibitors Transactivate RAF Dimers and ERK Signalling in Cells with Wild-Type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.B.; Macdonald, B.; Golitz, L.E.; LoRusso, P.; Sekulic, A. Cutaneous Adverse Effects of Targeted Therapies. J. Am. Acad. Dermatol. 2015, 72, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. ERK1/2 MAP Kinases: Structure, Function, and Regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Adjei, A.A. The Clinical Development of MEK Inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.T.; Pang, L.; Decker, S.J.; Bridges, A.J.; Saltiel, A.R. A Synthetic Inhibitor of the Mitogen-Activated Protein Kinase Cascade. Proc. Natl. Acad. Sci. USA 1995, 92, 7686–7689. [Google Scholar] [CrossRef] [PubMed]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 Inhibitors and Cancer Therapy: The Long and Winding Road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xie, L.; Bourne, P.E. Insights into the Binding Mode of MEK Type-III Inhibitors. A Step Towards Discovering and Designing Allosteric Kinase Inhibitors Across the Human Kinome. PLoS ONE 2017, 12, e0179936. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted Agents and Immunotherapies: Optimizing Outcomes in Melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Simeone, E.; Grimaldi, A.M.; Festino, L.; Vanella, V.; Palla, M.; Ascierto, P.A. Combination Treatment of Patients with BRAF-Mutant Melanoma: A New Standard of Care. BioDrugs 2017, 31, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, O.; ElHalawi, H.; Ahmed, H. Risk of Selected Dermatologic Toxicities in Cancer Patients Trated with MEK Inhibitors: A Comparative Systematic Review and Meta-Analysis. Future Oncol. 2015, 11, 3307–3319. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Lulli, D.; Girolomoni, G. Epidermal Growth Factor Receptor Signalling in Keratinocyte Biology: Implications for Skin Toxicity of Tyrosine Kinase Inhibitors. Arch. Toxicol. 2014, 88, 1189–1203. [Google Scholar] [CrossRef] [PubMed]
- Lulli, D.; Carbone, M.L.; Pastore, S. Epidermal Growth Factor Receptor Inhibitors Trigger a Type I Interferon Response in Human Skin. Oncotarget 2016, 7, 47777–47793. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer Cell-Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate Immune Signaling and Regulation in Cancer Immunotherapy. Cell Res. 2017, 27, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, X.; Wang, W.; Wang, Y.; Yin, Y.; Laan, L.J.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. IFN Regulatory Factor 1 Restricts Hepatitis E Virus Replication by Activating STAT1 to Induce Antiviral IFN-Stimulated Genes. FASEB J. 2016, 30, 3352–3367. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Derwish, L.; Hirasawa, K. IRF1 Downregulation by Ras/MEK is Independent of Translational Control of IRF1 mRNA. PLoS ONE 2016, 11, e0160529. [Google Scholar] [CrossRef] [PubMed]
- Fritsche-Guenther, R.; Witzel, F.; Sieber, A.; Herr, R.; Schmidt, N.; Braun, S.; Brummer, T.; Sers, C.; Blüthgen, N. Strong Negative Feedback from Erk to Raf Confers Robustness to MAPK Signalling. Mol. Syst. Biol. 2011, 7, 489. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Michal, J.J.; Zhang, L.; Ding, B.; Lunney, J.K.; Liu, B.; Jiang, Z. Interferon Induced IFIT Family Genes in Host Antiviral Defense. Int. J. Biol. Sci. 2013, 9, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Carlos, G.; Anforth, R.; Clements, A.; Menzies, A.M.; Carlino, M.S.; Chou, S.; Fernandez-Peñas, P. Cutaneous Toxic Effects of BRAF Inhibitors Alone and in Combination with MEK Inhibitors for Metastatic Melanoma. JAMA Dermatol. 2015, 151, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Müller, M.; Decker, T. The Regulation of Inflammation by Interferons and Their STATs. JAKSTAT 2013, 2, e23820. [Google Scholar] [CrossRef] [PubMed]
- Albanesi, C.; Scarponi, C.; Bosisio, D.; Sozzani, S.; Girolomoni, G. Immune Functions and Recruitment of Plasmacytoid Dendritic Cells in Psoriasis. Autoimmunity 2010, 43, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Korl-Mäurer, A.; Goebeler, M.; Mäurer, M. Cutaneous Adverse Events Associated with Interferon-Beta Treatments of Multiple Sclerosis. Int. J. Mol. Sci. 2015, 16, 14951–14960. [Google Scholar] [CrossRef] [PubMed]
- López de Padilla, C.M.; Niewold, T.B. The Type I Interferons: Basic Concepts and Clinical Relevance in Immune-Mediated Inflammatory Diseases. Gene 2016, 576, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Delaney, T.A.; Morehouse, C.; Brohawn, P.Z.; Groves, C.; Colonna, M.; Yao, Y.; Sanjuan, M.; Coyle, A.J. Type I IFNs Regulate Inflammation, Vasculopathy, and Fibrosis in Chronic Cutaneous Graft-Versus-Host Disease. J. Immunol. 2016, 197, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Stannard, J.N.; Reed, T.J.; Myes, E.; Lowe, L.; Sarkar, M.K.; Xing, X.; Gudjonsson, J.E.; Kahlenberg, J.M. Lupus Skin is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. J. Investig. Dermatol. 2017, 137, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Wörenkämper, E.; Freutel, S.; Henze, S.; Haller, O.; Bieber, T.; Tüting, T. Enhanced Type I Interferon Signalling Promotes Th1-Biased Inflammation in Cutaneous Lupus Erythematosus. J. Pathol. 2005, 205, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Bekisch, B.; Uerlich, M.; Haller, O.; Bieber, T.; Tüting, T. Type I Interferon-Associated Recruitment of Cytotoxic Lymphocytes: A Common Mechanism in Regressive Melanocytic Lesions. Am. J. Clin. Pathol. 2005, 124, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Chemokines and Chemokine Receptors Required for Optimal Responses to Anticancer Chemotherapy. OncoImmunology 2014, 3, e27663. [Google Scholar] [CrossRef] [PubMed]
- Lança, T.; Costa, M.F.; Gonçalves-Sousa, N.; Rei, M.; Gross, A.R.; Penido, C.; Silva-Santo, B. Protective Role of the Inflammatory CCR2/CCL2 Chemokine Pathway through Recruitment of Type 1 Cytotoxic γδ T Lymphocytes to Tumor Beds. J. Immunol. 2013, 190, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine Expression in Melanoma Metastases Associated with CD8+ T-Cell Recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skizki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-Redundant Requirement for CXCR3 Signalling during Tumoricidal T-Cell Trafficking across Tumour Vascular Checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Goldszmid, R.S.; Dzutsev, A.; Trinchieri, G. Host Immune Response to Infection and Cancer: Unexpected Commonalities. Cell Host Microbe 2014, 15, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I Interferon Response and Innate Immune Sensing of Cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects and Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 14, 690–714. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Sistigu, A.; Proietti, E.; Moschella, F. The Added Value of Type I Interferons to Cytotoxic Treatments of Cancer. Cytokine Growth Factor Rev. 2017, 36, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Fanales-Belasio, E.; Albanesi, C.; Chinni, L.M.; Giannetti, A.; Girolomoni, G. Granulocyte Macrophage Colony-Stimulating Factor is Overproduced by Keratinocytes in Atopic Dermatitis. Implications for Sustained Dendritic Cell Activation in the Skin. J. Clin. Investig. 1997, 99, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lulli, D.; Carbone, M.L.; Pastore, S. The MEK Inhibitors Trametinib and Cobimetinib Induce a Type I Interferon Response in Human Keratinocytes. Int. J. Mol. Sci. 2017, 18, 2227. https://doi.org/10.3390/ijms18102227
Lulli D, Carbone ML, Pastore S. The MEK Inhibitors Trametinib and Cobimetinib Induce a Type I Interferon Response in Human Keratinocytes. International Journal of Molecular Sciences. 2017; 18(10):2227. https://doi.org/10.3390/ijms18102227
Chicago/Turabian StyleLulli, Daniela, Maria Luigia Carbone, and Saveria Pastore. 2017. "The MEK Inhibitors Trametinib and Cobimetinib Induce a Type I Interferon Response in Human Keratinocytes" International Journal of Molecular Sciences 18, no. 10: 2227. https://doi.org/10.3390/ijms18102227
APA StyleLulli, D., Carbone, M. L., & Pastore, S. (2017). The MEK Inhibitors Trametinib and Cobimetinib Induce a Type I Interferon Response in Human Keratinocytes. International Journal of Molecular Sciences, 18(10), 2227. https://doi.org/10.3390/ijms18102227