
Obatoclax, a Pan-BCL-2 Inhibitor, Targets Cyclin D1 for Degradation to Induce Antiproliferation in Human Colorectal Carcinoma Cells



Abstract
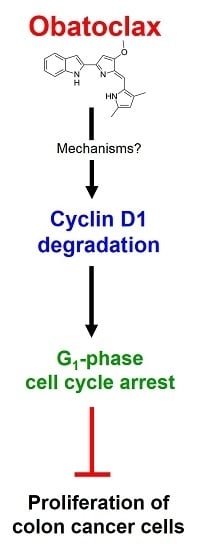
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
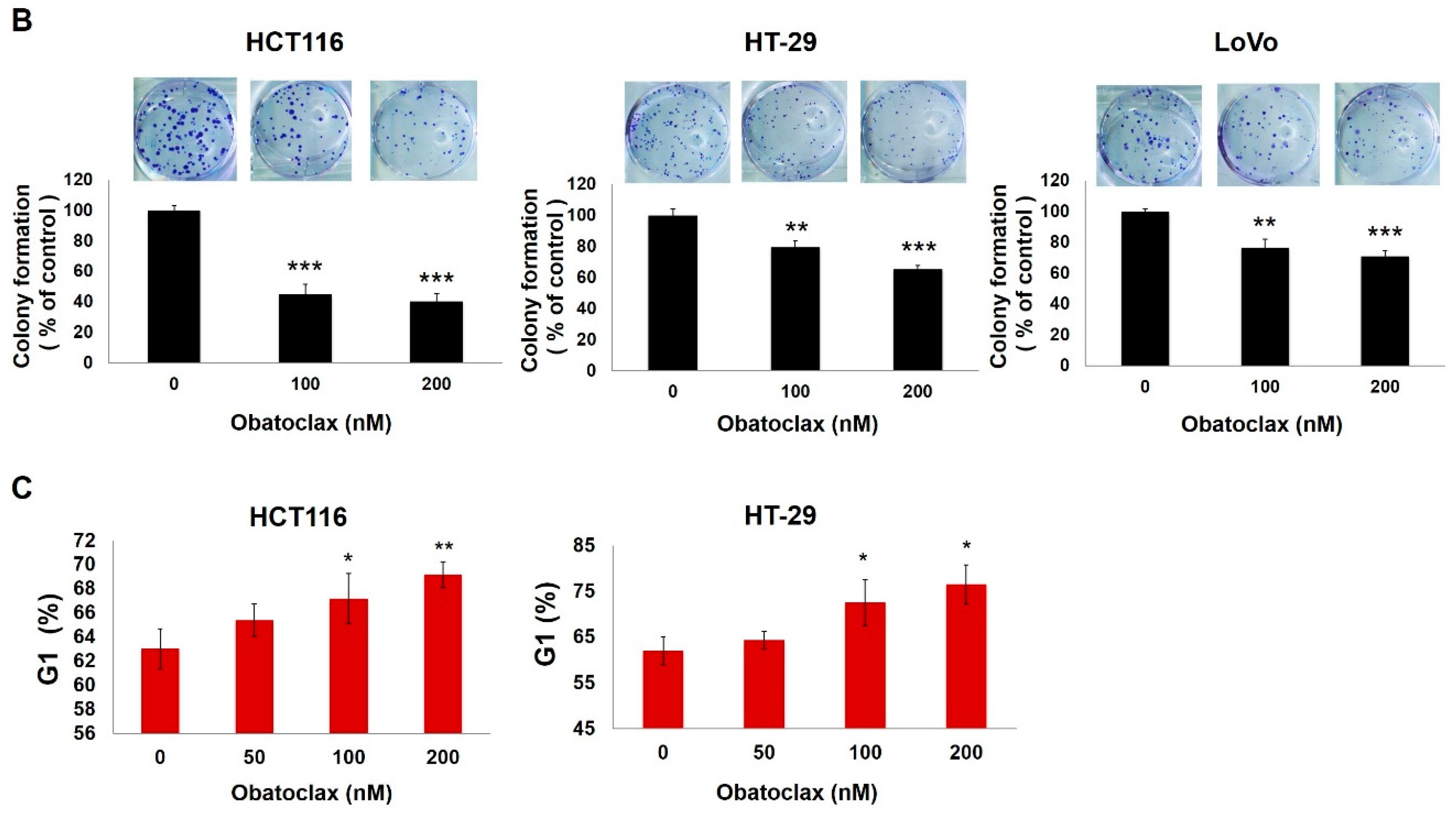
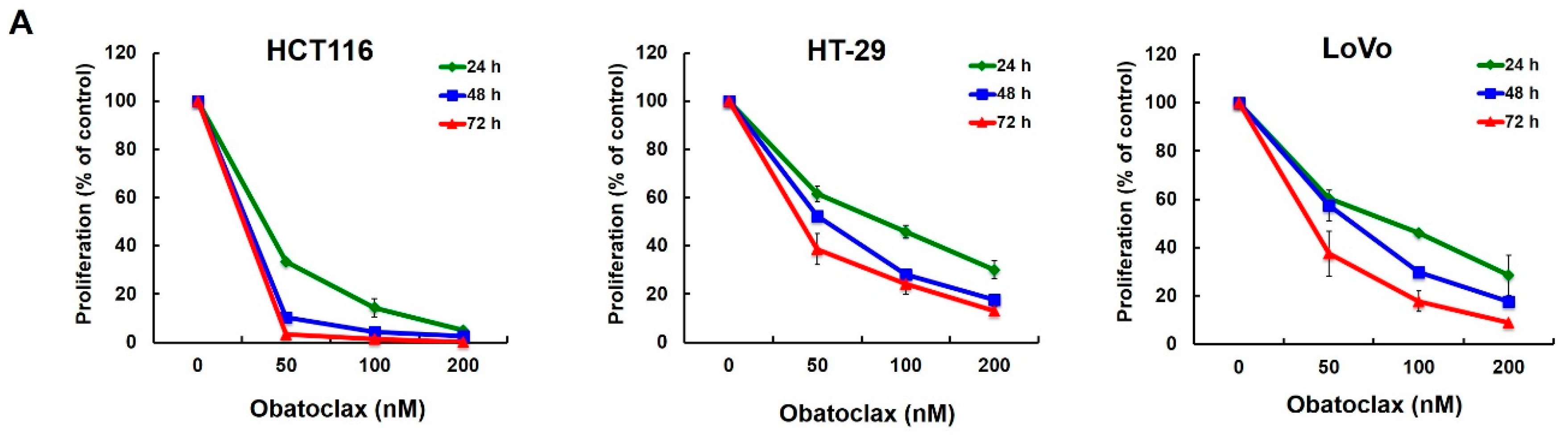
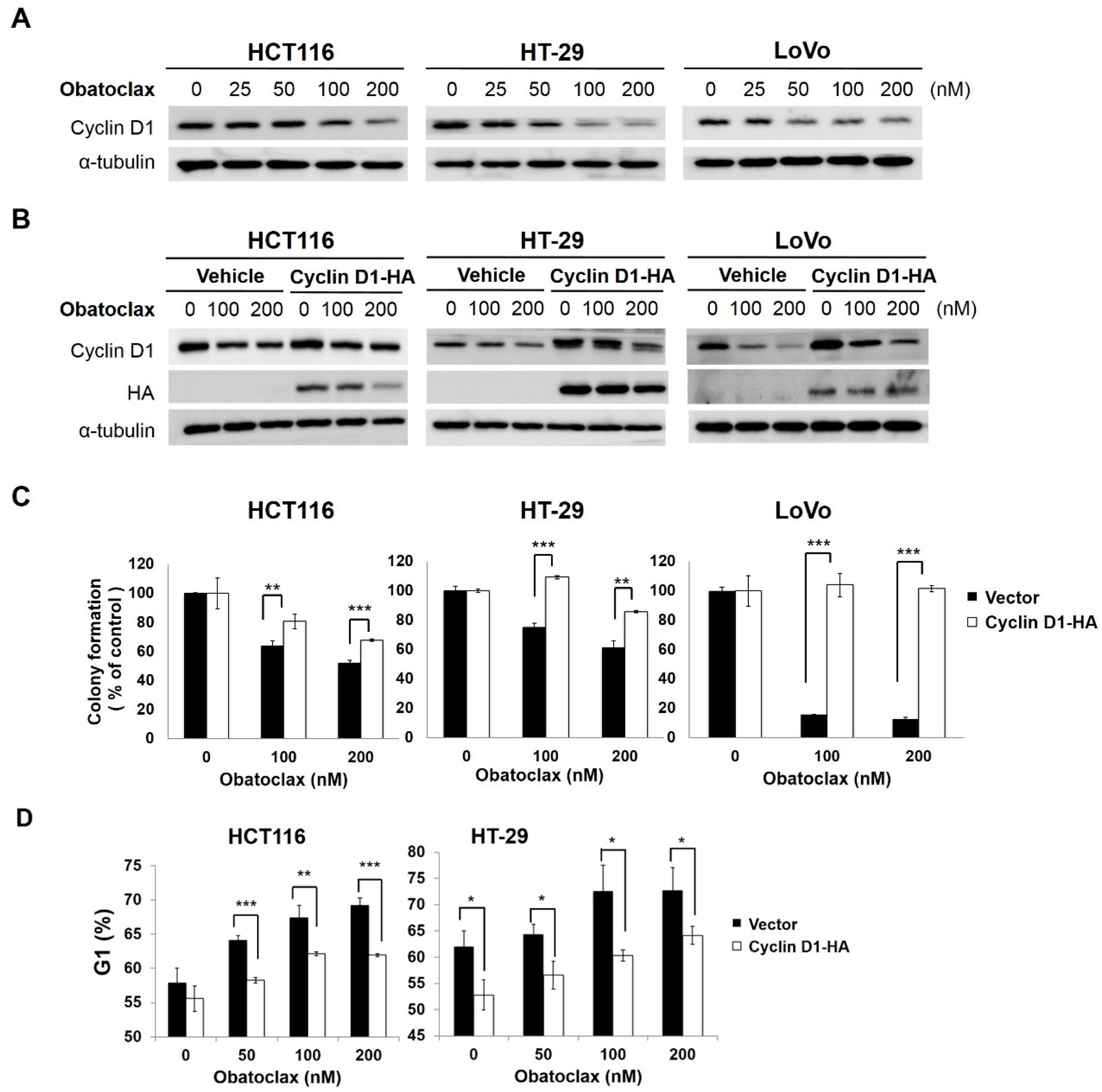
2.1. Obatoclax Inhibits Cell Proliferation and Induces G1 Cell-Cycle Arrest in a Panel of Human Colorectal Cancer Cell Lines
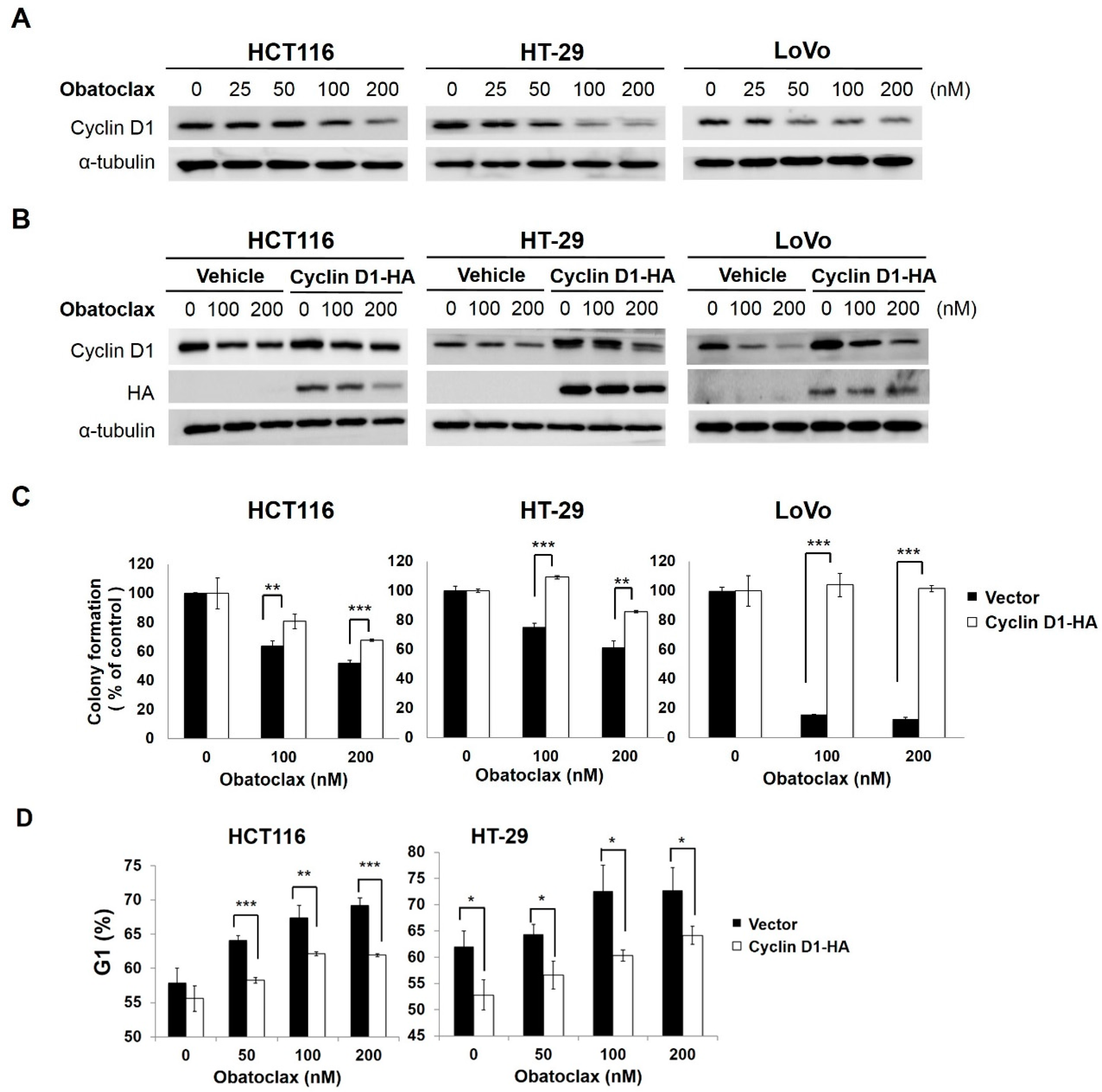
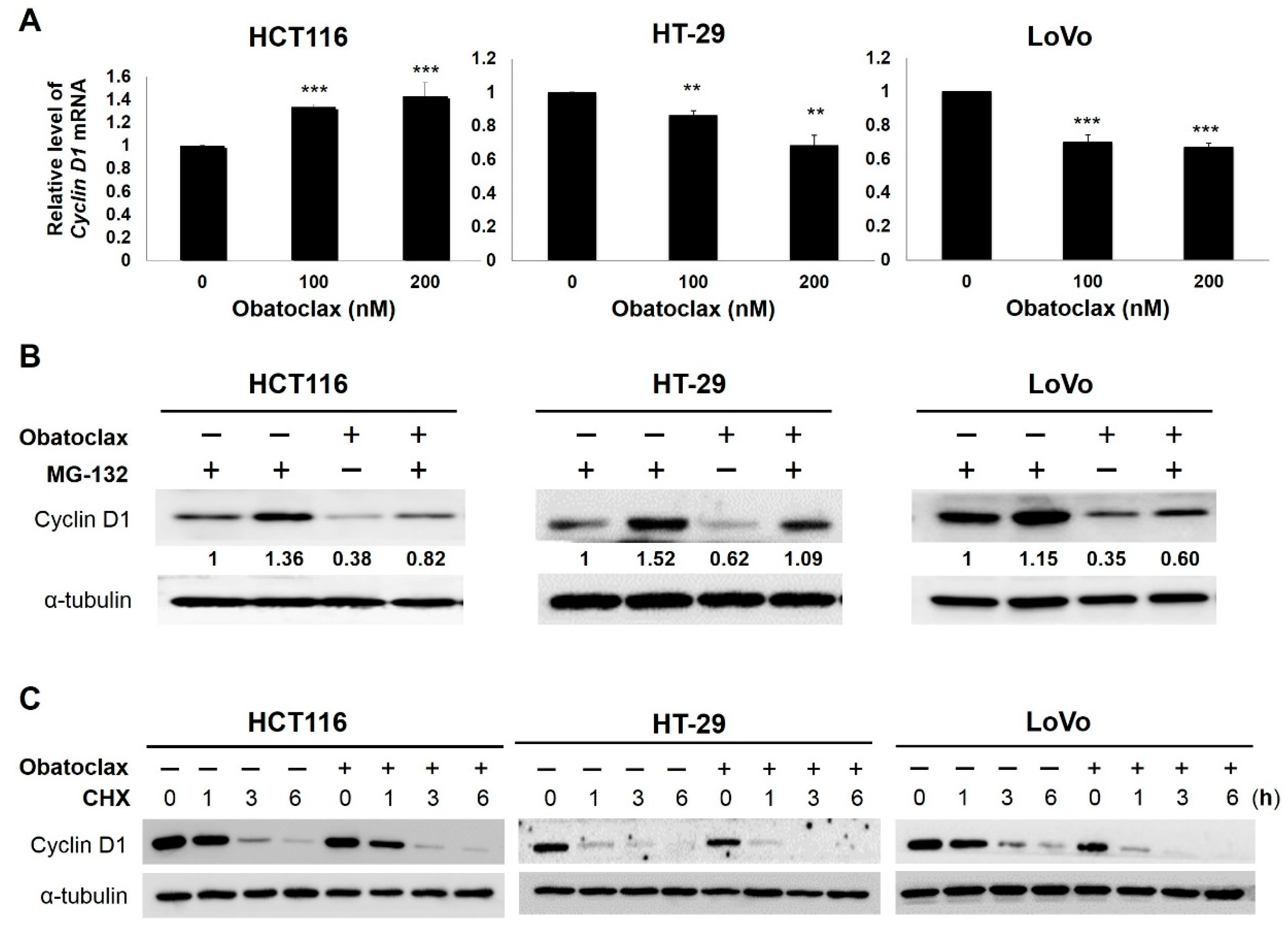
2.2. Cyclin D1 Downregulation Is Fundamental for Obatoclax-Induced Antiproliferation
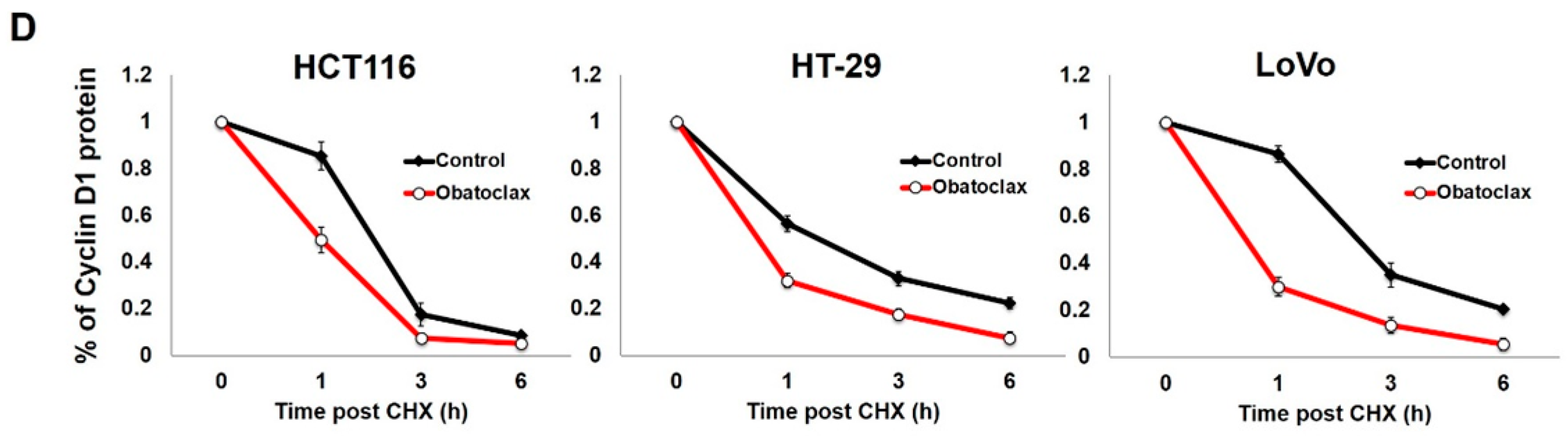
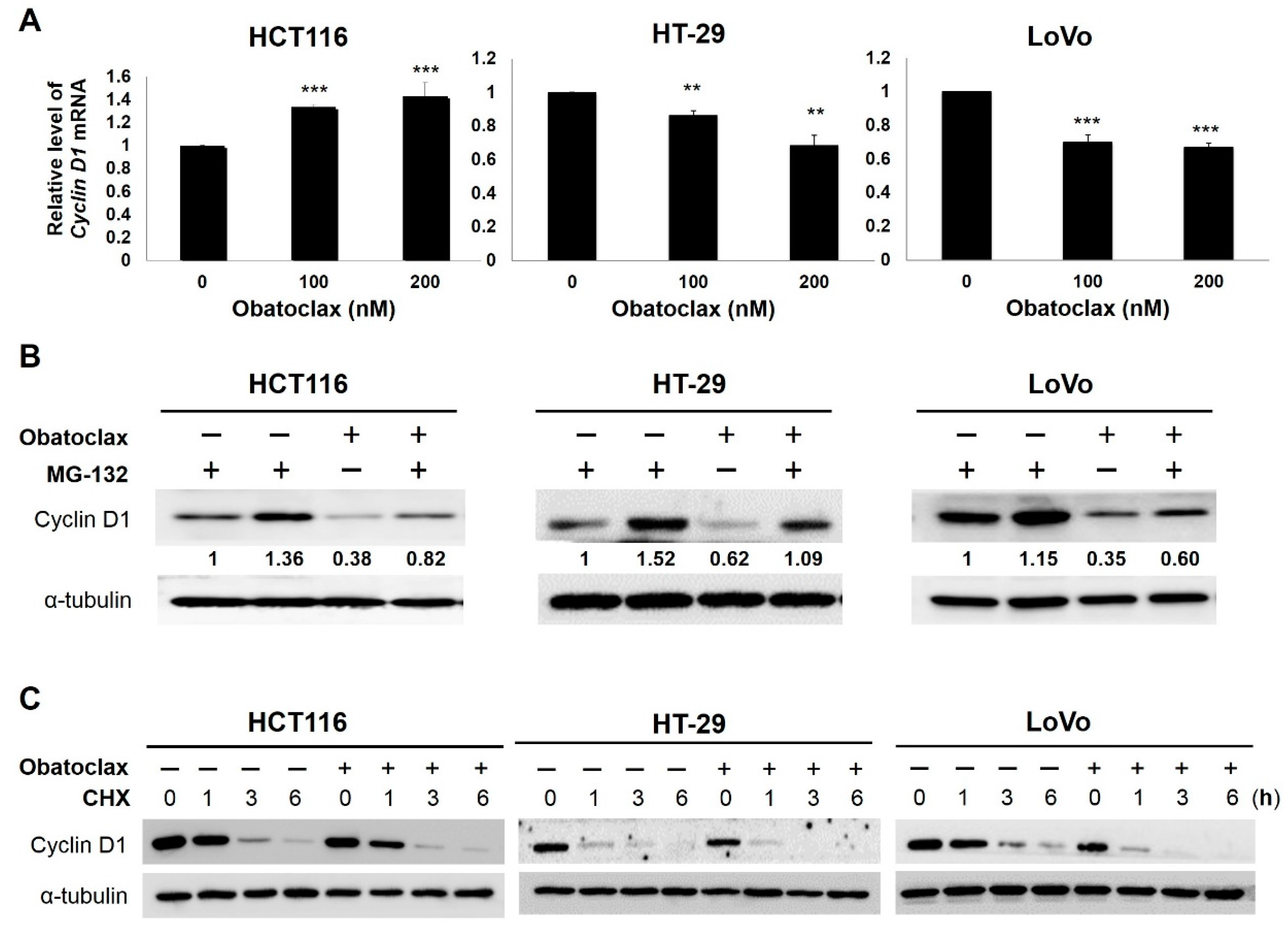
2.3. Obatoclax Downregulates Cyclin D1 Primarily through Inducing Cyclin D1 Protein Degradation
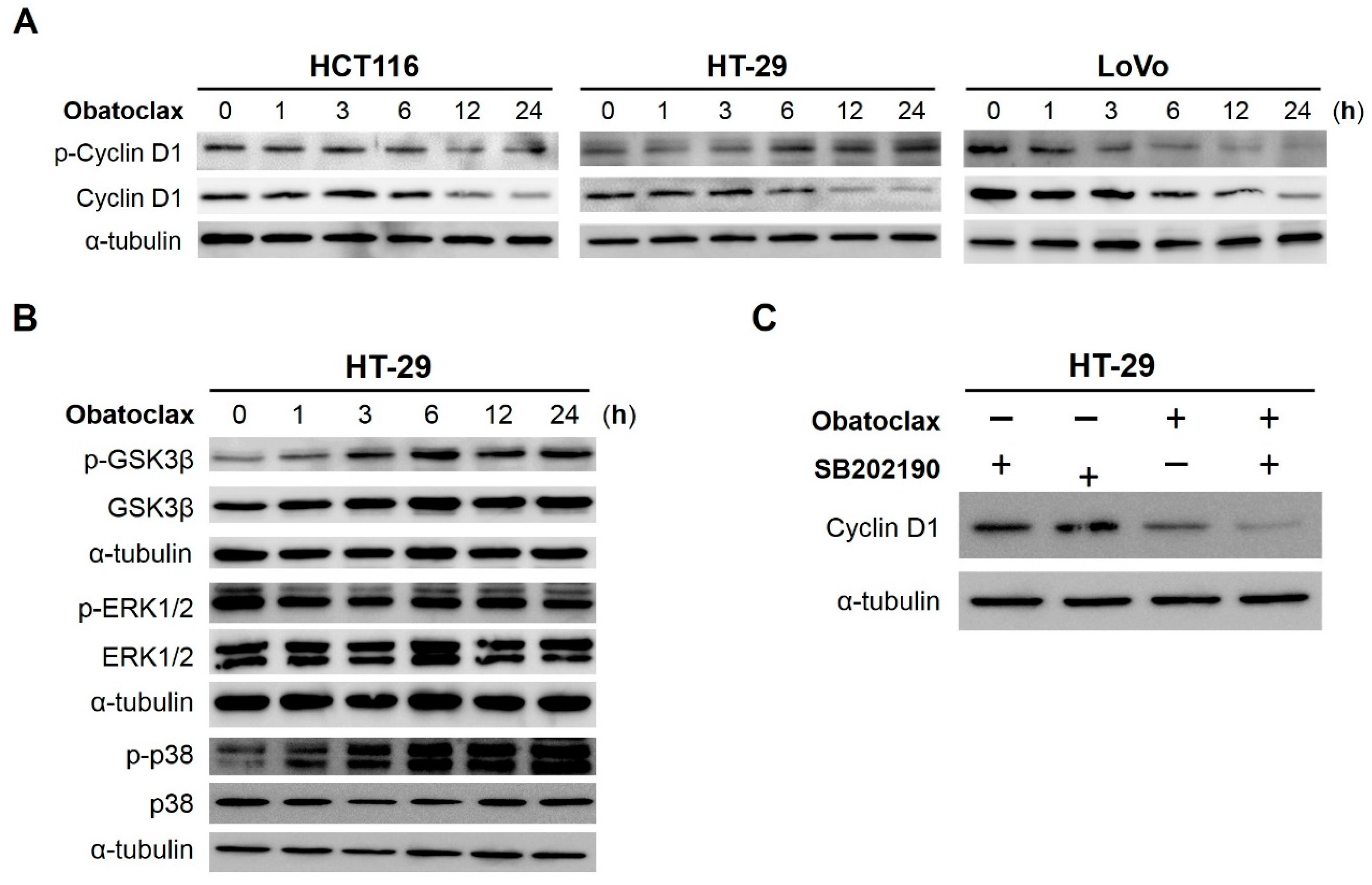
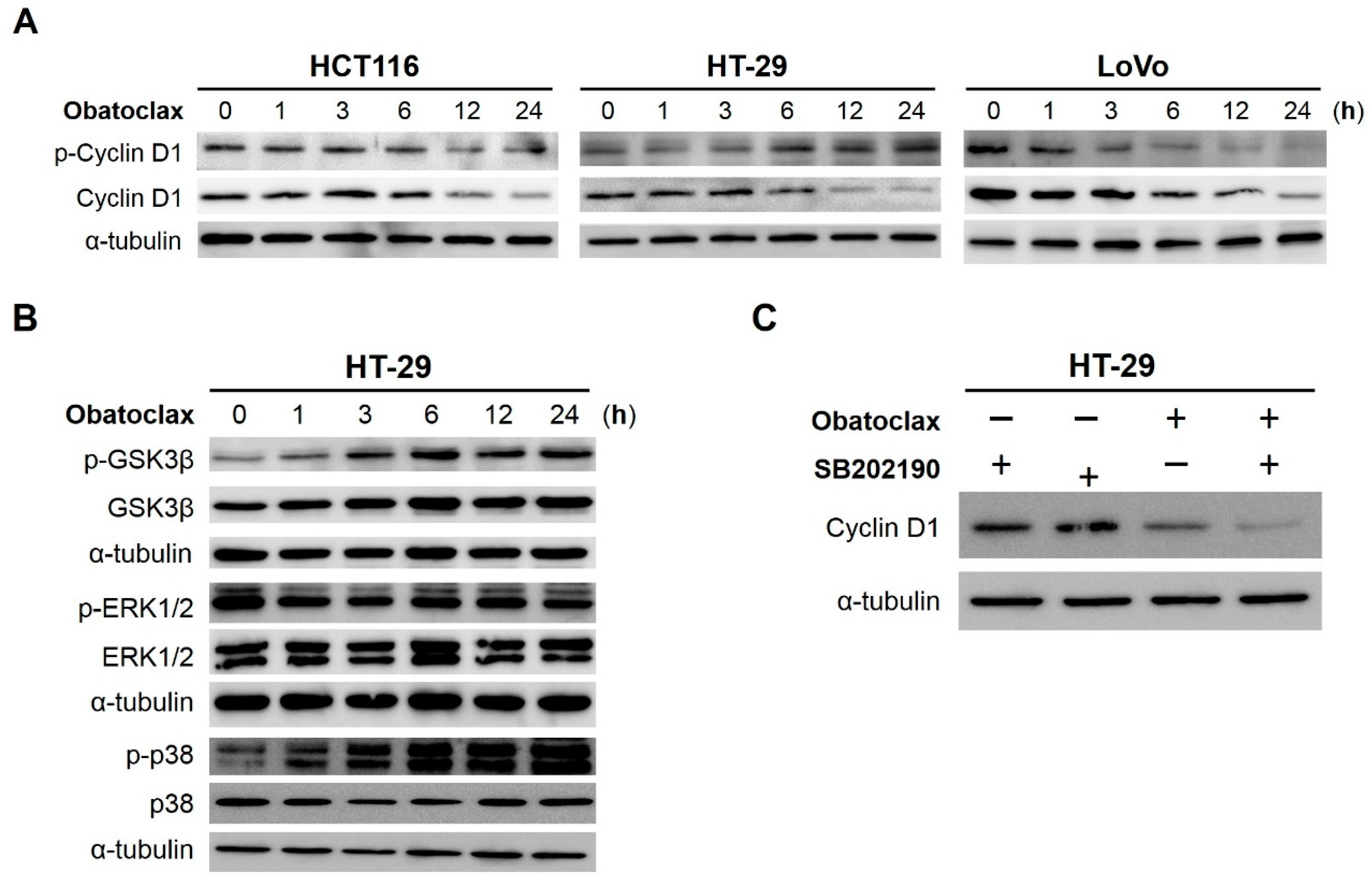
2.4. Obatoclax Induces Cyclin D1 Degradation in a T286 Phosphorylation-Dependent or -Independent Manners
2.5. GSK3β (Glycogen Synthase Kinase 3β), ERK1/2 (Extracellular Signal–Regulated Kinases 1/2), and p38MAPK (p38Mitogen-Activated Protein Kinase) Are Dispensable for Obatoclax-Induced Cyclin D1 Degradation in HT-29 Cells
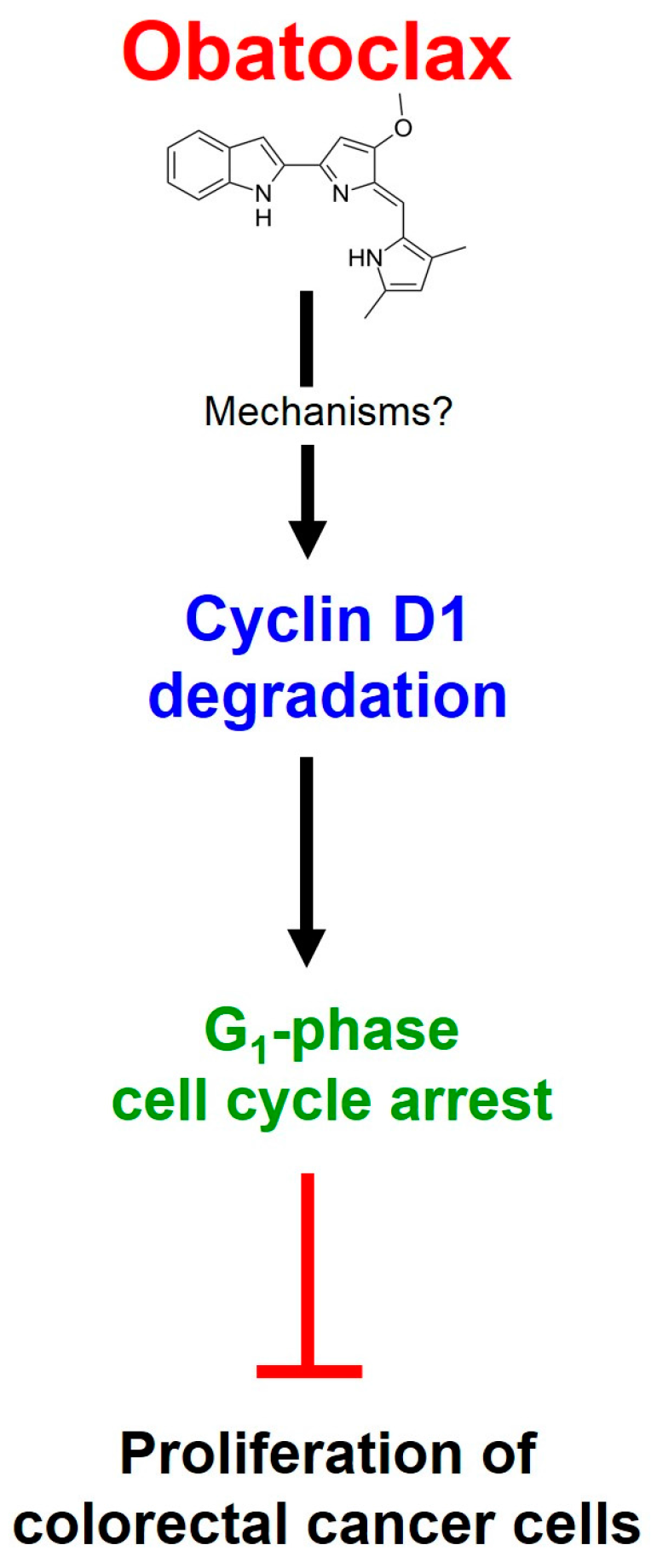
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Colony Formation Assay
4.5. Cell Cycle Analysis by Flow Cytometry
4.6. Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.7. Immunoblot Analysis
4.8. Construction of the Retroviral Vector pBabe-Based Cyclin D1-Expressing Plasmid for Stable Clone Generation
4.9. Cycloheximide Chase Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9–14. [Google Scholar] [PubMed]
- Sharma, R.A.; Manson, M.M.; Gescher, A.; Steward, W.P. Colorectal cancer chemoprevention: Biochemical targets and clinical development of promising agents. Eur. J. Cancer 2001, 37, 12–22. [Google Scholar] [CrossRef]
- Labianca, R.; Nordlinger, B.; Beretta, G.D.; Brouquet, A.; Cervantes, A. On behalf of the ESMO Guidelines Working Group. Primary colon cancer: ESMO Clinical Practice Guidelines for diagnosis, adjuvant treatment and follow-up. Ann. Oncol. 2010, 21, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.; Lee, S.H.; Meng, X.W.; Vincelette, N.D.; Knorr, K.L.; Ding, H.; Nowakowski, G.S.; Dai, H.; Kaufmann, S.H. Emerging understanding of BCL-2 biology: Implications for neoplastic progression and treatment. Biochim. Biophys. Acta 2015, 1853, 1658–1671. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Bergkamen, H.; Ehrenberg, R.; Hickmann, L.; Vick, B.; Urbanik, T.; Schimanski, C.C.; Berger, M.R.; Schad, A.; Weber, A.; Heeger, S.; et al. BCL-xL and Myeloid cell leukaemia-1 contribute to apoptosis resistance of colorectal cancer cells. World J. Gastroenterol. 2008, 14, 3829–3840. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Wang, Z.; Lv, C.-Y.; Liang, X.L.; Sun, M.; Guo, Y.-Y.; De, W. Prognostic significance of BCL-xL gene expression in human colorectal cancer. Acta Histochem. 2011, 113, 810–814. [Google Scholar]
- Henderson-Jackson, E.B.; Helm, J.; Ghayouri, M.; Hakam, A.; Nasir, A.; Leon, M.; Bui, M.; Yeatman, T.; Coppola, D. Correlation between Mcl-1 and pAKT protein expression in colorectal cancer. Int. J. Clin. Exp. Pathol. 2010, 3, 768–774. [Google Scholar] [PubMed]
- Goard, C.A.; Schimmer, A.D. An evidence-based review of obatoclax mesylate in the treatment of hematological malignancies. Core Evid. 2013, 8, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.D.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Prasad, S.; Tyagi, A.K.; Deb, L.; Huang, J.; Karelia, D.N.; Amin, S.G.; Aggarwal, B.B. Targeting cell survival proteins for cancer cell death. Pharmaceuticals 2016, 9, E11. [Google Scholar] [CrossRef] [PubMed]
- Lieber, J.; Armeanu-Ebinger, S.; Fuchs, J. The role of BH3-mimetic drugs in the treatment of pediatric hepatoblastoma. Int. J. Mol. Sci. 2015, 16, 4190–4208. [Google Scholar] [CrossRef] [PubMed]
- Vela, L.; Marzo, I. BCL-2 family of proteins as drug targets for cancer chemotherapy: The long way of BH3 mimetics from bench to bedside. Curr. Opin. Pharmacol. 2015, 23, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M.; Reed, J.C. Finally, an apoptosis-targeting therapeutics for cancer. Cancer Res. 2016, 76, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Watt, J.; Contractor, R.; Tsao, T.; Harris, D.; Estrov, Z.; Bornmann, W.; Kantarjian, H.; Viallet, J.; Samudio, I.; et al. Mechanisms of antileukemic activity of the novel BCL-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res. 2008, 68, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C., Jr.; Ferreira-Gonzalez, A.; Grant, S. Inhibition of BCL-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 2012, 119, 6089–6098. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Edwards, H.; Caldwell, J.T.; Wang, G.; Taub, J.W.; Ge, Y. Obatoclax potentiates the cytotoxic effect of cytarabine on acute myeloid leukemia cells by enhancing DNA damage. Mol. Oncol. 2015, 9, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Madiraju, S.R.M.; Goulet, D.; Viallet, J.; Bélec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef] [PubMed]
- Koehler, B.C.; Jassowicz, A.; Scherr, A.L.; Lorenz, S.; Radhakrishnan, P.; Kautz, N.; Elssner, C.; Weiss, J.; Jaeger, D.; Schneider, M.; et al. Pan-BCL-2 inhibitor obatoclax is a potent late stage autophagy inhibitor in colorectal cancer cells independent of canonical autophagy signaling. BMC Cancer 2015, 15, 919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz-Roberts, J.L.; Shajahan, A.N.; Cook, K.L.; Wärri, A.; Abu-Asab, M.; Clarke, R. GX15-070 (obatoclax) induces apoptosis and inhibits cathepsin d- and l-mediated autophagosomal lysis in antiestrogen-resistant breast cancer cells. Mol. Cancer Ther. 2013, 12, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Stamelos, V.A.; Fisher, N.; Bamrah, H.; Voisey, C.; Price, J.C.; Farrell, W.E.; Redman, C.W.; Richardson, A. The BH3 mimetic obatoclax accumulates in lysosomes and causes their alkalinization. PLoS ONE 2016, 11, e0150696. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Gu, C.; Shi, L.; Xun, T.; Li, X.; Liu, S.; Yu, L. Obatoclax induces G1/G0-phase arrest via p38/p21waf1/Cip1 signaling pathway in human esophageal cancer cells. J. Cell Biochem. 2014, 115, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Koehler, B.C.; Scherr, A.L.; Lorenz, S.; Elssner, C.; Kautz, N.; Welte, S.; Jaeger, D.; Urbanik, T.; Schulze-Bergkamen, H. Pan-BCL-2 inhibitor obatoclax delays cell cycle progression and blocks migration of colorectal cancer cells. PLoS ONE 2014, 9, e106571. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Assoian, R.K. Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell Sci. 2008, 121, 3853–3857. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Lee, S.H.; Phuchareon, J.; Albertson, D.G.; McCormick, F.; Tetsu, O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE 2006, 1, e128. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Park, G.H.; Eo, H.J.; Song, H.M.; Lee, J.W.; Kwon, M.J.; Koo, J.S.; Jeong, J.B. Tanshinone I induces cyclin D1 proteasomal degradation in an ERK1/2 dependent way in human colorectal cancer cells. Fitoterapia 2015, 101, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007, 67, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Tetsu, O.; McCormick, F. β-Catenin regulates expression of cyclinD1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar] [PubMed]
- Chandra, S.H.; Wacker, I.; Appelt, U.K.; Behrens, J.; Schneikert, J. A common role for various human truncated adenomatous polyposis coli isoforms in the control of β-catenin activity and cell proliferation. PLoS ONE 2012, 7, e34479. [Google Scholar] [CrossRef] [PubMed]
- Violette, S.; Poulain, L.; Dussaulx, E.; Pepin, D.; Faussat, A.M.; Chambaz, J.; Lacorte, J.M.; Staedel, C.; Lesuffleur, T. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of BCL-2 and BCL-xL in addition to BAX and p53 status. Int. J. Cancer 2002, 98, 498–504. [Google Scholar] [CrossRef] [PubMed]
- LaBonte, M.J.; Wilson, P.M.; Fazzone, W.; Russell, J.; Louie, S.G.; El-Khoueiry, A.; Lenz, H.J.; Ladner, R.D. The dual EGFR/HER2 inhibitor lapatinib synergistically enhances the antitumor activity of the histone deacetylase inhibitor panobinostat in colorectal cancer models. Cancer Res. 2011, 71, 3635–3648. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mallampalli, R.K. Small molecule therapeutics targeting F-box proteins in cancer. Semin. Cancer Biol. 2016, 36, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.A.; Elder, J.; McCloud, J.M.; Hall, C.; Deakin, M.; Fryer, A.A.; Elder, J.B.; Hoban, P.R. Subcellular localisation of cyclin D1 protein in colorectal tumours is associated with p21WAF1/CIP1 expression and correlates with patient survival. Int. J. Cancer 2001, 95, 302–306. [Google Scholar] [CrossRef]
- Bahnassy, A.A.; Zekri, A.R.; El-Houssini, S.; El-Shehaby, A.M.; Mahmoud, M.R.; Abdallah, S.; El-Serafi, M. Cyclin A and cyclin D1 as significant prognostic markers in colorectal cancer patients. BMC Gastroenterol. 2004, 23, 22–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wei, J.; Xu, C.; Zhao, Z.; You, T. Prognostic significance of cyclin D1 expression in colorectal cancer: A meta-analysis of observational studies. PLoS ONE 2014, 9, e94508. [Google Scholar] [CrossRef] [PubMed]
- Biliran, H., Jr.; Wang, Y.; Banerjee, S.; Xu, H.; Heng, H.; Thakur, A.; Bollig, A.; Sarkar, F.H.; Liao, J.D. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin. Cancer Res. 2005, 11, 6075–6086. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Zhao, W.; Gu, W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol. Cell 2009, 36, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Pragani, R.; Fox, J.T.; Shen, M.; Parmar, K.; Gaudiano, E.F.; Liu, L.; Tanega, C.; McGee, L.; Hall, M.D.; et al. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.T.; Li, R.; Becerra, C.R.; Tripathy, D.; Frenkel, E.P.; Verma, U.N. IκB kinase α regulates subcellular distribution and turnover of cyclin D1 by phosphorylation. J. Biol. Chem. 2005, 280, 33945–33952. [Google Scholar] [CrossRef] [PubMed]
- Eo, H.J.; Park, G.H.; Song, H.M.; Lee, J.W.; Kim, M.K.; Lee, M.H.; Lee, J.R.; Koo, J.S.; Jeong, J.B. Silymarin induces cyclin D1 proteasomal degradation via its phosphorylation of threonine-286 in human colorectal cancer cells. Int. Immunopharmacol. 2015, 24, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ewton, D.Z.; Deng, X.; Mercer, S.E.; Friedman, E. Mirk/Dyrk1B kinase destabilizes cyclin D1 by phosphorylation at threonine 288. J. Biol. Chem. 2004, 279, 27790–27798. [Google Scholar] [CrossRef] [PubMed]
- Agami, R.; Bernards, R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 2000, 102, 55–66. [Google Scholar] [CrossRef]
- Su, J.C.; Chang, J.H.; Huang, J.W.; Chen, P.P.; Chen, K.F.; Tseng, P.H.; Shiau, C.W. Copper-obatoclax derivative complexes mediate DNA cleavage and exhibit anti-cancer effects in hepatocellular carcinoma. Chem. Biol. Interact. 2015, 228, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Koehler, B.C.; Scherr, A.L.; Lorenz, S.; Urbanik, T.; Kautz, N.; Elssner, C.; Welte, S.; Bermejo, J.L.; Jäger, D.; Schulze-Bergkamen, H. Beyond cell death—Antiapoptotic BCL-2 proteins regulate migration and invasion of colorectal cancer cells in vitro. PLoS ONE 2013, 8, e76446. [Google Scholar] [CrossRef] [PubMed]
- Ligueros, M.; Jeoung, D.; Tang, B.; Hochhauser, D.; Reidenberg, M.M.; Sonenberg, M. Gossypol inhibition of mitosis, cyclin D1 and Rb protein in human mammary cancer cells and cyclin-D1 transfected human fibrosarcoma cells. Br. J. Cancer 1997, 76, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Wang, L.S.; Chang, H.L.; Ye, W.; Sugimoto, Y.; Dowd, M.K.; Wan, P.J.; Lin, Y.C. Effects of serum on (−)-gossypol-suppressed growth in human prostate cancer cells. Anticancer Res. 2006, 26, 3613–3620. [Google Scholar] [PubMed]
- Wang, Z.; Azmi, A.S.; Ahmad, A.; Banerjee, S.; Wang, S.; Sarkar, F.H.; Mohammad, R.M. TW-37, a small-molecule inhibitor of BCL-2, inhibits cell growth and induces apoptosis in pancreatic cancer: Involvement of Notch-1 signaling pathway. Cancer Res. 2009, 69, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.F.; Peng, Y.T.; Chuang, S.M.; Lin, S.C.; Feng, B.L.; Lu, C.H.; Yu, W.J.; Chang, J.S.; Chang, C.C. Prodigiosin down-regulates survivin to facilitate paclitaxel sensitization in human breast carcinoma cell lines. Toxicol. Appl. Pharmacol. 2009, 235, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.Y.; Shieh, J.J.; Chen, C.J.; Pan, M.Y.; Yang, S.Y.; Lin, S.C.; Chang, J.S.; Lee, A.Y.; Chang, C.C. Prodigiosin down-regulates SKP2 to induce p27KIP1 stabilization and antiproliferation in human lung adenocarcinoma cells. Br. J. Pharmacol. 2012, 166, 2095–2108. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Or, C.-H.R.; Chang, Y.; Lin, W.-C.; Lee, W.-C.; Su, H.-L.; Cheung, M.-W.; Huang, C.-P.; Ho, C.; Chang, C.-C. Obatoclax, a Pan-BCL-2 Inhibitor, Targets Cyclin D1 for Degradation to Induce Antiproliferation in Human Colorectal Carcinoma Cells. Int. J. Mol. Sci. 2017, 18, 44. https://doi.org/10.3390/ijms18010044
Or C-HR, Chang Y, Lin W-C, Lee W-C, Su H-L, Cheung M-W, Huang C-P, Ho C, Chang C-C. Obatoclax, a Pan-BCL-2 Inhibitor, Targets Cyclin D1 for Degradation to Induce Antiproliferation in Human Colorectal Carcinoma Cells. International Journal of Molecular Sciences. 2017; 18(1):44. https://doi.org/10.3390/ijms18010044
Chicago/Turabian StyleOr, Chi-Hung R., Yachu Chang, Wei-Cheng Lin, Wee-Chyan Lee, Hong-Lin Su, Muk-Wing Cheung, Chang-Po Huang, Cheesang Ho, and Chia-Che Chang. 2017. "Obatoclax, a Pan-BCL-2 Inhibitor, Targets Cyclin D1 for Degradation to Induce Antiproliferation in Human Colorectal Carcinoma Cells" International Journal of Molecular Sciences 18, no. 1: 44. https://doi.org/10.3390/ijms18010044