
Estrogen Enhances the Expression of the Multidrug Transporter Gene ABCG2—Increasing Drug Resistance of Breast Cancer Cells through Estrogen Receptors


 , ,
, ,
Abstract
:
1. Introduction
2. Results
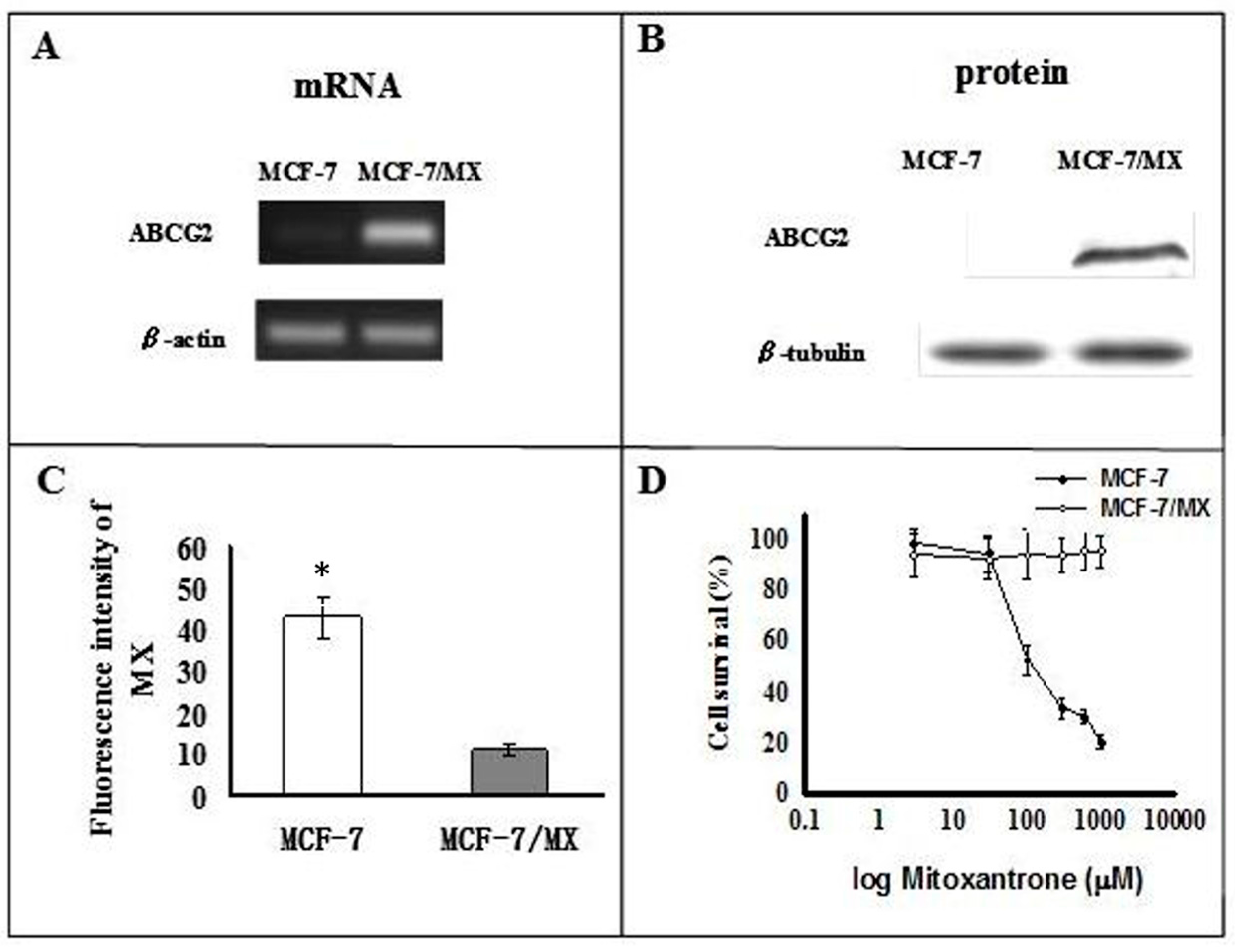
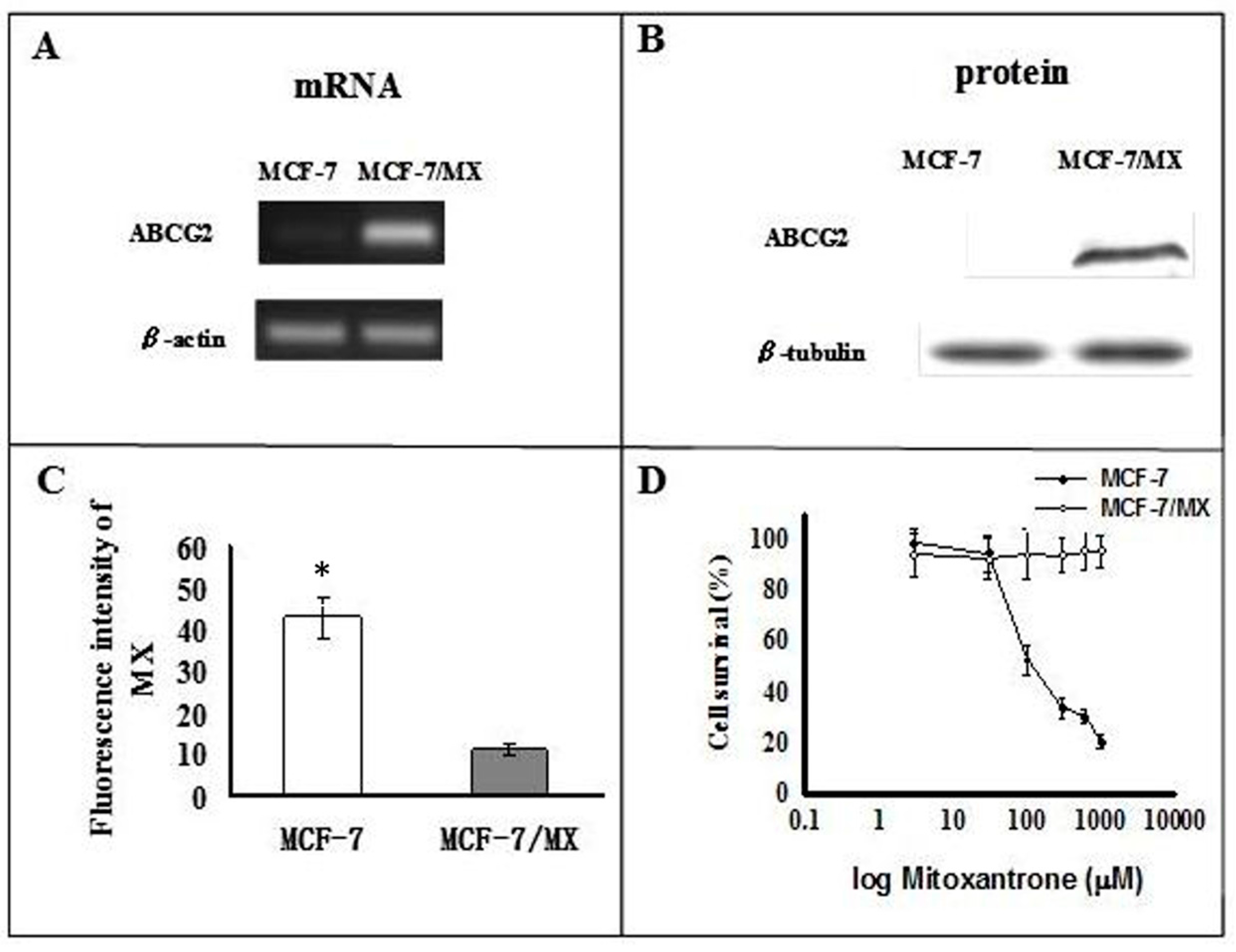
2.1. Characterization of MCF-7 and MCF-7/MX Cells
2.2. Microarray Analysis
2.3. Expression of Mitochondrial-Related Genes
2.4. Assessment of Mitochondrial Function by Hypoxia Treatment
2.5. The Production of ROS under Hypoxia
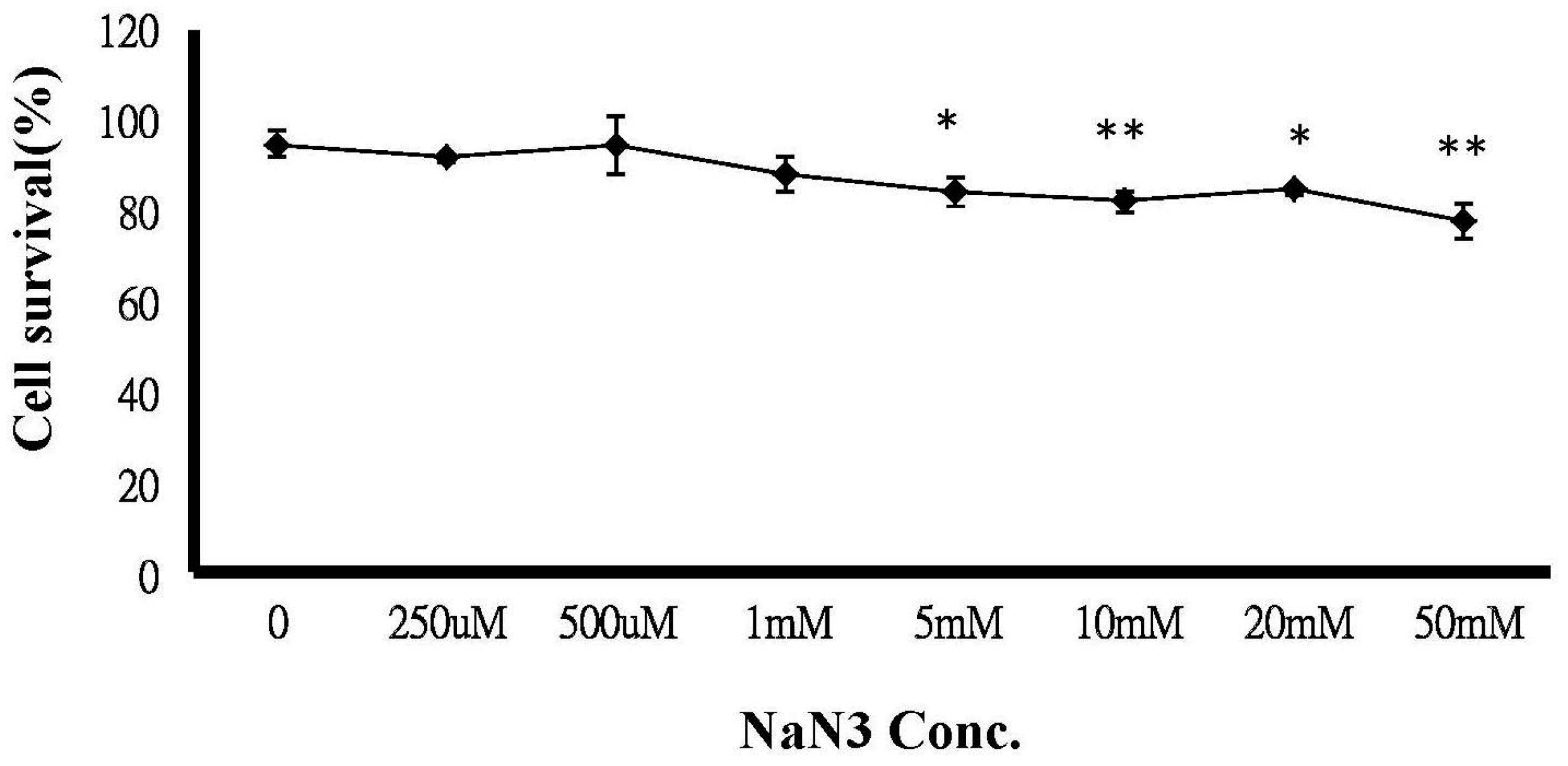
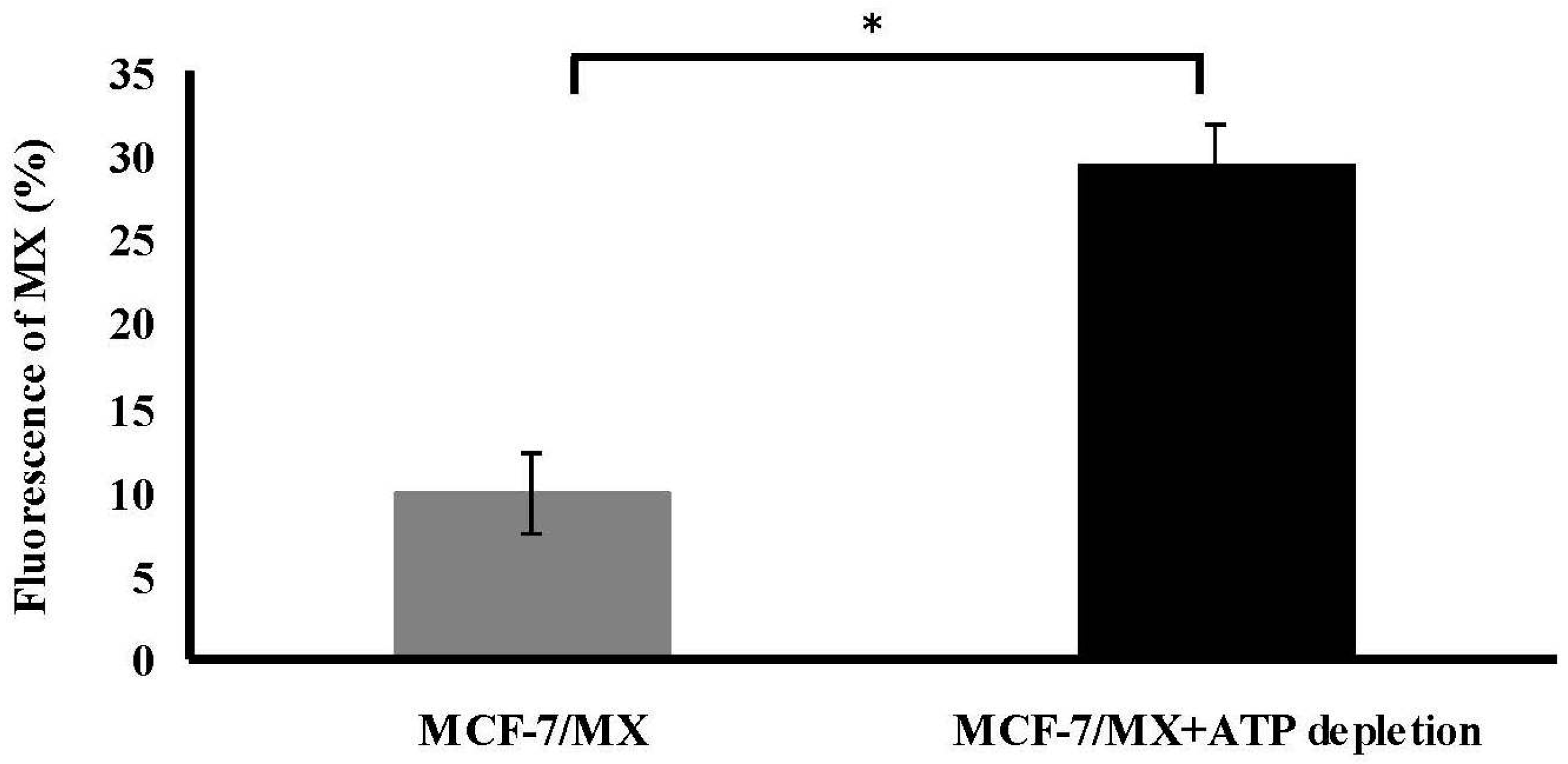
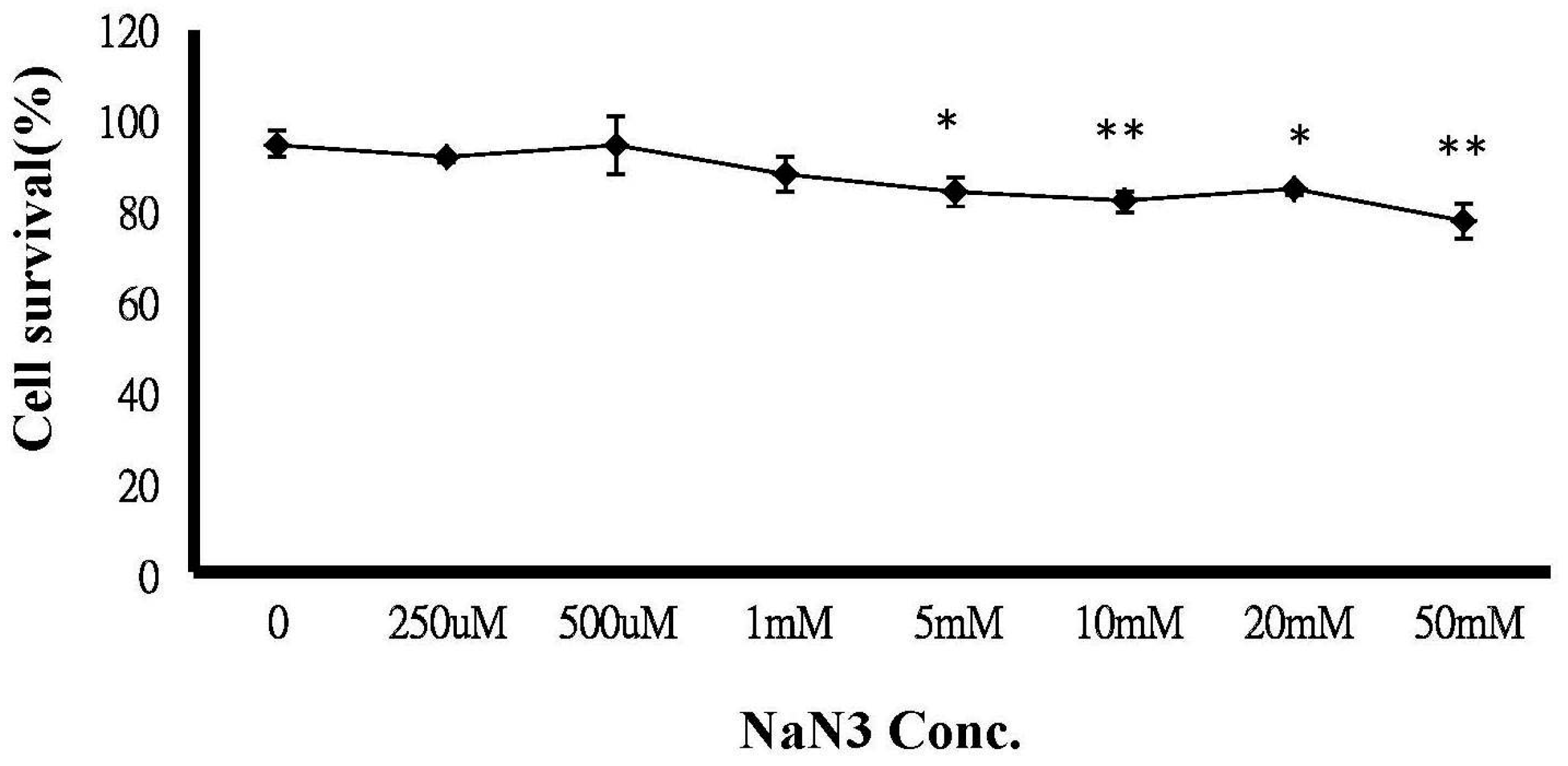
2.6. ATP Depletion Increased the Sensitivity of MCF-7/MX Cells to MX
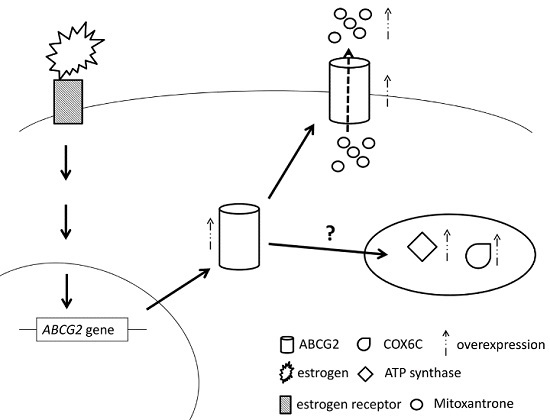
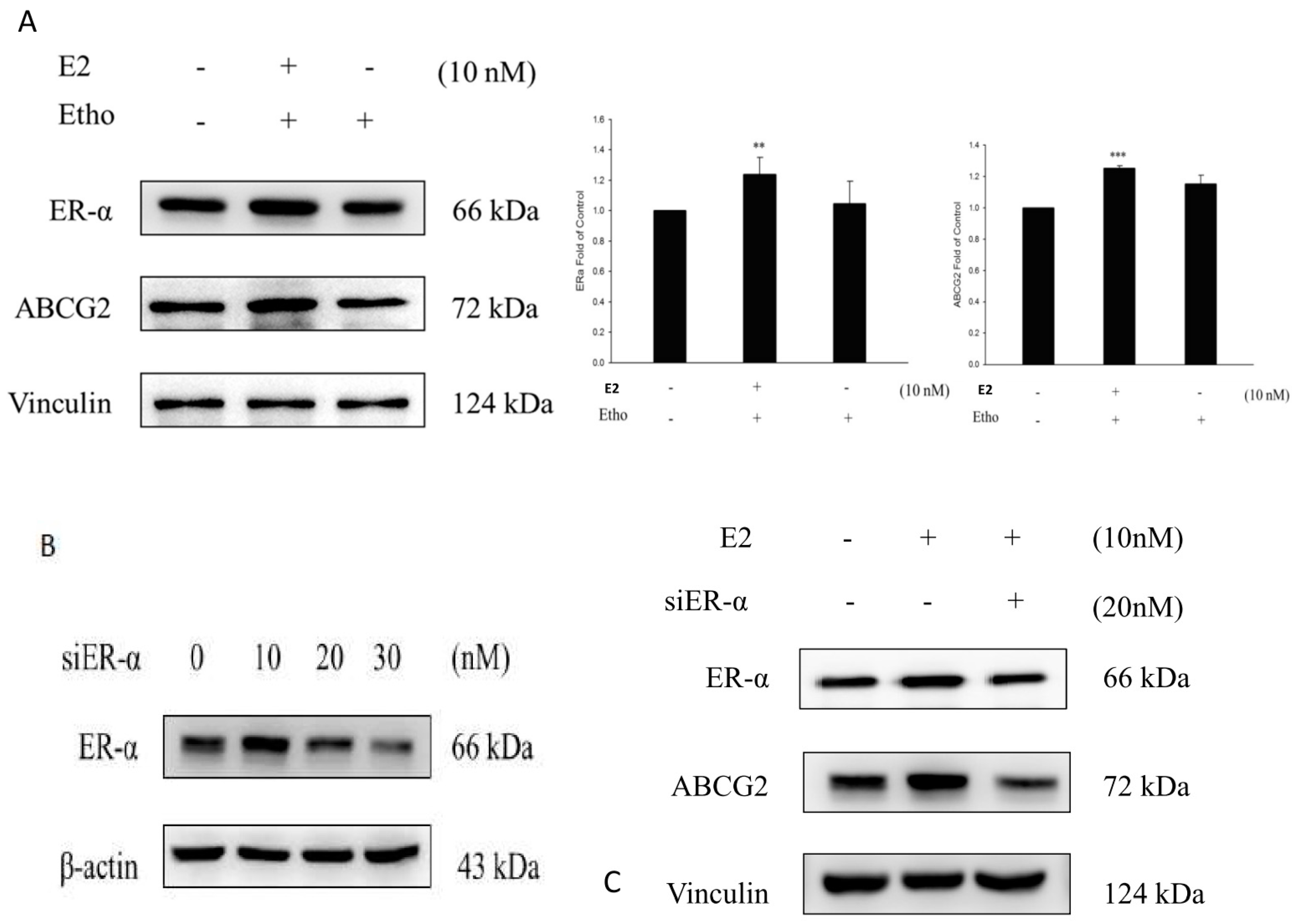
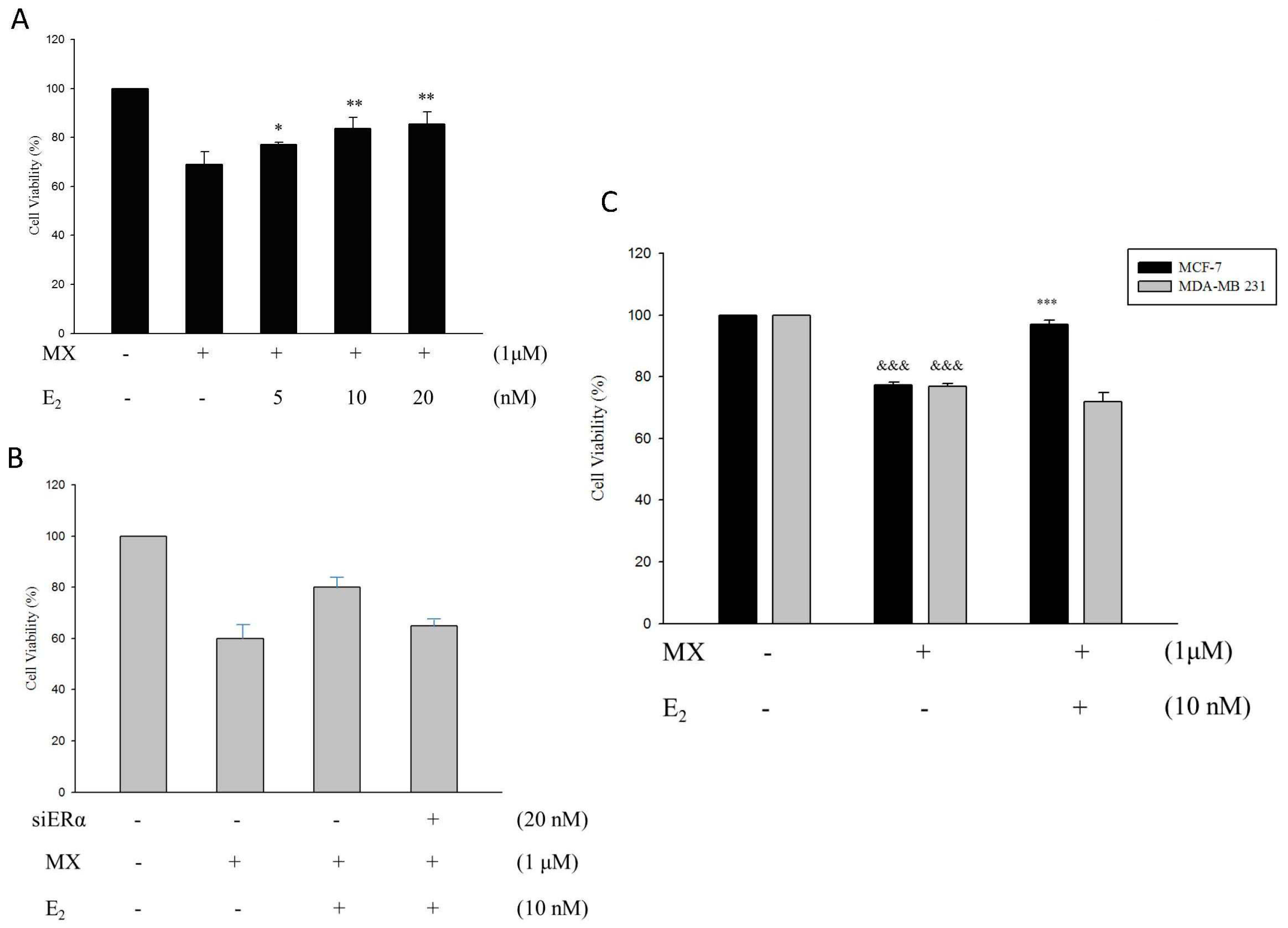
2.7. E2 Increased the Drug Resistance to MX by Enhancing the Expression of ABCG2 through ERα in MCF7 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cytotoxicity Assays
4.4. Flow Cytometric Detection of Functional Drug Efflux and Accumulation
4.5. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.6. Western Blot Analysis
4.7. cDNA Microarray
4.7.1. Preparation of the cDNA Glass Array
4.7.2. Preparation of Fluorescence-Labeled Targets and cDNA Microarray Hybridisation and Array Analysis
4.8. Mitochondrial Membrane Potential (MMP) Assay
4.9. Intracellular ROS Analysis
4.10. ATP Depletion
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Kroger, N.; Achterrath, W.; Hegewisch-Becker, S.; Mross, K.; Zander, A.R. Current options in treatment of anthracycline-resistant breast cancer. Cancer Treat. Rev. 1999, 25, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Lage, H. An overview of cancer multidrug resistance: A still unsolved problem. Cell. Mol. Life Sci. 2008, 65, 3145–3167. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel-Eibrink, M.M.; Wiemer, E.A.; Prins, A.; Meijerink, J.P.; Vossebeld, P.J.; van der Holt, B.; Pieters, R.; Sonneveld, P. Increased expression of the breast cancer resistance protein (BCRP) in relapsed or refractory acute myeloid leukemia (AML). Leukemia 2002, 16, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Pakos, E.E.; Ioannidis, J.P. The association of P-glycoprotein with response to chemotherapy and clinical outcome in patients with osteosarcoma. A meta-analysis. Cancer 2003, 98, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Haber, M.; Smith, J.; Bordow, S.B.; Flemming, C.; Cohn, S.L.; London, W.B.; Marshall, G.M.; Norris, M.D. Association of high-level MRP1 expression with poor clinical outcome in a large prospective study of primary neuroblastoma. J. Clin. Oncol. 2006, 24, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.; Chang, E.; Suh, K.; Lee, C.; Jee, J.; Shin, H. Prognostic Factors in Patients with Stage II/III Breast Cancer Treated with Adjuvant Extension of Neoadjuvant Chemotherapy: A Retrospective Cohort Study with Ten-Years of Follow-Up Data. J. Breast Cancer 2011, 14, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Kumpulainen, E.J.; Hirvikoski, P.P.; Johansson, R.T. Long-term outcome of adjuvant chemotherapy cyclophosphamide, mitoxantrone, and fluorouracil in women with breast cancer. Acta Oncol. 2008, 47, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Salerno, S.; Da, S.F.; Taliani, S.; Simorini, F.; La Motta, C.; Fornaciari, G.; Marini, A.M. Recent advances in the development of dual topoisomerase I and II inhibitors as anticancer drugs. Curr. Med. Chem. 2010, 17, 4270–4290. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, L.; Carpaneto, A.; Alterini, R.; Carrai, V.; Bernardi, F.; Bellesi, G.; Longo, G.; Bosi, A.; Rossi Ferrini, P. Treatment of large cell lymphoma in elderly patients with a mitoxantrone, cyclophosphamide, etoposide, and prednisone regimen: Long-term follow-up results. Cancer 2003, 97, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.A.; Reese, D.M. Treatment options in hormone-refractory prostate cancer: Current and future approaches. Drugs 2001, 61, 2177–2192. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.A.; Argilagos, R.F.; Emmons, M.; Boulware, D.; Beam, C.A.; Sullivan, D.M.; Dalton, W.S. Cell adhesion to fibronectin (CAM-DR) influences acquired mitoxantrone resistance in U937 cells. Cancer Res. 2006, 66, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Waters, R.; Leighton, C.; Hancock, J.; Sutton, R.; Moorman, A.V.; Ancliff, P.; Morgan, M.; Masurekar, A.; Goulden, N.; et al. Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): An open-label randomised trial. Lancet 2010, 376, 2009–2017. [Google Scholar] [CrossRef]
- Henderson, I.C.; Allegra, J.C.; Woodcock, T.; Wolff, S.; Bryan, S.; Cartwright, K.; Dukart, G.; Henry, D. Randomized clinical trial comparing mitoxantrone with doxorubicin in previously treated patients with metastatic breast cancer. J. Clin. Oncol. 1989, 7, 560–571. [Google Scholar] [PubMed]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T.; et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: Demonstration of homology to ABC transport genes. Cancer Res. 1999, 59, 8–13. [Google Scholar] [PubMed]
- Allikmets, R.; Schriml, L.M.; Hutchinson, A.; Romano-Spica, V.; Dean, M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998, 58, 5337–5339. [Google Scholar] [PubMed]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005, 7, E118–E133. [Google Scholar] [CrossRef] [PubMed]
- Ejendal, K.F.; Hrycyna, C.A. Multidrug resistance and cancer: The role of the human ABC transporter ABCG2. Curr. Protein Pept. Sci. 2002, 3, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Smit, J.W.; Brinkhuis, R.F.; Maliepaard, M.; Beijnen, J.H.; Schellens, J.H.; Schinkel, A.H. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J. Natl. Cancer Inst. 2000, 92, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van de Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar] [PubMed]
- Wang, F.L.; Wang, Y.; Wong, W.K.; Liu, Y.; Addivinola, F.J.; Liang, P.; Chen, L.B.; Kantoff, P.W.; Pardee, A.B. Two differentially expressed genes in normal human prostate tissue and in carcinoma. Cancer Res. 1996, 56, 3634–3637. [Google Scholar] [PubMed]
- Komatani, H.; Kotani, H.; Hara, Y.; Nakagawa, R.; Matsumoto, M.; Arakawa, H.; Nishimura, S. Identification of breast cancer resistant protein/mitoxantrone resistance/placenta-specific, ATP-binding cassette transporter as a transporter of NB-506 and J-107088, topoisomerase I inhibitors with an indolocarbazole structure. Cancer Res. 2001, 61, 2827–2832. [Google Scholar] [PubMed]
- Hohenberger, P.; Felgner, C.; Haensch, W.; Schlag, P.M. Tumor oxygenation correlates with molecular growth determinants in breast cancer. Breast Cancer Res. Treat. 1998, 48, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A.; Briest, S.; Hockel, M. Oxygenation gain factor: A novel parameter characterizing the association between hemoglobin level and the oxygenation status of breast cancers. Cancer Res. 2003, 63, 7634–7637. [Google Scholar] [PubMed]
- Papkovsky, D.B.; Hynes, J.; Will, Y. Respirometric Screening Technology for ADME-Tox studies. Expert Opin. Drug Metab. Toxicol. 2006, 2, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Amuthan, G.; Biswas, G.; Ananadatheerthavarada, H.K.; Vijayasarathy, C.; Shephard, H.M.; Avadhani, N.G. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 2002, 21, 7839–7849. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Tyleckova, J.; Hrabakova, R.; Mairychova, K.; Halada, P.; Radova, L.; Dzubak, P.; Hajduch, M.; Gadher, S.J.; Kovarova, H. Cancer cell response to anthracyclines effects: Mysteries of the hidden proteins associated with these drugs. Int. J. Mol. Sci. 2012, 13, 15536–15564. [Google Scholar] [CrossRef] [PubMed]
- Kule, C.; Ondrejickova, O.; Verner, K. Doxorubicin, daunorubicin, and mitoxantrone cytotoxicity in yeast. Mol. Pharmacol. 1994, 46, 1234–1240. [Google Scholar] [PubMed]
- Pan, J.; Sun, L.-C.; Tao, Y.-F.; Zhou, Z.; Du, X.; Peng, L.; Feng, X.; Wang, J.; Li, Y.; Liu, L.; et al. ATP synthase ecto-α-subunit: A novel therapeutic target for breast cancer. J. Transl. Med. 2011, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.; Nakanishi, T.; Ross, D.; Fenselau, C. Alterations in the mitochondrial proteome of adriamycin resistant MCF-7 breast cancer cells. J. Proteome Res. 2006, 5, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Huttemann, M.; Arnold, S.; Lee, I.; Bender, E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic. Biol. Med. 2000, 29, 211–221. [Google Scholar] [CrossRef]
- Grandjean, F.; Bremaud, L.; Robert, J.; Ratinaud, M.H. Alterations in the expression of cytochrome c oxidase subunits in doxorubicin-resistant leukemia K562 cells. Biochem. Pharmacol. 2002, 63, 823–831. [Google Scholar] [CrossRef]
- Zinkewich-Peotti, K.; Andrews, P.A. Loss of cis-diamminedichloroplatinum(II) resistance in human ovarian carcinoma cells selected for rhodamine 123 resistance. Cancer Res. 1992, 52, 1902–1906. [Google Scholar] [PubMed]
- Oliva, C.R.; Moellering, D.R.; Gillespie, G.Y.; Griguer, C.E. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS ONE 2011, 6, e24665. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Reers, M.; Smith, T.W.; Chen, L.B. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 1991, 30, 4480–4486. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GeneBank/UniGene | Gene Description | Fold Increase | Map |
|---|---|---|---|
| BC008987 | Major histocomp atibility complex class II, DR β4 | 8.3125105 | 6p21.3 |
| AB009303 | Matrix metalloproteinase 16 | 7.8706665 | 8q21 |
| AL048744 | Clusterin | 7.168563 | 8p21-p12 |
| NM_002456 | Mucin 1 | 5.0657783 | 1q21 |
| X14787 | Thrombospondin 1 | 5.054978 | 15q15 |
| BC020968 | Chemokine (C-X-C motif) receptor 4 | 4.8163495 | 2q21 |
| BC000879 | Kynureninase | 4.7721705 | 2q22.3 |
| AI028272 | Lymphorid nuclear protein related to AF4 | 4.69689 | 2q11.2-q12 |
| X17033 | Integrin, α2 | 3.9517348 | 5q23-q31 |
| AI0411381 | Choline phosphotransferase | 3.8714666 | 12q |
| NM_003380 | Vimentin | 3.5217342 | 10p13 |
| J05633 | Integrin, β5 | 3.2270584 | 3q21.2 |
| AI025170 | Cytochrome c oxidase subunit VIc | 3.56 | 8q22-q23 |
| NM_005147 | ATP synthase | 3.472383 | 10p15.1 |
| BC015513 | Glutathione S-transferase M4 | 3.2115557 | 1p13.3 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, F.-W.; Fan, H.-C.; Liu, J.-M.; Fan, T.-P.; Jing, J.; Yang, C.-L.; Hsu, R.-J. Estrogen Enhances the Expression of the Multidrug Transporter Gene ABCG2—Increasing Drug Resistance of Breast Cancer Cells through Estrogen Receptors. Int. J. Mol. Sci. 2017, 18, 163. https://doi.org/10.3390/ijms18010163
Chang F-W, Fan H-C, Liu J-M, Fan T-P, Jing J, Yang C-L, Hsu R-J. Estrogen Enhances the Expression of the Multidrug Transporter Gene ABCG2—Increasing Drug Resistance of Breast Cancer Cells through Estrogen Receptors. International Journal of Molecular Sciences. 2017; 18(1):163. https://doi.org/10.3390/ijms18010163
Chicago/Turabian StyleChang, Fung-Wei, Hueng-Chuen Fan, Jui-Ming Liu, Tai-Ping Fan, Jin Jing, Chia-Ling Yang, and Ren-Jun Hsu. 2017. "Estrogen Enhances the Expression of the Multidrug Transporter Gene ABCG2—Increasing Drug Resistance of Breast Cancer Cells through Estrogen Receptors" International Journal of Molecular Sciences 18, no. 1: 163. https://doi.org/10.3390/ijms18010163