
Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
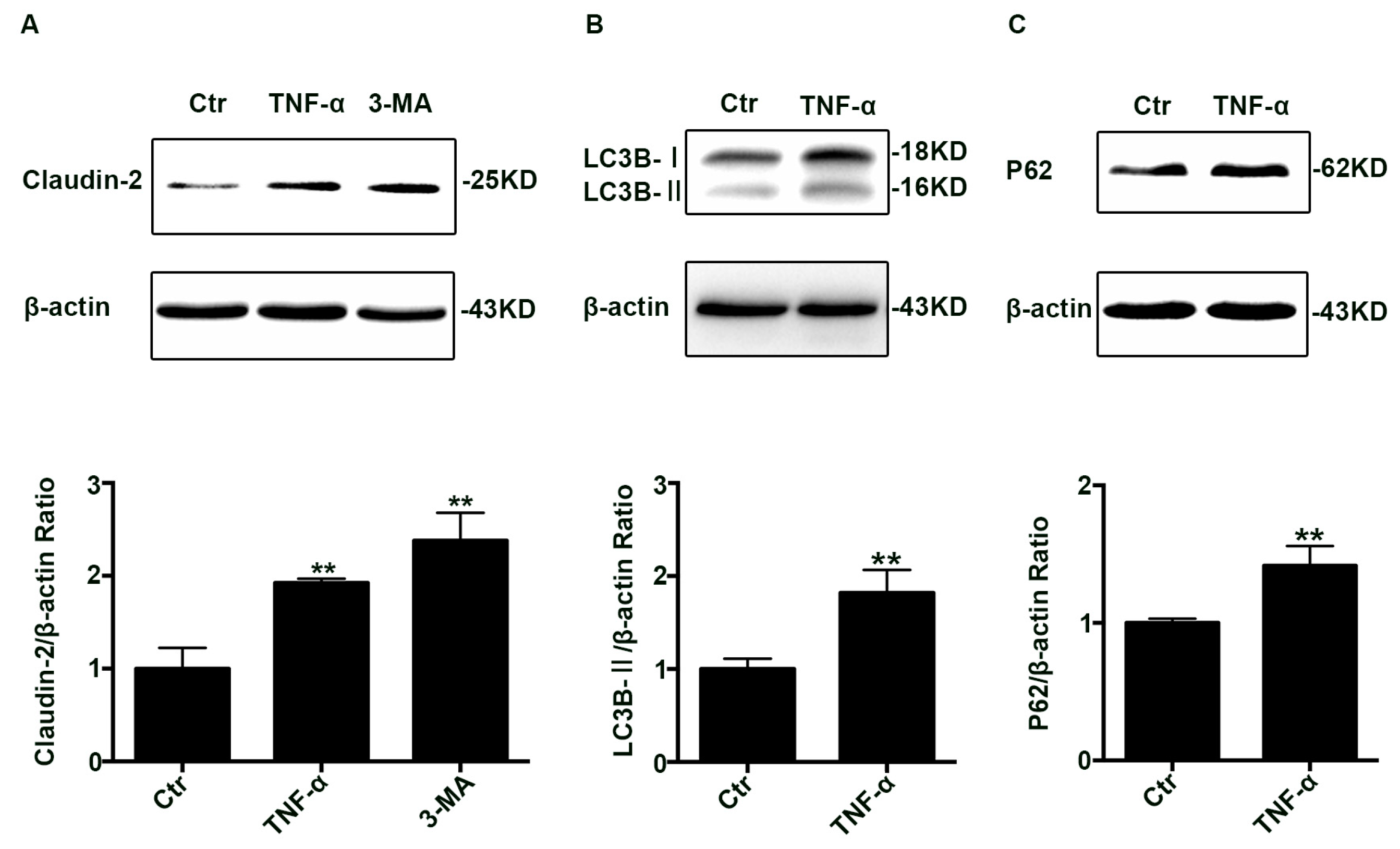
2.1. The Claudin-2, LC3B-II, and P62 Expression Increased in TNF-α Treated Caco-2 Cell Monolayers
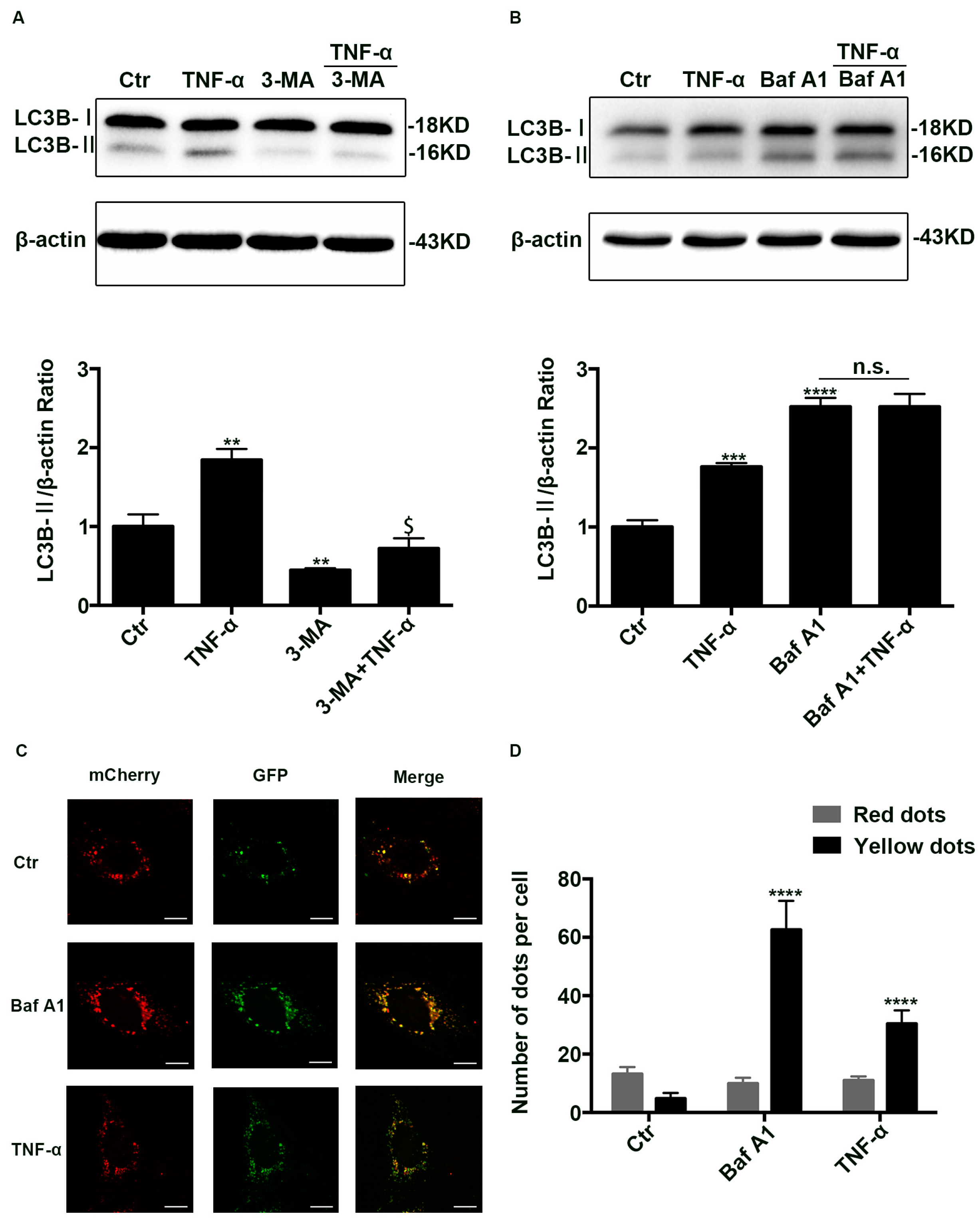
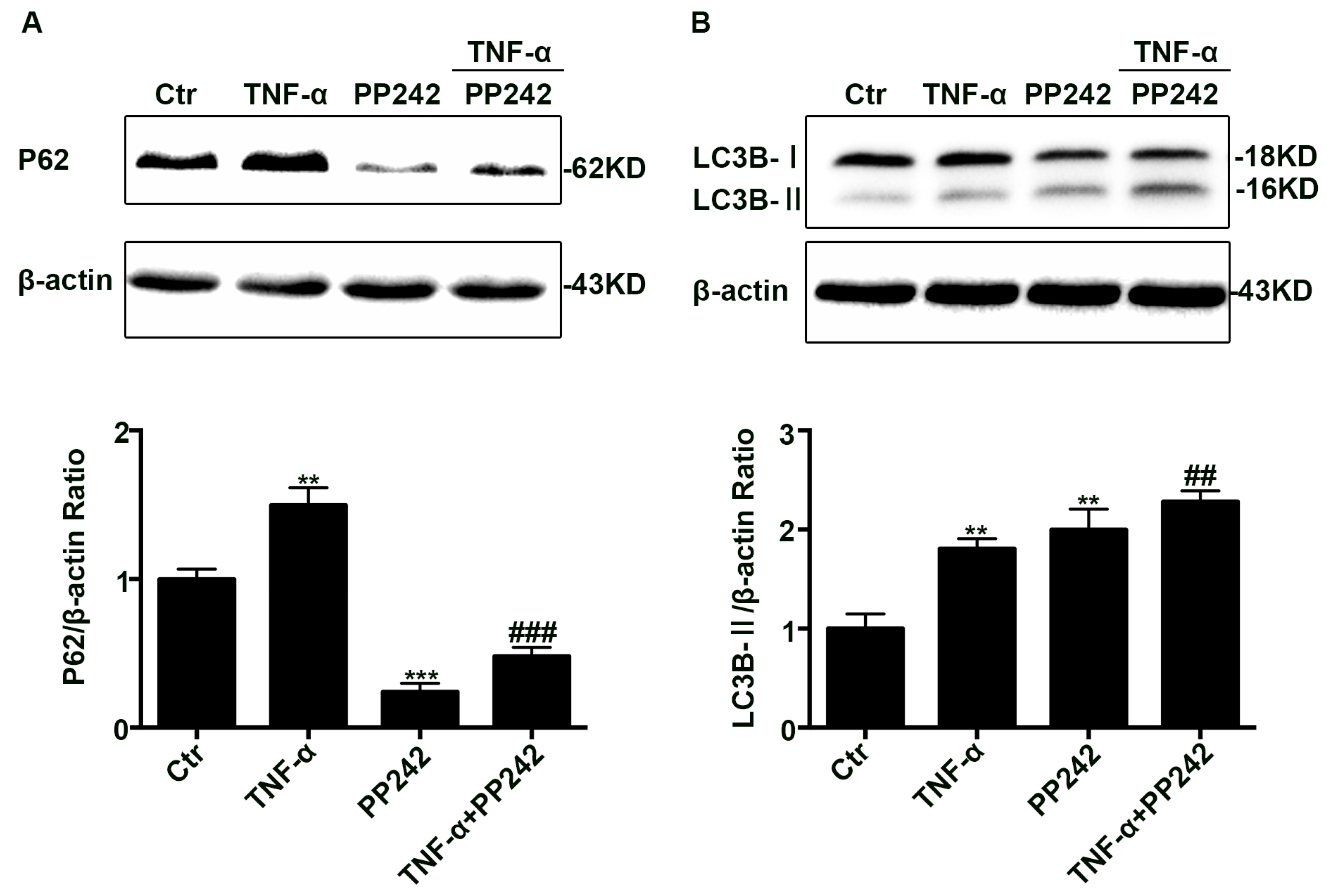
2.2. Autophagic Degradation Was Inhibited in TNF-α Treated Caco-2 Cells
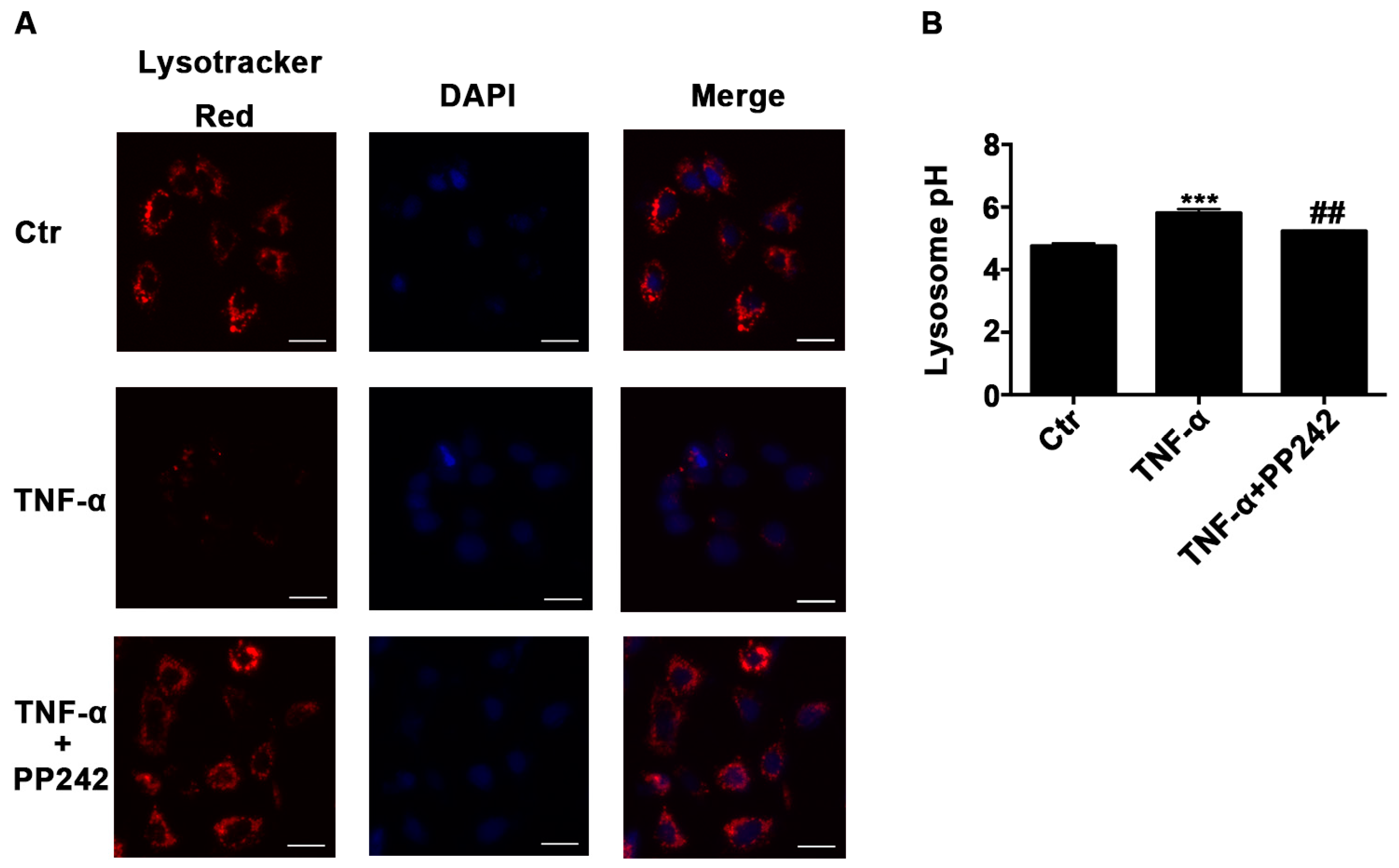
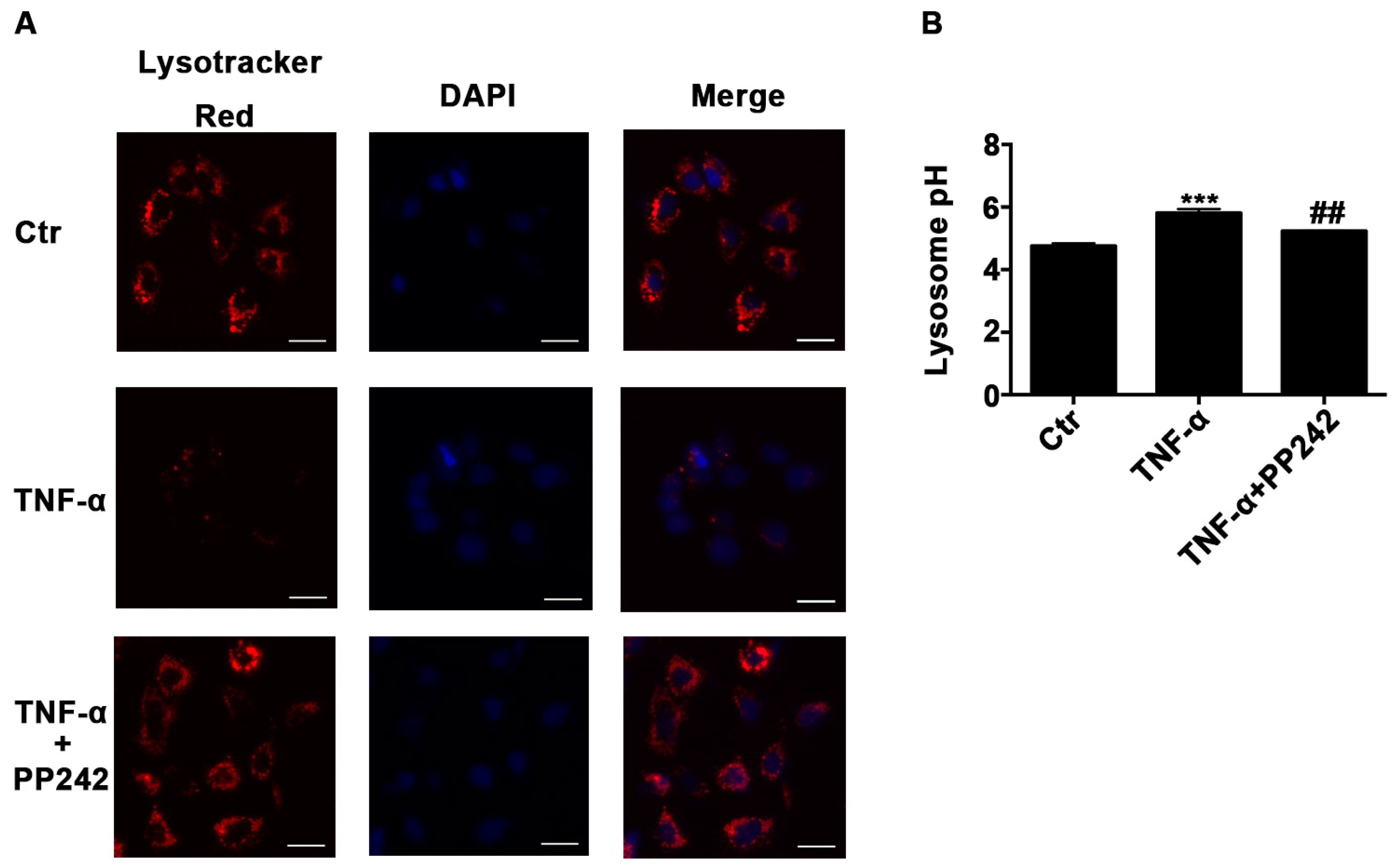
2.3. Lysosomal Acidic Environment Was Compromised and PP242 Rescued It in TNF-α Treated Caco-2 Cells
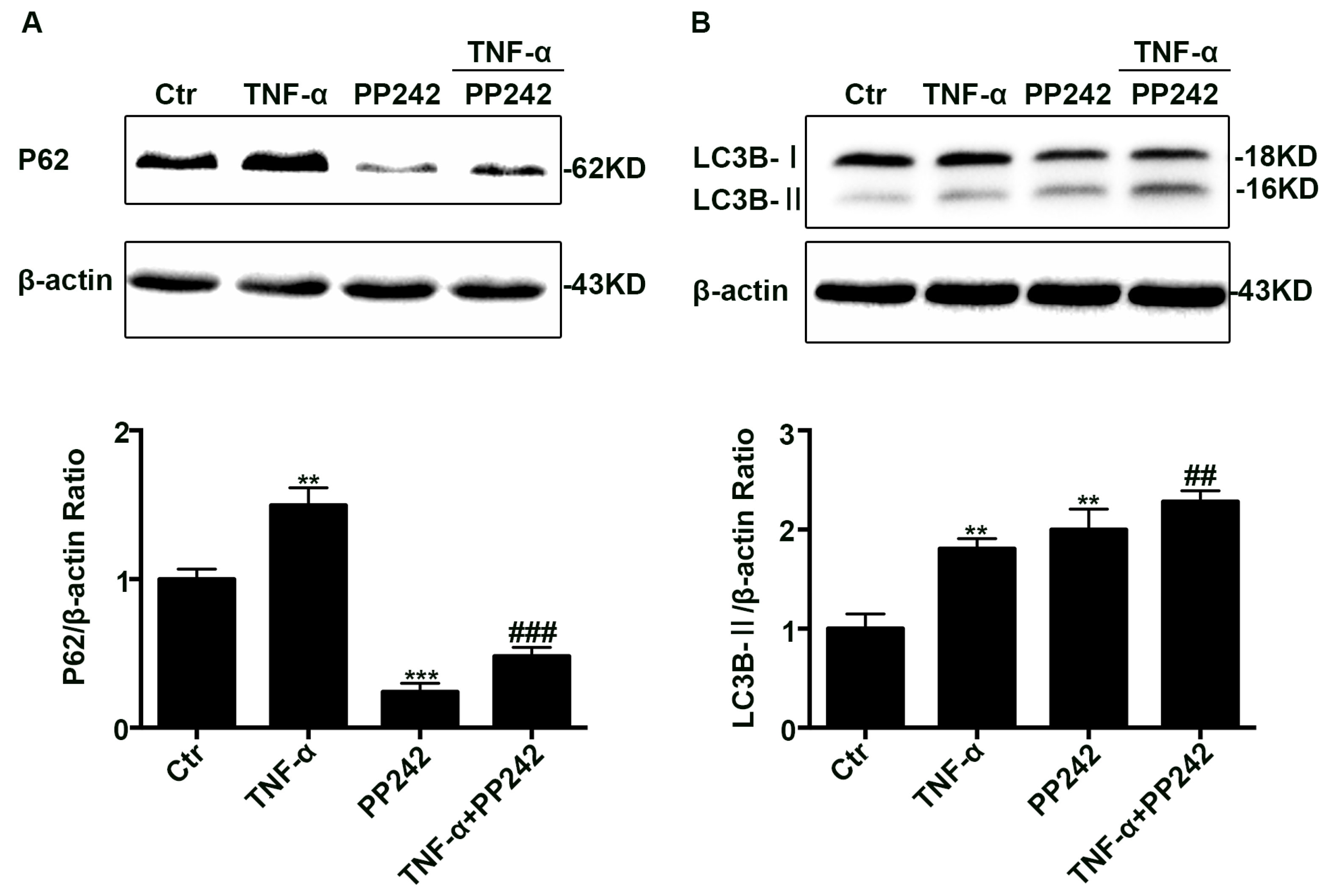
2.4. PP242 Relieved the Change of Autophagy Flux Induced by TNF-α
2.5. PP242 Partly Relieved the Change of Claudin-2 Expression and Intestinal Epithelial Tight Junction Function Induced by TNF-α
2.6. Repeated Experiments in IEC-6 Cells Confirmed PP242 Alleviated the Change of Lysosome pH, Autophagic Flux, Claudin-2 Protein, and Intestinal Epithelial Tight Junction Function Induced by TNF-α
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Western Blot Analysis
4.3. Ad-mCherry-GFP-LC3B Transfection
4.4. Lysosomal pH Measurement
4.5. Immunofluorescence Staining
4.6. Measurement of TER and Permeability
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luissint, A.C.; Parkos, C.A.; Nusrat, A. Inflammation and the intestinal barrier: Leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology 2016, 151, 616–632. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Miwa, H. Gastrointestinal mucosal barrier function and diseases. J. Gastroenterol. 2016, 51, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins-2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Mankertz, J.; Hillenbrand, B.; Tavalali, S.; Huber, O.; Fromm, M.; Schulzke, J.D. Functional crosstalk between Wnt signaling and Cdx-related transcriptional activation in the regulation of the claudin-2 promoter activity. Biochem. Biophys. Res. Commun. 2004, 314, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, K.R.; Liu, S.X.; Tian, R.; Kushnir, A.; Turner, J.R.; Li, H.L.; Chou, P.M.; Weber, C.R.; de Plaen, I.G. Bifidobacteria stabilize claudins at tight junctions and prevent intestinal barrier dysfunction in mouse necrotizing enterocolitis. Am. J. Pathol. 2013, 182, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.T.; Yan, W.H.; Cao, Y.; Yan, J.K.; Cai, W. Neutralization of IL-6 and TNF-α ameliorates intestinal permeability in DSS-induced colitis. Cytokine 2016, 83, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Zhu, J.; Zuo, S.; Zhang, J.L.; Chen, Z.Y.; Chen, G.W.; Wang, X.; Pan, Y.S.; Liu, Y.C.; Wang, P.Y. Protective effect of hydrogen sulfide on TNF-α and IFN-γ-induced injury of intestinal epithelial barrier function in Caco-2 monolayers. Inflamm. Res. 2015, 64, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, E.; Castagnetti, F.; Bianconi, V.; Madaro, L.; de Bardi, M.; Nazio, F.; D’Amico, A.; Bertini, E.; Cecconi, F.; Puri, P.L.; et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ. 2016, 23, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Kim, J.; Choi, M. Autophagy mediates phase transitions from cell death to life. Heliyon 2015, 1, e00027. [Google Scholar] [CrossRef] [PubMed]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J. Biol. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Liu, W.; Xue, P.; Zhang, Y.; Wang, Q.; Jin, Y. Increased autophagy is required to protect periodontal ligament stem cells from apoptosis in inflammatory microenvironment. J. Clin. Periodontol. 2016, 43, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Francois, A.; Terro, F.; Quellard, N.; Fernandez, B.; Chassaing, D.; Janet, T.; Rioux Bilan, A.; Paccalin, M.; Page, G. Impairment of autophagy in the central nervous system during lipopolysaccharide-induced inflammatory stress in mice. Mol. Brain 2014, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, D.; Wang, W.; Piao, S.; Zhou, J.; Saiyin, W.; Zheng, C.; Sun, H.; Li, Y. Inhibition of autophagy by 3-MA enhances IL-24-induced apoptosis in human oral squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. 2015, 34, 97. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Q.; Xie, W.J.; Pan, D.; Chen, H.; Zhang, L. Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity of gastric cancer cells. Tumour Biol. 2016, 37, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, S.H.; Nicolas, V.; Bauvy, C.; Yang, N.D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, P.; Pittet, V.; Vader, J.P.; Burnand, B.; Gonvers, J.J.; de Saussure, P.; Mottet, C.; Seibold, F.; Rogler, G.; Sagmeister, M.; et al. Infliximab for Crohn’s disease in the Swiss IBD Cohort Study: Clinical management and appropriateness. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Tayman, C.; Aydemir, S.; Yakut, I.; Serkant, U.; Ciftci, A.; Arslan, E.; Koc, O. TNF-α blockade efficiently reduced severe intestinal damage in necrotizing enterocolitis. J. Investig. Surg. 2016, 29, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Fowler, E.V.; Doecke, J.; Simms, L.A.; Zhao, Z.Z.; Webb, P.M.; Hayward, N.K.; Whiteman, D.C.; Florin, T.H.; Montgomery, G.W.; Cavanaugh, J.A.; et al. ATG16L1 T300A shows strong associations with disease subgroups in a large Australian IBD population: Further support for significant disease heterogeneity. Am. J. Gastroenterol. 2008, 103, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Katsuno, T.; Nakagawa, T.; Sato, T.; Noguchi, Y.; Sazuka, S.; Saito, K.; Arai, M.; Yokote, K.; Yokosuka, O. Intestinal epithelial cells with impaired autophagy lose their adhesive capacity in the presence of TNF-α. Dig. Dis. Sci. 2012, 57, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xiao, L.; Yin, Y.; Wei, L. Ligustilide inhibits tumour necrosis factor-α-induced autophagy during C2C12 cells differentiation. Biomed. Pharmacother. Biomed. Pharmacother. 2015, 69, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Cheng, X.Y.; Jin, M.; Cao, Y.L.; Yang, Y.P.; Wang, J.D.; Li, Q.; Wang, F.; Hu, L.F.; Liu, C.F. TNF compromises lysosome acidification and reduces α-synuclein degradation via autophagy in dopaminergic cells. Exp. Neurol. 2015, 271, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Opperman, C.M.; Sishi, B.J. Tumor necrosis factor α stimulates p62 accumulation and enhances proteasome activity independently of ROS. Cell Biol. Toxicol. 2015, 31, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Martin-Martin, N.; Ryan, G.; McMorrow, T.; Ryan, M.P. Sirolimus and cyclosporine A alter barrier function in renal proximal tubular cells through stimulation of ERK1/2 signaling and claudin-1 expression. Am. J. Physiol. Ren. Physiol. 2010, 298, F672–F682. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, L.; Citi, S. Cingulin regulates claudin-2 expression and cell proliferation through the small GTPase RhoA. Mol. Biol. Cell 2006, 17, 3569–3577. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Ahmad, R.; Chaturvedi, R.; Smith, J.J.; Midha, R.; Mittal, M.K.; Krishnan, M.; Chen, X.; Eschrich, S.; Yeatman, T.J.; et al. Claudin-2 expression increases tumorigenicity of colon cancer cells: Role of epidermal growth factor receptor activation. Oncogene 2011, 30, 3234–3247. [Google Scholar] [CrossRef] [PubMed]
- Amoozadeh, Y.; Dan, Q.; Xiao, J.; Waheed, F.; Szaszi, K. Tumor necrosis factor α induces a biphasic change in claudin-2 expression in tubular epithelial cells: Role in barrier functions. Am. J. Physiol. Cell Physiol. 2015, 309, C38–C50. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Holmes, J.; Bridges, A.; Anderson, J.M. Claudin-2-dependent changes in noncharged solute flux are mediated by the extracellular domains and require attachment to the PDZ-scaffold. Ann. N. Y. Acad. Sci. 2009, 1165, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Holmes, J.; Bridges, A.; Gookin, J.L.; Coccaro, M.R.; Proctor, W.; Colegio, O.R.; Anderson, J.M. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J. Cell Sci. 2008, 121, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Squillantea, E.; Gillespieb, M.; Shao, J. Transepithelial electrical resistance is not a reliable measurement of the Caco-2 monolayer integrity in Transwell. Drug Deliv. 2004, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Khatib, K.; Guo, S.; Ye, D.; Youssef, M.; Ma, T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1054–G1064. [Google Scholar] [CrossRef] [PubMed]
- Luettig, J.; Rosenthal, R.; Barmeyer, C.; Schulzke, J.D. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 2015, 3, e977176. [Google Scholar] [CrossRef] [PubMed]
- Amasheh, M.; Fromm, A.; Krug, S.M.; Amasheh, S.; Andres, S.; Zeitz, M.; Fromm, M.; Schulzke, J.D. TNF-α-induced and berberine-antagonized tight junction barrier impairment via tyrosine kinase, Akt and NFκB signaling. J. Cell Sci. 2010, 123, 4145–4155. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Zhang, L.; Wen, X.; Wang, X.; Cheng, X.; Du, H.; Hu, Y.; Li, L.; Dong, B.; Li, Z.; et al. PP242 suppresses cell proliferation, metastasis, and angiogenesis of gastric cancer through inhibition of the PI3K/AKT/mTOR pathway. Anticancer Drugs 2014, 25, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, Y.X.; Tsao, T.; Qiu, Y.; Kornblau, S.M.; Baggerly, K.A.; Liu, W.; Jessen, K.; Liu, Y.; Kantarjian, H.; et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012, 120, 2679–2689. [Google Scholar] [CrossRef] [PubMed]
- Mutalib, M.; Borrelli, O.; Blackstock, S.; Kiparissi, F.; Elawad, M.; Shah, N.; Lindley, K. The use of sirolimus (rapamycin) in the management of refractory inflammatory bowel disease in children. J. Crohn’s Colitis 2014, 8, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Su, M.; Xie, F.; Ai, J.; Ren, Y.; Zhang, J.; Guan, R.; He, W.; Gong, Y.; Guo, Y. Tetrandrine blocks autophagic flux and induces apoptosis via energetic impairment in cancer cells. Cell Death Dis. 2014, 5, e1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, W.; Guha, S.; Baltazar, G.C.; Coffey, E.E.; Laties, A.M.; Rubenstein, R.C.; Reenstra, W.W.; Mitchell, C.H. Cystic fibrosis transmembrane conductance regulator contributes to reacidification of alkalinized lysosomes in RPE cells. Am. J. Physiol. Cell Physiol. 2012, 303, C160–C169. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, W.; Reigada, D.; Nguyen, J.; Laties, A.M.; Mitchell, C.H. Restoration of lysosomal pH in RPE cells from cultured human and ABCA4−/− mice: Pharmacologic approaches and functional recovery. Investig. Ophthalmol. Vis. Sci. 2008, 49, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.D.; Chen, W.; Sun, L.H.; Wang, W.S.; Zhou, S.W.; Yang, H. The protective effect of enteric glial cells on intestinal epithelial barrier function is enhanced by inhibiting inducible nitric oxide synthase activity under lipopolysaccharide stimulation. Mol. Cell. Neurosci. 2011, 46, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Harada, T.; Li, J.; Uchiyama, T.; Han, Y.; Englert, J.A.; Fink, M.P. Bile modulates intestinal epithelial barrier function via an extracellular signal related kinase 1/2 dependent mechanism. Intensiv. Care Med. 2005, 31, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Jin, G.; Du, L.; Yang, Q. Modulation of intestinal folate absorption by erythropoietin in vitro. Mol. Pharm. 2014, 11, 358–366. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Yan, J.; Xiao, Y.; Shen, Y.; Wang, J.; Ge, W.; Chen, Y. Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers. Int. J. Mol. Sci. 2017, 18, 157. https://doi.org/10.3390/ijms18010157
Zhang C, Yan J, Xiao Y, Shen Y, Wang J, Ge W, Chen Y. Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers. International Journal of Molecular Sciences. 2017; 18(1):157. https://doi.org/10.3390/ijms18010157
Chicago/Turabian StyleZhang, Cong, Junkai Yan, Yongtao Xiao, Yujie Shen, Jiazheng Wang, Wensong Ge, and Yingwei Chen. 2017. "Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers" International Journal of Molecular Sciences 18, no. 1: 157. https://doi.org/10.3390/ijms18010157