
Indoxyl Sulfate as a Mediator Involved in Dysregulation of Pulmonary Aquaporin-5 in Acute Lung Injury Caused by Acute Kidney Injury

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
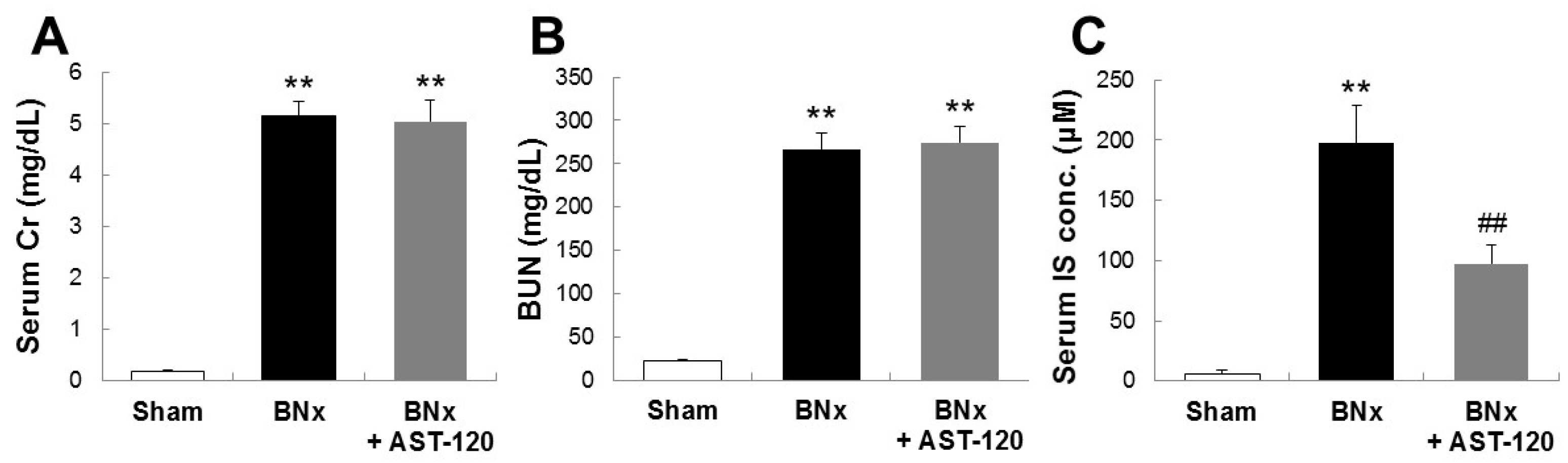
2.1. SCr, BUN and Serum Accumulations of IS
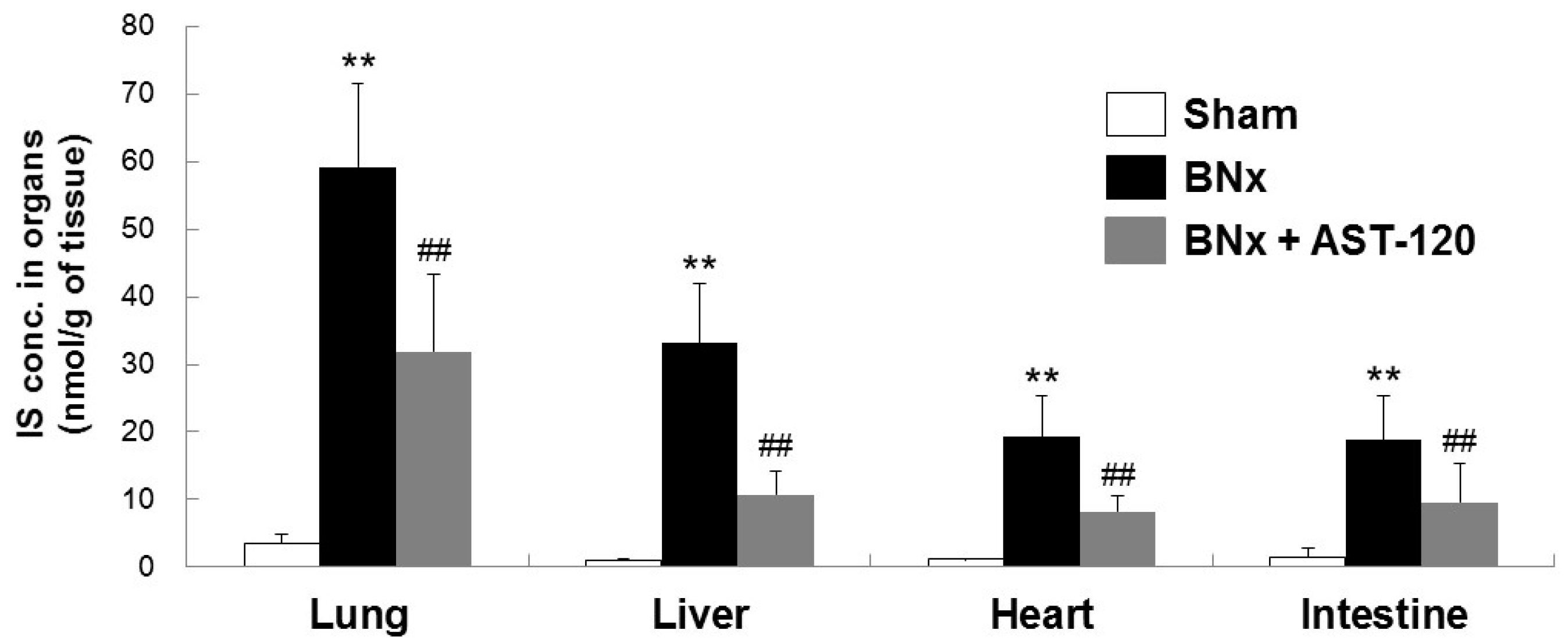
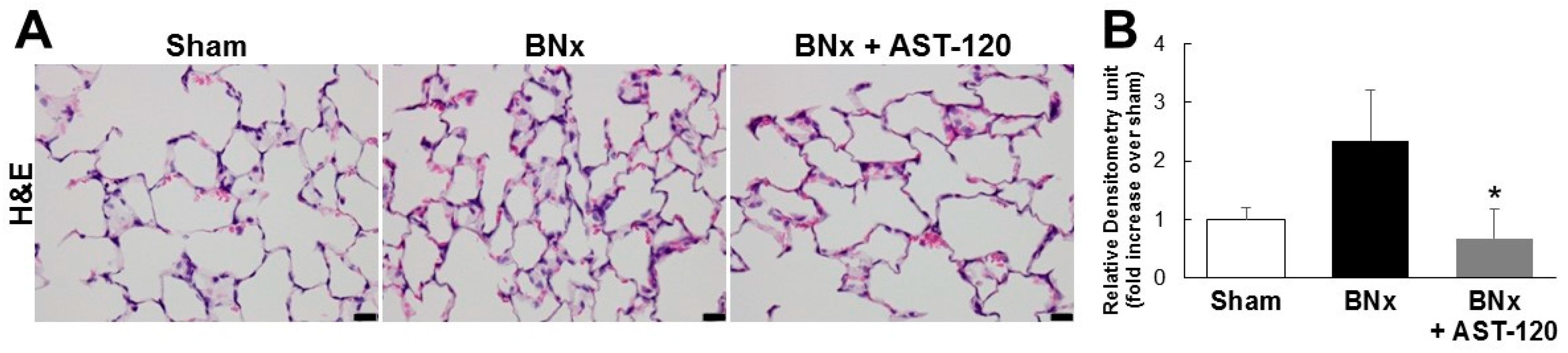
2.2. Organ Accumulation of IS and Histological Changes of Lung Tissue
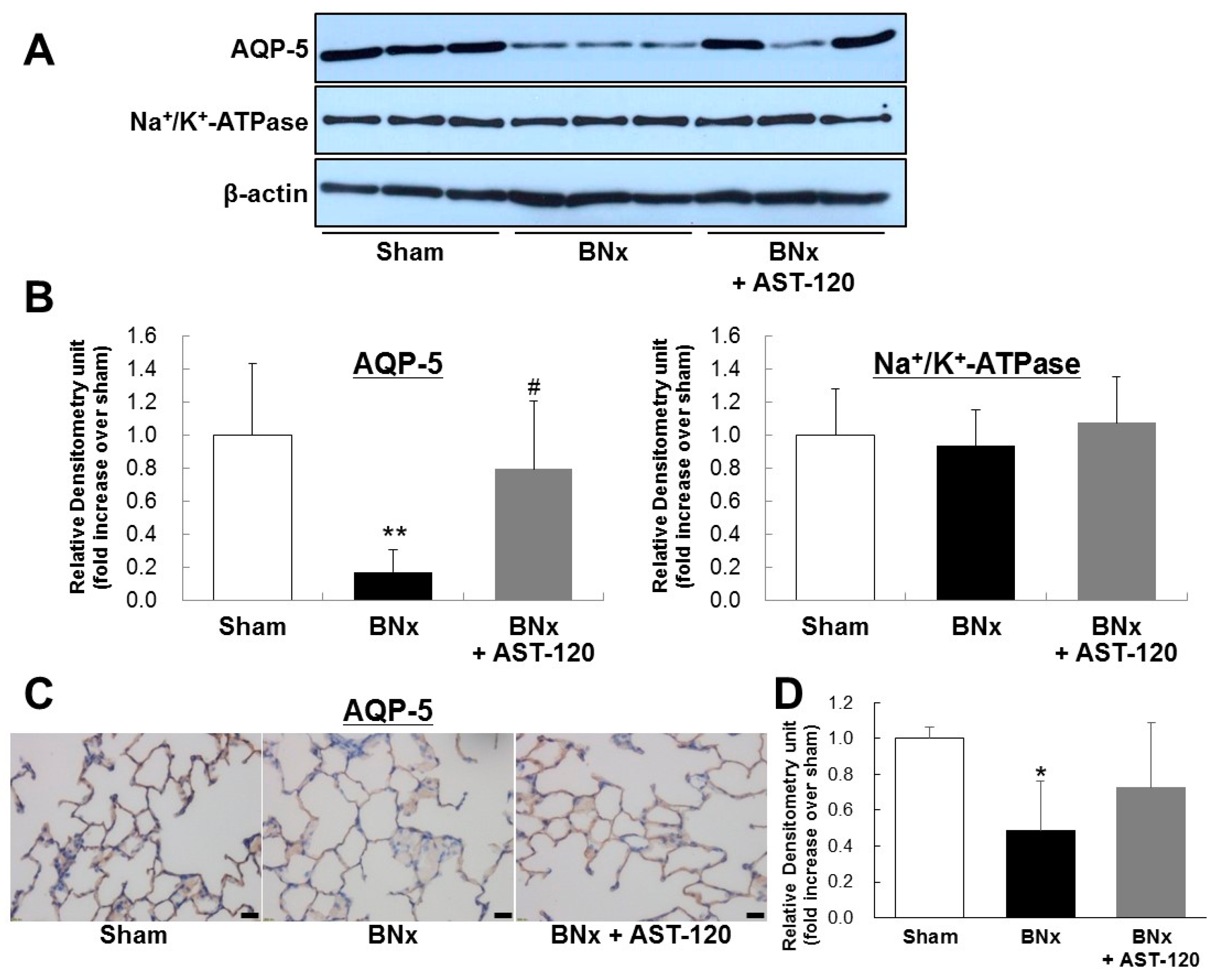
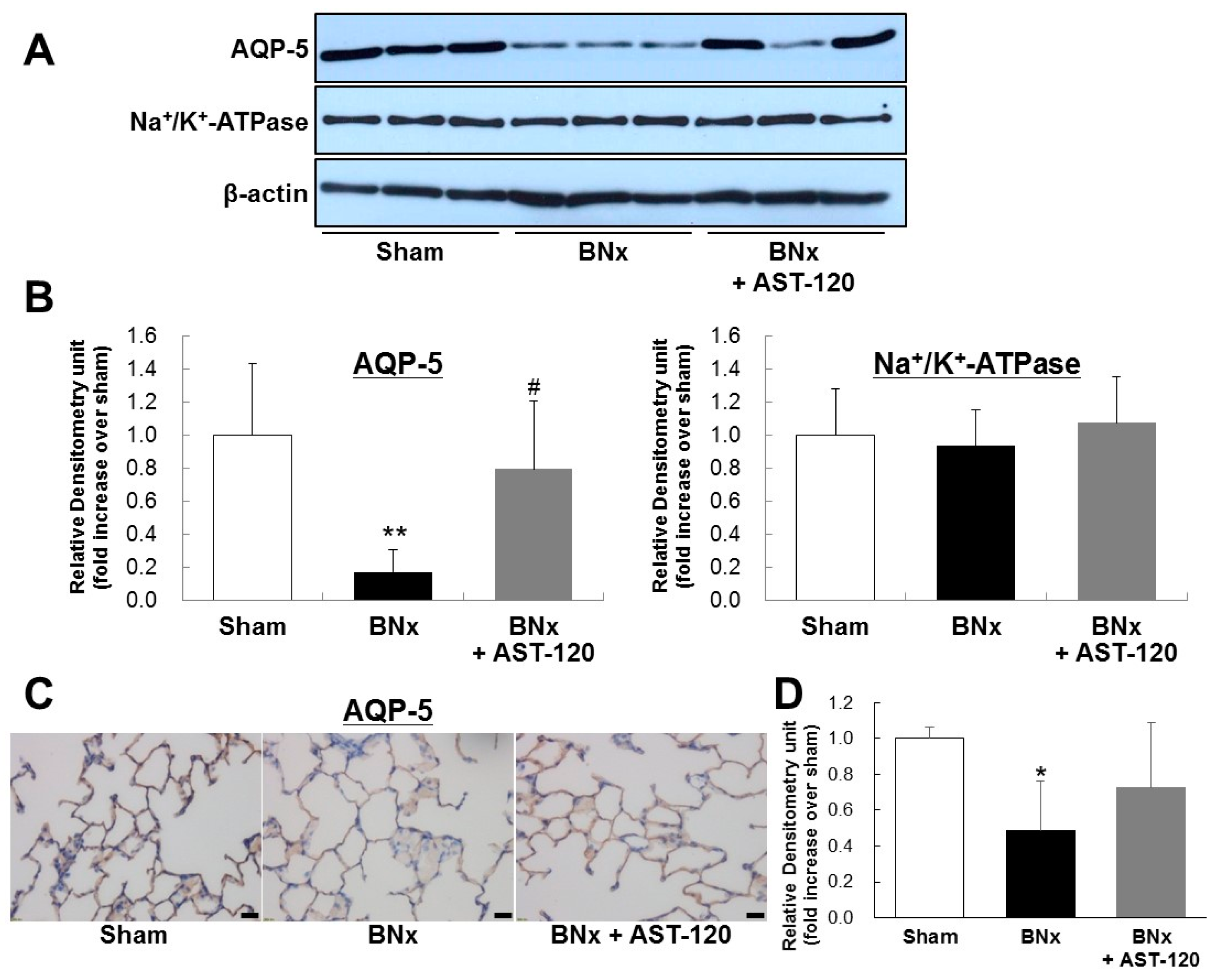
2.3. AQP-5 and Na+/K+-ATPase Protein Expressions of the Lung
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animal Experiments
4.3. High-Performance Liquid Chromatography Determination of IS Concentration
4.4. Western Blot Analysis
4.5. Histochemical Staining
4.6. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute renal failure. N. Engl. J. Med. 1996, 334, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Okusa, M.D. Distant organ injury following acute kidney injury. Am. J. Physiol. Ren. Physiol. 2007, 293, F28–F29. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Chamoun, F.; Hotchkiss, J. Molecular mechanisms underlying combined kidney-lung dysfunction during acute renal failure. Contrib. Nephrol. 2001, 132, 41–52. [Google Scholar]
- Wheeler, A.P.; Bernard, G.R. Acute lung injury and the acute respiratory distress syndrome: A clinical review. Lancet 2007, 369, 1553–1564. [Google Scholar] [CrossRef]
- Liu, K.D.; Matthay, M.A. Advances in critical care for the nephrologist: Acute lung injury/ARDS. Clin. J. Am. Soc. Nephrol. 2008, 3, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Krane, C.M.; Fortner, C.N.; Hand, A.R.; McGraw, D.W.; Lorenz, J.N.; Wert, S.E.; Towne, J.E.; Paul, R.J.; Whitsett, J.A.; Menon, A.G. Aquaporin 5-deficient mouse lungs are hyperresponsive to cholinergic stimulation. Proc. Natl. Acad. Sci. USA 2001, 98, 14114–14119. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Fukuda, N.; Song, Y.; Matthay, M.A.; Verkman, A.S. Lung fluid transport in aquaporin-5 knockout mice. J. Clin. Investig. 2000, 105, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Wang, Z.; Nemoto, T.; Hotchkiss, J.; Yokota, N.; Soleimani, M. Acute renal failure leads to dysregulation of lung salt and water channels. Kidney Int. 2003, 63, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; de Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.C.; Young, G.H.; Huang, P.H.; Lo, S.C.; Wang, K.C.; Sun, C.Y.; Liang, C.J.; Huang, T.M.; Chen, J.H.; Chang, F.C.; et al. In acute kidney injury, indoxyl sulfate impairs human endothelial progenitor cells: Modulation by statin. Angiogenesis 2013, 16, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Banoglu, E.; King, R.S. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2002, 27, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Banoglu, E.; Jha, G.G.; King, R.S. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, T.; Matsuzaki, T.; Yokoo, K.; Kusumoto, M.; Iwata, K.; Hamada, A.; Saito, H. Regulation of renal organic ion transporters in cisplatin-induced acute kidney injury and uremia in rats. Pharm. Res. 2008, 25, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Watanabe, H.; Morisaki, T.; Matsuzaki, T.; Ohmura, T.; Hamada, A.; Saito, H. Involvement of indoxyl sulfate in renal and central nervous system toxicities during cisplatin-induced acute renal failure. Pharm. Res. 2007, 24, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Saigo, C.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Matsunaga, R.; Jono, H.; Nishi, K.; Saito, H. Meclofenamate elicits a nephropreventing effect in a rat model of ischemic acute kidney injury by suppressing indoxyl sulfate production and restoring renal organic anion transporters. Drug Des. Dev. Ther. 2014, 8, 1073–1082. [Google Scholar]
- Niwa, T. Uremic toxicity of indoxyl sulfate. Nagoya J. Med. Sci. 2010, 72, 1–11. [Google Scholar] [PubMed]
- Fujita, K.; Sugiura, T.; Okumura, H.; Umeda, S.; Nakamichi, N.; Watanabe, Y.; Suzuki, H.; Sunakawa, Y.; Shimada, K.; Kawara, K.; et al. Direct inhibition and down-regulation by uremic plasma components of hepatic uptake transporter for SN-38, an active metabolite of irinotecan, in humans. Pharm. Res. 2014, 31, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, M.; Kamobayashi, H.; Sato, D.; Komori, M.; Yoshimura, M.; Hamada, A.; Kohda, Y.; Tomita, K.; Saito, H. Alleviation of cisplatin-induced acute kidney injury using phytochemical polyphenols is accompanied by reduced accumulation of indoxyl sulfate in rats. Clin. Exp. Nephrol. 2011, 15, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Nakamura, M.; Tsutsumi, Y.; Suenaga, A.; Otagiri, M. Pharmacokinetics and tissue distribution of uraemic indoxyl sulphate in rats. Biopharm. Drug Dispos. 2003, 24, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Shibahara, N.; Takagi, S.; Inoue, T.; Katsuoka, Y. AST-120 treatment in pre-dialysis period affects the prognosis in patients on hemodialysis. Ren. Fail. 2008, 30, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Nakai, K.; Fukagawa, M. Role of oxidative stress and indoxyl sulfate in progression of cardiovascular disease in chronic kidney disease. Ther. Apher. Dial. 2011, 15, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-β 1, TIMP-1 and pro-α 1(I) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, S15–S22. [Google Scholar]
- Miyazaki, T.; Ise, M.; Hirata, M.; Endo, K.; Ito, Y.; Seo, H.; Niwa, T. Indoxyl sulfate stimulates renal synthesis of transforming growth factor-β 1 and progression of renal failure. Kidney Int. Suppl. 1997, 63, S211–S214. [Google Scholar] [PubMed]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar] [PubMed]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-κB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Liu, Z. Function of aquaporin 1 and α-epithelial Na+ channel in rat acute lung injury induced by acute ischemic kidney injury. Int. Urol. Nephrol. 2013, 45, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Bolati, D.; Higashiyama, Y.; Nishijima, F.; Shimizu, K.; Niwa, T. Indoxyl sulfate upregulates renal expression of MCP-1 via production of ROS and activation of NF-κB, p53, ERK, and JNK in proximal tubular cells. Life Sci. 2012, 90, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Hai-hua, C.; Xian-long, Z.; Yu-lu, S.; Jiong, Y. Roles of p38 MAPK and JNK in TGF-β1-induced human alveolar epitherial to mesenchymal transition. Arch. Med. Res. 2013, 44, 93–98. [Google Scholar]
- Kreda, S.M.; Gynn, M.C.; Fenstermacher, D.A.; Boucher, R.C.; Gabriel, S.E. Expression and localization of epithelial aquaporins in the adult human lung. Am. J. Respir. Cell Mol. Biol. 2001, 24, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.L.; Hoke, T.S.; Fang, W.F.; Altmann, C.J.; Douglas, I.S.; Faubel, S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int. 2008, 74, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Hoke, T.S.; Douglas, I.S.; Klein, C.L.; He, Z.; Fang, W.; Thurman, J.M.; Tao, Y.; Dursun, B.; Voelkel, N.F.; Edelstein, C.L.; et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J. Am. Soc. Nephrol. 2007, 18, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Andres-Hernando, A.; Altmann, C.; Bhargava, R.; Bacalja, J.; Webb, R.G.; He, Z.; Edelstein, C.L.; Faubel, S. Circulating IL-6 mediates lung injury via CXCL1 production after acute kidney injury in mice. Am. J. Physiol. Ren. Physiol. 2012, 303, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Yoshimura, M.; Saigo, C.; Komori, M.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Wakida, A.; Chuman, E.; Nishi, K.; et al. Hepatic sulfotransferase as a nephropreventing target by suppression of the uremic toxin indoxyl sulfate accumulation in ischemic acute kidney injury. Toxicol. Sci. 2014, 141, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Watanabe, H.; Yoshitome, K.; Morisaki, T.; Hamada, A.; Nonoguchi, H.; Kohda, Y.; Tomita, K.; Inui, K.; Saito, H. Downregulation of organic anion transporters in rat kidney under ischemic/reperfusion-induced acute renal failure. Kidney Int. 2007, 71, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Seidel, D.; Knels, L.; Morishima, N.; Neisser, A.; Bramke, S.; Koslowski, R. Early signs of lung fibrosis after in vitro treatment of rat lung slices with CdCl2 and TGF-β1. Histochem. Cell Biol. 2004, 121, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, Y.; Terada, N.; Saitoh, S.; Ohno, N.; Jin, T.; Ohno, S. Histochemical analyses and quantum dot imaging of microvascular blood flow with pulmonary edema in living mouse lungs by “in vivo cryotechnique”. Histochem. Cell Biol. 2012, 137, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Grams, M.E.; Rabb, H. The distant organ effects of acute kidney injury. Kidney Int. 2012, 81, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, A.; Masutomi, H.; Handa, S.; Maruyama, N. Age-associated decrease of senescence marker protein-30/gluconolactonase in individual mouse liver cells: Immunohistochemistry and immunofluorescence. Geriatr. Gerontol. Int. 2015, 15, 804–810. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yabuuchi, N.; Sagata, M.; Saigo, C.; Yoneda, G.; Yamamoto, Y.; Nomura, Y.; Nishi, K.; Fujino, R.; Jono, H.; Saito, H. Indoxyl Sulfate as a Mediator Involved in Dysregulation of Pulmonary Aquaporin-5 in Acute Lung Injury Caused by Acute Kidney Injury. Int. J. Mol. Sci. 2017, 18, 11. https://doi.org/10.3390/ijms18010011
Yabuuchi N, Sagata M, Saigo C, Yoneda G, Yamamoto Y, Nomura Y, Nishi K, Fujino R, Jono H, Saito H. Indoxyl Sulfate as a Mediator Involved in Dysregulation of Pulmonary Aquaporin-5 in Acute Lung Injury Caused by Acute Kidney Injury. International Journal of Molecular Sciences. 2017; 18(1):11. https://doi.org/10.3390/ijms18010011
Chicago/Turabian StyleYabuuchi, Nozomi, Masataka Sagata, Chika Saigo, Go Yoneda, Yuko Yamamoto, Yui Nomura, Kazuhiko Nishi, Rika Fujino, Hirofumi Jono, and Hideyuki Saito. 2017. "Indoxyl Sulfate as a Mediator Involved in Dysregulation of Pulmonary Aquaporin-5 in Acute Lung Injury Caused by Acute Kidney Injury" International Journal of Molecular Sciences 18, no. 1: 11. https://doi.org/10.3390/ijms18010011