
Roles of Voltage-Gated Tetrodotoxin-Sensitive Sodium Channels NaV1.3 and NaV1.7 in Diabetes and Painful Diabetic Neuropathy


{kind=link}
Abstract
:1. Introduction
2. Main Cells Involved in Diabetes and Painful Diabetic Neuropathy
3. Review of Voltage-Gated Sodium Channels
4. Roles of TTX-S NaV1.3 and NaV1.7 Channels in Painful Diabetic Neuropathy
5. NaV1.3
6. NaV1.7
7. Roles of TTX-S NaV1.3 and NaV1.7 Channels in Diabetes
8. Pancreatic α Cells
9. Pancreatic β Cells
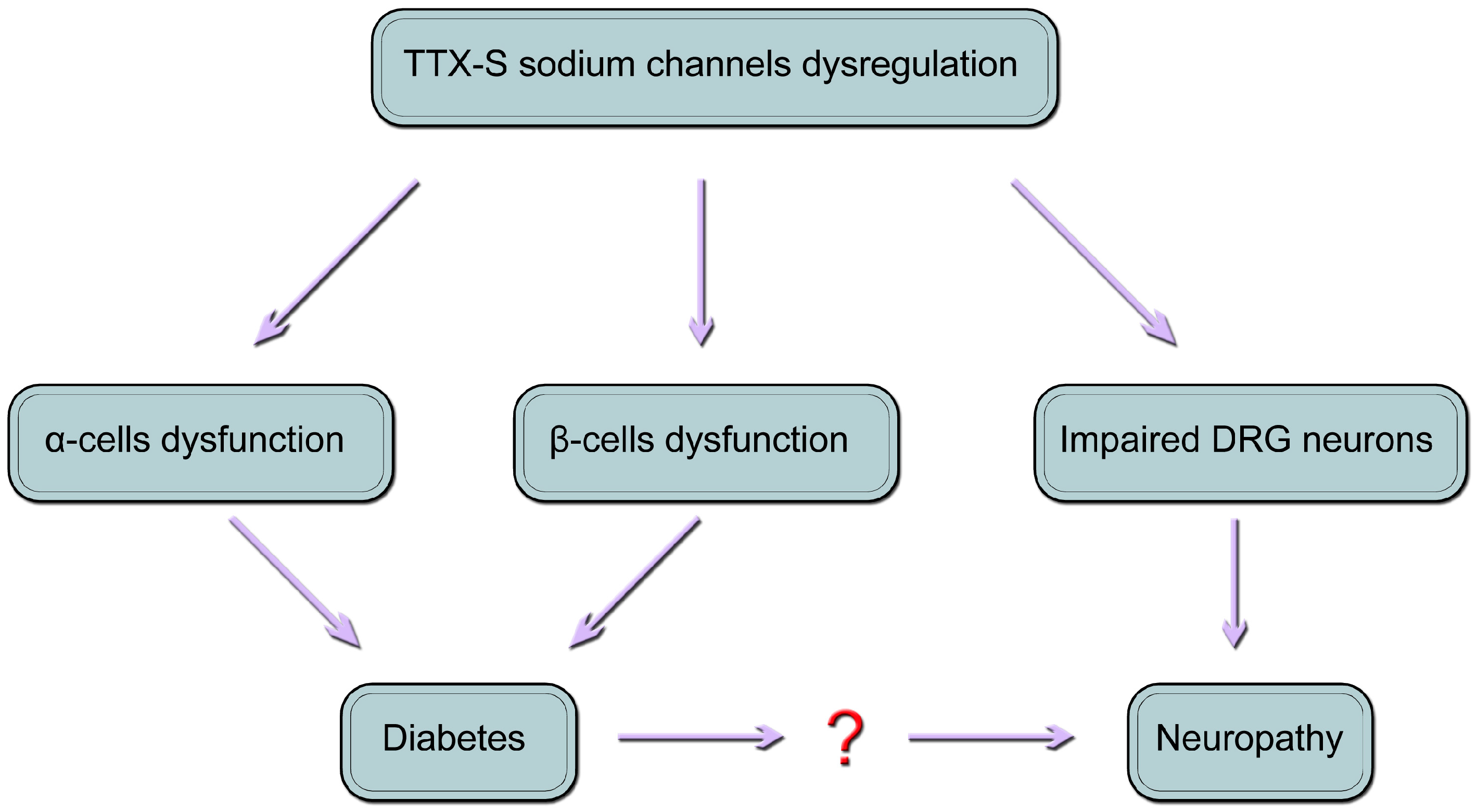
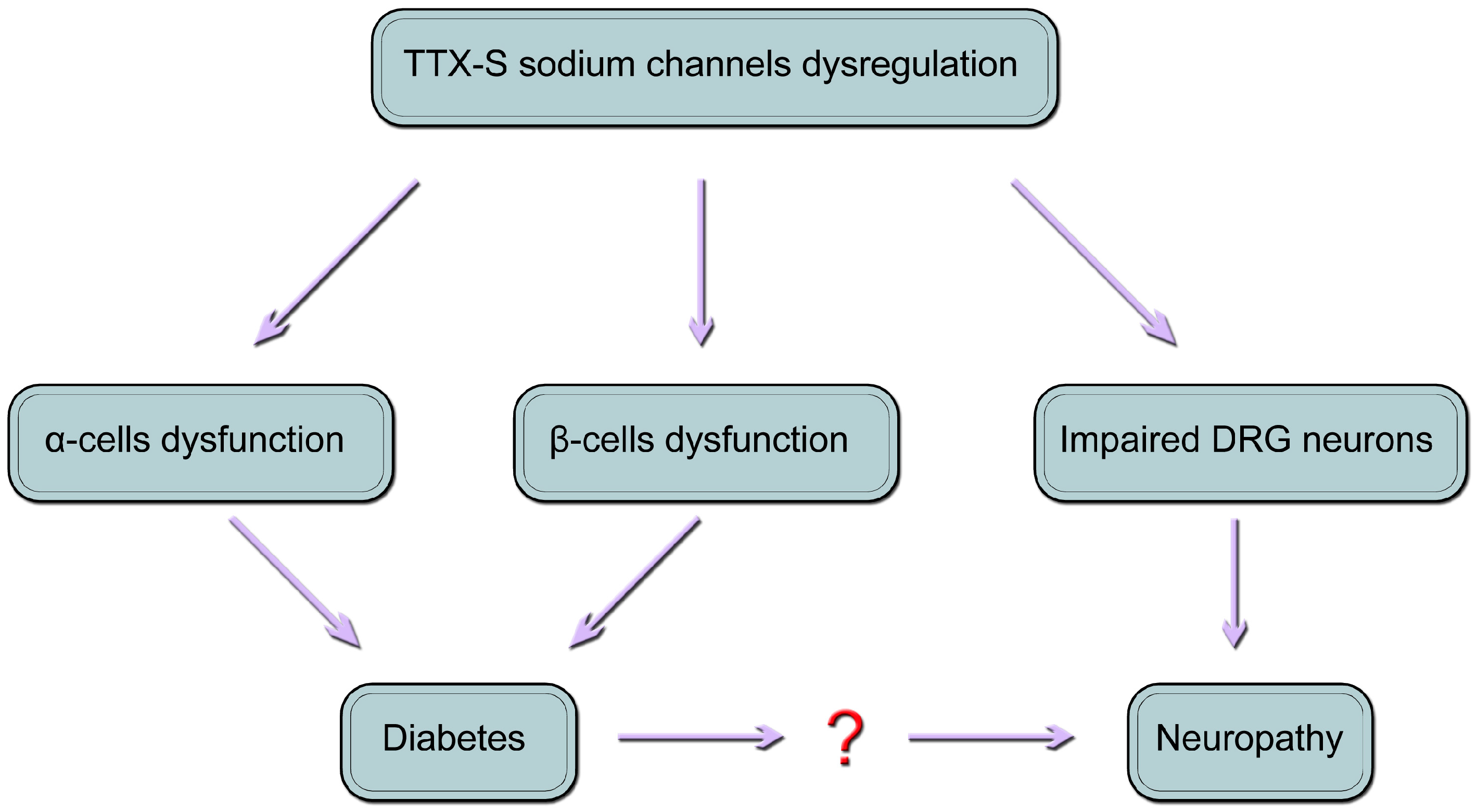
10. Conclusions
Author Contributions
Conflicts of Interest
References
- Guariguata, L.; Whiting, D.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 2014, 103, 137–149. [Google Scholar] [CrossRef] [PubMed]
- IDF Diabetes Atlas Group. Update of mortality attributable to diabetes for the IDF Diabetes Atlas: Estimates for the year 2013. Diabetes Res. Clin. Pract. 2015, 109, 461–465. [Google Scholar]
- Abbott, C.A.; Malik, R.A.; van Ross, E.R.; Kulkarni, J.; Boulton, A.J. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the UK. Diabetes Care 2011, 34, 2220–2224. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Brophy, S.; Williams, R.; Taylor, A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care 2006, 29, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Spallone, V.; Greco, C. Painful and painless diabetic neuropathy: One disease or two? Curr. Diabetes Rep. 2013, 13, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, K.; Bouhassira, D.; de Bacquer, D.; Weiss, S.; Matthys, K.; Raemen, H.; Mathieu, C.; Colin, I.M. Prevalence and impact on quality of life of peripheral neuropathy with or without neuropathic pain in type 1 and type 2 diabetic patients attending hospital outpatients clinics. Diabetes Metab. 2009, 35, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Asbury, A.K. Diabetic neuropathy. Ann. Neurol. 1984, 15, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Gooch, C.; Podwall, D. The diabetic neuropathies. Neurologist 2004, 10, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Pabbidi, R.M.; Cao, D.-S.; Parihar, A.; Pauza, M.E.; Premkumar, L.S. Direct role of streptozotocin in inducing thermal hyperalgesia by enhanced expression of transient receptor potential vanilloid 1 in sensory neurons. Mol. Pharmacol. 2008, 73, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Cappelleri, J.C.; Joshi, A.V. Association between pain severity and health care resource use, health status, productivity and related costs in painful diabetic peripheral neuropathy patients. Pain Med. 2011, 12, 799–807. [Google Scholar]
- Ritzwoller, D.P.; Ellis, J.L.; Korner, E.J.; Hartsfield, C.L.; Sadosky, A. Comorbidities, healthcare service utilization and costs for patients identified with painful DPN in a managed-care setting. Curr. Med. Res. Opin. 2009, 25, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. KATP channels and islet hormone secretion: New insights and controversies. Nat. Rev. Endocrinol. 2013, 9, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Eliasson, L.; Kanno, T.; Zhang, Q.; Gopel, S. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog. Biophys. Mol. Biol. 2011, 107, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Grabauskas, G.; Heldsinger, A.; Wu, X.; Xu, D.; Zhou, S.; Owyang, C. Diabetic visceral hypersensitivity is associated with activation of mitogen-activated kinase in rat dorsal root ganglia. Diabetes 2011, 60, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Said, G. Diabetic neuropathy: An update. J. Neurol. 1996, 243, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. Voltage-Gated Sodium Channels: Therapeutic Targets for Pain. Pain Med. 2009, 10, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L. Diversity of Mammalian Voltage-Gated Sodium Channels. Ann. N. Y. Acad. Sci. 1999, 868, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G. Painful Na-channelopathies: An expanding universe. Trends Mol. Med. 2013, 19, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G. Neuroscience: Channelopathies have many faces. Nature 2011, 472, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Wilson, M.J.; Gajewiak, J.; Rivier, J.E.; Bulaj, G.; Olivera, B.M.; Yoshikami, D. Pharmacological fractionation of tetrodotoxin-sensitive sodium currents in rat dorsal root ganglion neurons by μ-conotoxins. Br. J. Pharmacol. 2013, 169, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Kocsis, J.D.; Waxman, S.G. Slow sodium conductances of dorsal root ganglion neurons: Intraneuronal homogeneity and interneuronal heterogeneity. J. Neurophysiol. 1995, 72, 2796–2815. [Google Scholar]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G.; Cummins, T.R.; Dib-Hajj, S.D.; Black, J.A. Voltage-gated sodium channels and the molecular pathogenesis of pain: A review. J. Rehabil. Res. Dev. 2000, 37, 517–528. [Google Scholar] [PubMed]
- Cummins, T.R.; Sheets, P.L.; Waxman, S.G. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain 2007, 131, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G.; Kocsis, J.D.; Black, J.A. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is re-expressed following axotomy. J. Neurophysiol. 1994, 72, 466–470. [Google Scholar] [PubMed]
- Black, J.A.; Liu, S.; Tanaka, M.; Cummins, T.R.; Waxman, S.G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 2004, 108, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Hains, B.C.; Klein, J.P.; Saab, C.Y.; Craner, M.J.; Black, J.A.; Waxman, S.G. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 2003, 23, 8881–8892. [Google Scholar] [PubMed]
- Black, J.A.; Cummins, T.R.; Plumpton, C.; Chen, Y.H.; Hormuzdiar, W.; Clare, J.J.; Waxman, S.G. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J. Neurophysiol. 1999, 82, 2776–2785. [Google Scholar] [PubMed]
- Yin, R.; Liu, D.; Chhoa, M.; Li, C.M.; Luo, Y.; Zhang, M.; Lehto, S.G.; Immke, D.C.; Moyer, B.D. Voltage-gated sodium channel function and expression in injured and uninjured rat dorsal root ganglia neurons. Int. J. Neurosci. 2016, 126, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Waxman, S.G. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 1997, 17, 3503–3514. [Google Scholar] [PubMed]
- Lai, J.; Gold, M.S.; Kim, C.S.; Di, B.A.; Ossipov, M.H.; Porreca, F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain 2002, 95, 143–152. [Google Scholar] [CrossRef]
- Lindia, J.A.; Köhler, M.G.; Martin, W.J.; Abbadie, C. Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain 2005, 117, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Sleeper, A.A.; Cummins, T.R.; Dib-Hajj, S.D.; Hormuzdiar, W.; Tyrrell, L.; Waxman, S.G.; Black, J.A. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J. Neurosci. 2000, 20, 7279–7289. [Google Scholar] [PubMed]
- Waxman, S.G.; Hains, B.C. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci. 2006, 29, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Aglieco, F.; Renganathan, M.; Herzog, R.I.; Dib-Hajj, S.D.; Waxman, S.G. Nav1.3 sodium channels: Rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 2001, 21, 5952–5961. [Google Scholar] [PubMed]
- Craner, M.J.; Klein, J.P.; Renganathan, M.; Black, J.A.; Waxman, S.G. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann. Neurol. 2002, 52, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.S.; Gonzalez, M.I.; Bramwell, S.; Pinnock, R.D.; Lee, K.; Dixon, A.K. β3, a novel auxiliary subunit for the voltage gated sodium channel is upregulated in sensory neurones following streptozocin induced diabetic neuropathy in rat. Neurosci. Lett. 2001, 309, 1–4. [Google Scholar] [CrossRef]
- Cheng, K.I.; Wang, H.C.; Chuang, Y.T.; Chou, C.W.; Tu, H.P.; Yu, Y.C.; Chang, L.L.; Lai, C.S. Persistent mechanical allodynia positively correlates with an increase in activated microglia and increased P-p38 mitogen-activated protein kinase activation in streptozotocin-induced diabetic rats. Eur. J. Pain 2014, 18, 162–173. [Google Scholar] [CrossRef] [PubMed]
- He, X.-H.; Zang, Y.; Chen, X.; Pang, R.-P.; Xu, J.-T.; Zhou, X.; Wei, X.H.; Li, Y.Y.; Xin, W.J.; Qin, Z.H.; et al. TNF-α contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain 2010, 151, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.M.; Cummins, T.R.; Waxman, S.G. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 2007, 579, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Samad, O.A.; Tan, A.M.; Cheng, X.; Foster, E.; Dib-Hajj, S.D.; Waxman, S.G. Virus-mediated shRNA knockdown of Nav1.3 in rat dorsal root ganglion attenuates nerve injury-induced neuropathic pain. Mol. Ther. 2013, 21, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.M.; Samad, O.A.; Dib-Hajj, S.D.; Waxman, S.G. Virus-Mediated Knockdown of Nav1.3 in Dorsal Root Ganglia of STZ-Induced Diabetic Rats Alleviates Tactile Allodynia. Mol. Med. 2015, 21, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Langworthy, K.; Hinson, A.W.; Dib-Hajj, S.D.; Waxman, S.G. NGF has opposing effects on Na+ channel III and SNS gene expression in spinal sensory neurons. Neuroreport 1997, 8, 2331–2335. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Howe, J.R.; Waxman, S.G. Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 1998, 18, 9607–9619. [Google Scholar] [PubMed]
- Waxman, S.G. Neurobiology: A channel sets the gain on pain. Nature 2006, 444, 831–832. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Morrow, T.J.; Paulson, P.E.; Isom, L.L.; Wiley, J.W. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and-resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 2004, 279, 29341–29350. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zang, Y.; Zhou, L.; Gui, W.; Liu, X.; Zhong, Y. The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem. Int. 2014, 75, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Zhou, Z.; Hao, S.; Mata, M.; Fink, D.J. Reduction of voltage gated sodium channel protein in DRG by vector mediated miRNA reduces pain in rats with painful diabetic neuropathy. Mol. Pain 2012, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Mata, M.; Fink, D.J. Vector-mediated release of GABA attenuates pain-related behaviors and reduces NaV1.7 in DRG neurons. Eur. J. Pain 2011, 15, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, M.; Mata, M.; Fink, D.J. Continuous δ-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy. J. Neurosci. 2008, 28, 6652–6658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Yang, J.P.; Zhang, J.-R.; Li, R.Q.; Wang, J.; Jan, J.J.; Zhuang, Q. Gabapentin reduces allodynia and hyperalgesia in painful diabetic neuropathy rats by decreasing expression level of Nav1.7 and p-ERK1/2 in DRG neurons. Brain Res. 2013, 1493, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Renganathan, M.; Cummins, T.R.; Waxman, S.G. Contribution of Nav1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 2001, 86, 629–640. [Google Scholar] [PubMed]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnölzer, M.; Lasitschka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.T.; Huang, P.Y.; Yen, C.T.; Chen, C.C.; Lee, M.J. A Novel SCN9A Mutation Responsible for Primary Erythromelalgia and Is Resistant to the Treatment of Sodium Channel Blockers. PLoS ONE 2013, 8, e55212. [Google Scholar] [CrossRef] [PubMed]
- Fertleman, C.; Ferrie, C. What’s in a name—Familial rectal pain syndrome becomes paroxysmal extreme pain disorder. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1294–1295. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Hoeijmakers, J.G.; Ahn, H.S.; Cheng, X.; Han, C.; Choi, J.S.; Estacion, M.; Lauria, G.; Vanhoutte, E.K.; Gerrits, M.M.; et al. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 2012, 71, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Sheynin, J.; Shorer, Z.; Reimann, F.; Nicholas, A.K.; Zubovic, L.; Baralle, M.; Wraige, E.; Manor, E.; Levy, J.; et al. Congenital insensitivity to pain: Novel SCN9A missense and in-frame deletion mutations. Hum. Mutat. 2010, 31, E1670–E1686. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.G.; Faber, C.G.; Merkies, I.S.; Waxman, S.G. Channelopathies, painful neuropathy, and diabetes: Which way does the causal arrow point? Trends Mol. Med. 2014, 20, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.K.; Nones, C.F.; Reis, R.C.; Chichorro, J.G.; Cunha, J.M. Diabetic neuropathic pain: Physiopathology and treatment. World J. Diabetes 2015, 6, 432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chibalina, M.V.; Bengtsson, M.; Groschner, L.N.; Ramracheya, R.; Rorsman, N.J.; Leiss, V.; Nassar, M.A.; Welling, A.; Gribble, F.M.; et al. Na+ current properties in islet α-and β-cells reflect cell-specific Scn3a and Scn9a expression. J. Physiol. 2014, 592, 4677–4696. [Google Scholar] [CrossRef] [PubMed]
- Vignali, S.; Leiss, V.; Karl, R.; Hofmann, F.; Welling, A. Characterization of voltage-dependent sodium and calcium channels in mouse pancreatic A-and B-cells. J. Physiol. 2006, 572, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Göpel, S.; Kanno, T.; Barg, S.; Weng, X.G.; Gromada, J.; Rorsman, P. Regulation of glucagon release in mouse α-cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J. Physiol. 2000, 528, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Ramracheya, R.; Ward, C.; Shigeto, M.; Walker, J.N.; Amisten, S.; Zhang, Q.; Johnson, P.R.; Rorsman, P.; Braun, M. Membrane potential-dependent inactivation of voltage-gated ion channels in α-cells inhibits glucagon secretion from human islets. Diabetes 2010, 59, 2198–2208. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Rupnik, M.S.; Karimian, N.; Herrera, P.L.; Gilon, P.; Feng, Z.P.; Gaisano, H.Y. In situ electrophysiological examination of pancreatic α cells in the streptozotocin-induced diabetes model, revealing the cellular basis of glucagon hypersecretion. Diabetes 2013, 62, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, A.K.; Yang, M.; Ning, Y.; Kahlig, K.M.; Krause, M.; Rajamani, S.; Belardinelli, L. Blockade of Na+ channels in pancreatic α-cells has antidiabetic effects. Diabetes 2014, 63, 3545–3556. [Google Scholar] [CrossRef] [PubMed]
- Dusaulcy, R.; Handgraaf, S.; Heddad-Masson, M.; Visentin, F.; Vesin, C.; Reimann, F.; Gribble, F.; Philippe, J.; Gosmain, Y. α-Cell Dysfunctions and Molecular Alterations in Male Insulinopenic Diabetic Mice Are Not Completely Corrected by Insulin. Endocrinology 2016, 157, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic β-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.E. Signal integration at the level of ion channel and exocytotic function in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1065–E1069. [Google Scholar] [CrossRef] [PubMed]
- Nita, I.I.; Hershfinkel, M.; Kantor, C.; Rutter, G.A.; Lewis, E.C.; Sekler, I. Pancreatic β-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J. 2014, 28, 3301–3312. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.C.; Vilin, Y.Y.; Roberge, M.; Kurata, H.T.; Johnson, J.D. Multiparameter Screening Reveals a Role for Na+ Channels in Cytokine-Induced β-Cell Death. Mol. Endocrinol. 2014, 28, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, V.; Esguerra, J.; Ofori, J.; Mollet, I.; Braun, M.; Stoffel, M.; Wendt, A.; Eliasson, L. Modulation of microRNA-375 expression alters voltage-gated Na+ channel properties and exocytosis in insulin-secreting cells. Acta Physiol. 2015, 213, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Szabat, M.; Modi, H.; Ramracheya, R.; Girbinger, V.; Chan, F.; Lee, J.T.; Piske, M.; Kamal, S.; Carol Yang, Y.H.; Welling, A. High-content screening identifies a role for Na+ channels in insulin production. R. Soc. Open Sci. 2015, 2, 150306. [Google Scholar] [CrossRef] [PubMed]
- Ernst, S.J.; Aguilar-Bryan, L.; Noebels, J.L. Sodium channel β1 regulatory subunit deficiency reduces pancreatic islet glucose-stimulated insulin and glucagon secretion. Endocrinology 2009, 150, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Hoeijmakers, J.G.; Liu, S.; Gerrits, M.M.; te Morsche, R.H.; Lauria, G.; Dib-Hajj, S.D.; Drenth, J.P.; Faber, C.G.; Merkies, I.S.; et al. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain 2012, 135, 2613–2628. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Li, Q.; Liu, X.; Liu, S. Roles of Voltage-Gated Tetrodotoxin-Sensitive Sodium Channels NaV1.3 and NaV1.7 in Diabetes and Painful Diabetic Neuropathy. Int. J. Mol. Sci. 2016, 17, 1479. https://doi.org/10.3390/ijms17091479
Yang L, Li Q, Liu X, Liu S. Roles of Voltage-Gated Tetrodotoxin-Sensitive Sodium Channels NaV1.3 and NaV1.7 in Diabetes and Painful Diabetic Neuropathy. International Journal of Molecular Sciences. 2016; 17(9):1479. https://doi.org/10.3390/ijms17091479
Chicago/Turabian StyleYang, Linlin, Quanmin Li, Xinming Liu, and Shiguang Liu. 2016. "Roles of Voltage-Gated Tetrodotoxin-Sensitive Sodium Channels NaV1.3 and NaV1.7 in Diabetes and Painful Diabetic Neuropathy" International Journal of Molecular Sciences 17, no. 9: 1479. https://doi.org/10.3390/ijms17091479