
A Genome-Wide Methylation Approach Identifies a New Hypermethylated Gene Panel in Ulcerative Colitis


Abstract
:
1. Introduction
2. Results
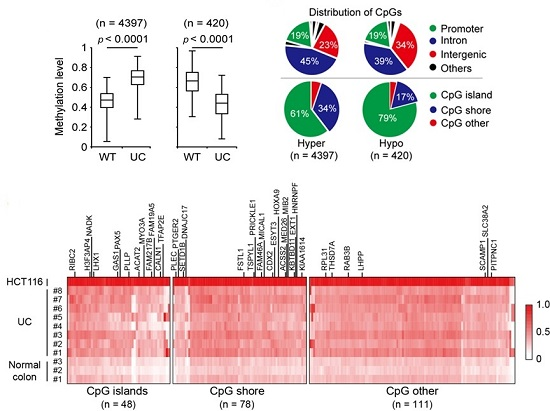
2.1. Genome-Wide DNA Methylation Changes in UC Patients
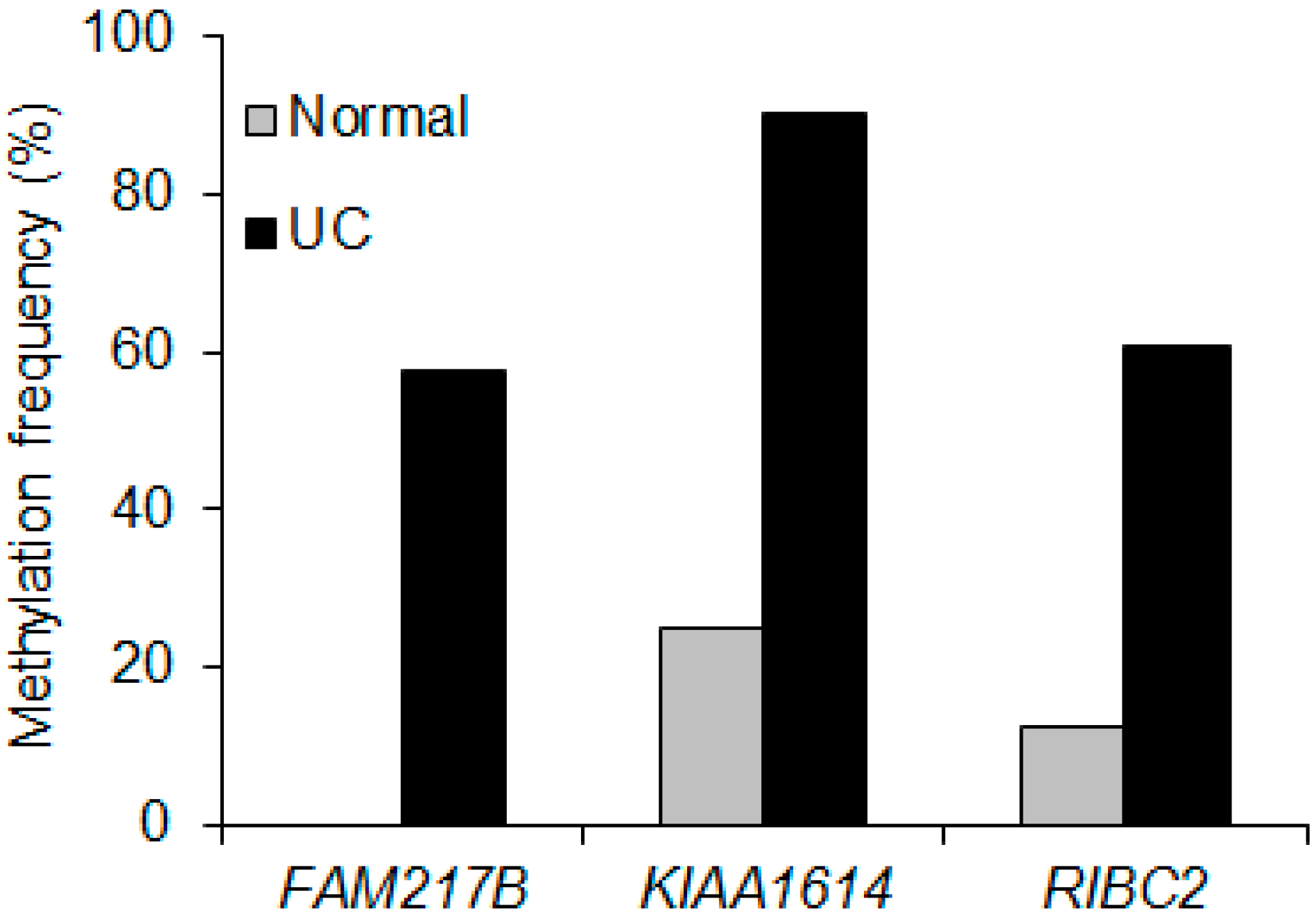
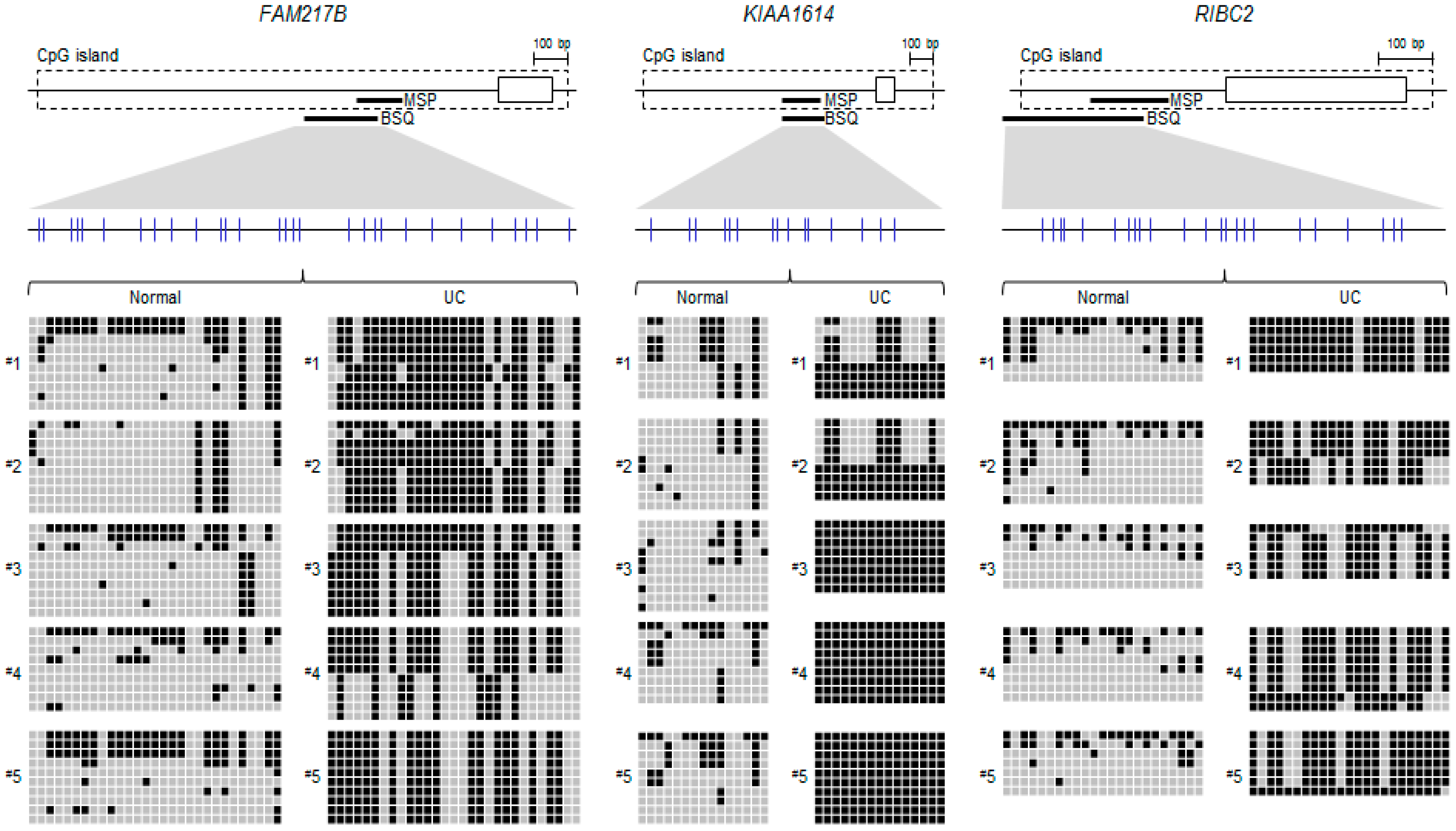
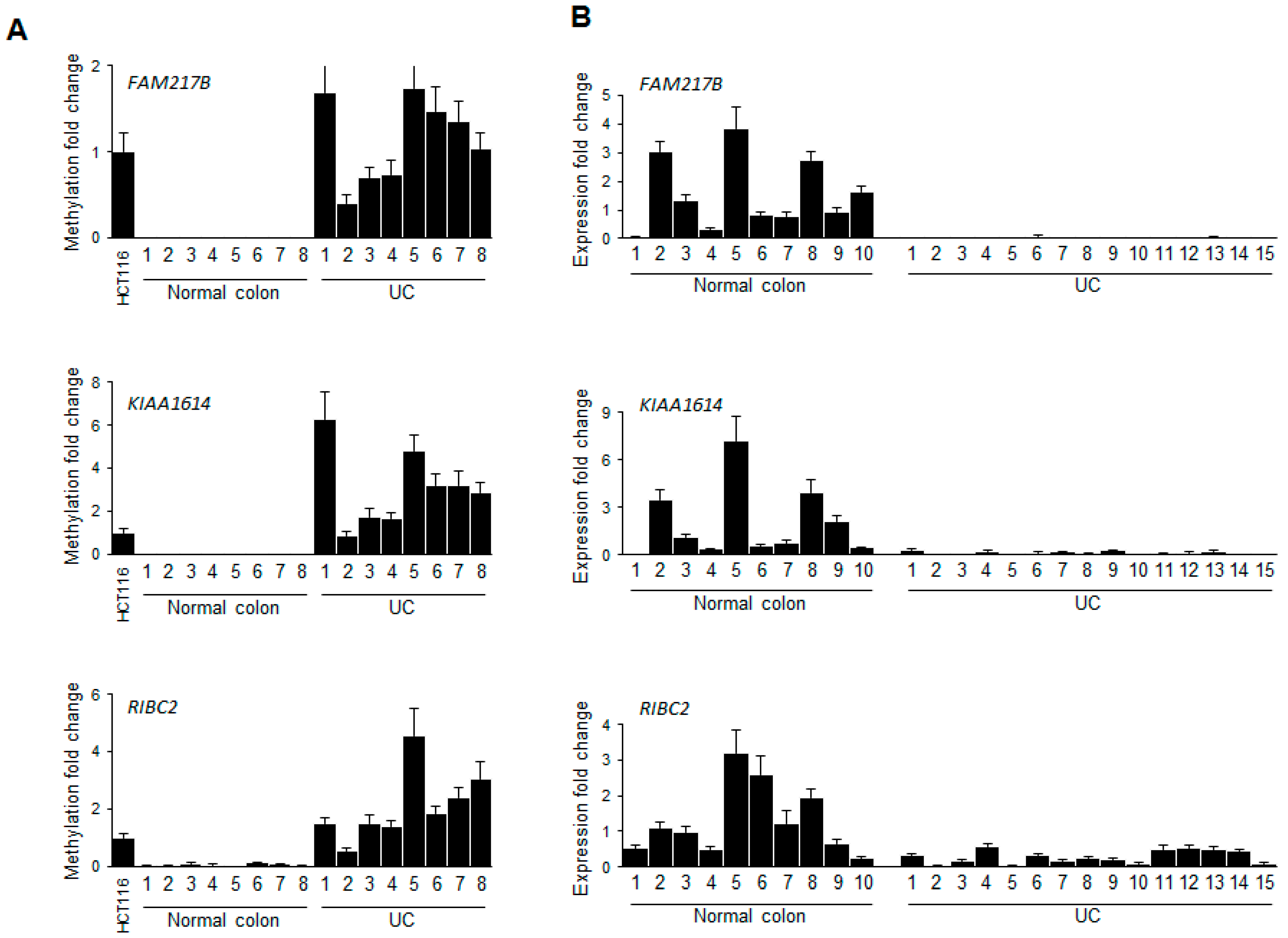
2.2. Validation of Selected Candidate Genes in UC Patient Samples
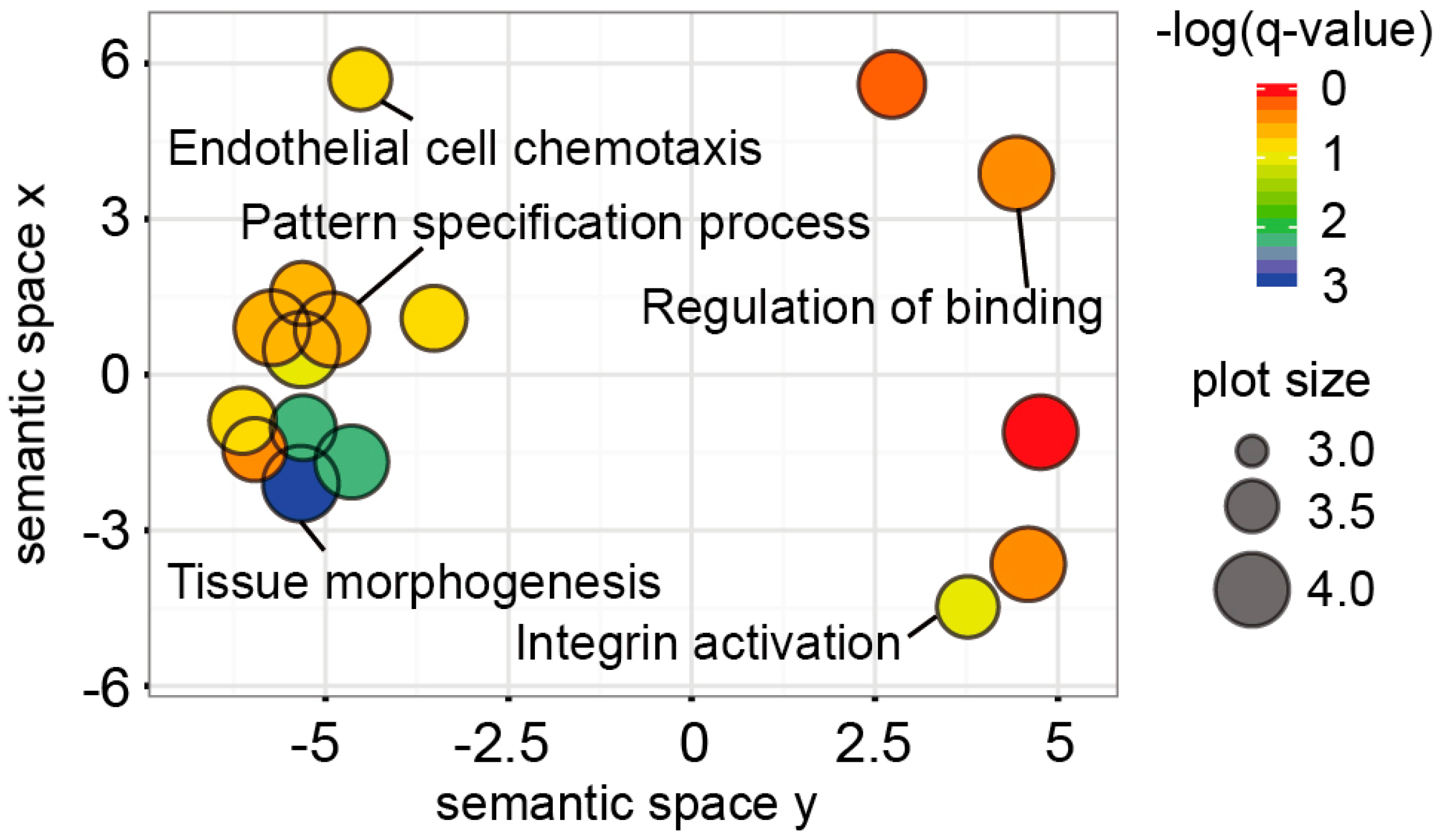
2.3. Functional Implications of the UC Hypermethylated Gene Panel
3. Discussion
4. Materials and Methods
4.1. Tissue Samples
4.2. Ethic Statement
4.3. Genome-Wide DNA Methylation Analysis
4.4. DNA Methylation Analysis
4.5. Quantitative Methylation-Specific PCR (qMSP)
4.6. Quantitative Real-Time RT-PCR (qRT-PCR)
4.7. Computational Analysis
4.8. Statistical Analysis Nel
4.9. Availability of Data and Materials
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Fiocchi, C. Inflammatory bowel disease: Etiology and pathogenesis. Gastroenterology 1998, 115, 182–205. [Google Scholar] [CrossRef]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Hedderich, J.; May, S.; Lu, T.; Schuldt, D.; Nikolaus, S.; Rosenstiel, P.; Krawczak, M.; et al. Replication of signals from recent studies of crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat. Genet. 2008, 40, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Philip Schumm, L.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M. High density DNA methylation array with single cpg site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Karatzas, P.S.; Gazouli, M.; Safioleas, M.; Mantzaris, G.J. DNA methylation changes in inflammatory bowel disease. Ann. Gastroenterol.: Quart. Pub. Hellenic Soc. Gastroenterol. 2014, 27, 125–132. [Google Scholar]
- Kim, J.-G.; Park, M.-T.; Heo, K.; Yang, K.-M.; Yi, J.M. Epigenetics meets radiation biology as a new approach in cancer treatment. Int. J. Mol. Sci. 2013, 14, 15059–15073. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-O.; Park, J.; Kang, M.J.; Lee, S.H.; Jee, S.R.; Rye, D.Y.; Yang, K.; Yi, J.M. DNA hypermethylation of a selective gene panel as a risk marker for colon cancer in patients with ulcerative colitis. Int. J. Mol. Sci. 2013, 31, 1255–1261. [Google Scholar]
- Yi, J.M.; Dhir, M.; van Neste, L.; Downing, S.R.; Jeschke, J.; Glöckner, S.C.; de Freitas Calmon, M.; Hooker, C.M.; Funes, J.M.; Boshoff, C.; et al. Genomic and epigenomic integration identifies a prognostic signature in colon cancer. Clin. Cancer Res. 2011, 17, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Petronis, A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010, 465, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Stoll, M.; Corneliussen, B.; Costello, C.M.; Waetzig, G.H.; Mellgard, B.; Koch, W.A.; Rosenstiel, P.; Albrecht, M.; Croucher, P.J.P.; Seegert, D.; et al. Genetic variation in dlg5 is associated with inflammatory bowel disease. Nat. Genet. 2004, 36, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of d4z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Ventham, N.T.; Kennedy, N.A.; Nimmo, E.R.; Satsangi, J. Beyond gene discovery in inflammatory bowel disease: The emerging role of epigenetics. Gastroenterology 2013, 145, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, L.; Egan, L.J. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis 2012, 33, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.J.; Klump, B.; Holzmann, K.; Borchard, F.; Gregor, M.; Porschen, R. Hypermethylation of the p16ink4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998, 58, 3942–3945. [Google Scholar] [PubMed]
- Wang, F.; Arisawa, T.; Tahara, T.; Takahama, K.; Watanabe, M.; Hirata, I.; Nakano, H. Aberrant DNA methylation in ulcerative colitis without neoplasia. Hepatogastroenterology 2008, 55, 62–65. [Google Scholar] [PubMed]
- Gould, N.J.D.; Davidson, K.L.M.; Nwokolo, C.U.; Arasaradnam, R.P. A systematic review of the role of DNA methylation on inflammatory genes in ulcerative colitis. Epigenomics 2016, 8, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Fraga, M.F.; Paz, M.F.; Campo, E.; Colomer, D.; Novo, F.J.; Calasanz, M.J.; Galm, O.; Guo, M.; Benitez, J.; et al. Cancer epigenetics and methylation. Science 2002, 297, 1807–1808. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Karatzas, P.S.; Mantzaris, G.J.; Safioleas, M.; Gazouli, M. DNA methylation profile of genes involved in inflammation and autoimmunity in inflammatory bowel disease. Medicine 2014, 93, e309. [Google Scholar] [CrossRef] [PubMed]
- Li Yim, A.Y.F.; Duijvis, N.W.; Zhao, J.; de Jonge, W.J.; D’Haens, G.R.A.M.; Mannens, M.M.A.M.; Mul, A.N.P.M.; te Velde, A.A.; Henneman, P. Peripheral blood methylation profiling of female crohn’s disease patients. Clin. Epigenet. 2016, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Taylor, W.R.; Aboelsoud, M.M.; Foote, P.H.; Yab, T.C.; Cao, X.; Smyrk, T.C.; Loftus, E.V., Jr.; Mahoney, D.W.; Ahlquist, D.A.; et al. DNA methylation and mutation of small colonic neoplasms in ulcerative colitis and crohn’s colitis: Implications for surveillance. Inflamm. Bowel Dis. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Papadia, C.; Louwagie, J.; del Rio, P.; Grooteclaes, M.; Coruzzi, A.; Montana, C.; Novelli, M.; Bordi, C.; de’ Angelis, G.L.; Bassett, P.; et al. Foxe1 and syne1 genes hypermethylation panel as promising biomarker in colitis-associated colorectal neoplasia. Inflamm. Bowel Dis. 2014, 20, 271–277. [Google Scholar] [CrossRef] [PubMed]
- DeVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grützmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating methylated sept9 DNA in plasma is a biomarker for colorectal cancer. Clin. Chem. 2009, 55, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Glockner, S.C.; Dhir, M.; Yi, J.M.; McGarvey, K.E.; van Neste, L.; Louwagie, J.; Chan, T.A.; Kleeberger, W.; de Bruïne, A.P.; Smits, K.M.; et al. Methylation of tfpi2 in stool DNA: A potential novel biomarker for the detection of colorectal cancer. Cancer Res. 2009, 69, 4691–4699. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, W.-D.; Papadopoulos, N.; Goodman, S.N.; Bjerregaard, N.C.; Laurberg, S.; Levin, B.; Juhl, H.; Arber, N.; Moinova, H.; et al. Sensitive digital quantification of DNA methylation in clinical samples. Nat. Biotechnol. 2009, 27, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Azarschab, P.; Porschen, R.; Gregor, M.; Blin, N.; Holzmann, K. Epigenetic control of the e-cadherin gene (CDH1) by CpG methylation in colectomy samples of patients with ulcerative colitis. Genes Chromosome Cancer 2002, 35, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Kato, J.; Hiraoka, S.; Horii, J.; Suzuki, H.; Higashi, R.; Kaji, E.; Kondo, Y.; Yamamoto, K. DNA methylation of colon mucosa in ulcerative colitis patients: Correlation with inflammatory status. Inflamm. Bowel Dis. 2011, 17, 1955–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Kibbe, W.A.; Lin, S.M. Lumi: A pipeline for processing illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Davis, S.; Bilke, T.; Triche, J.T.; Bootawalla, M. Methylumi: Handle illumina methylation data. Available online: https://www.scienceopen.com/document?vid=4d66aeae-d817-400f-a84c-0b0df0066f3e (accessed on 30 June 2015).
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in illumina infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-G.; Kim, T.-O.; Bae, J.-H.; Shim, J.-W.; Kang, M.J.; Yang, K.; Ting, A.H.; Yi, J.M. Epigenetically regulated mir941 and mir1247 target gastric cancer cell growth and migration. Epigenetics 2014, 9, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The genemania prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acid. Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number (Mean) |
|---|---|
| Total no. of patients | 79 |
| Age (years) median (range) | 42.4 (16–68) |
| Gender, n (%) | |
| Male | 48 (60.8) |
| Female | 31 (39.2) |
| Duration of disease | |
| ≤1 year | 41 (51.9) |
| 1–8 year | 24 (30.4) |
| >9 year | 14 (17.7) |
| Lesion location, n (%) | |
| Proctitis | 44 (55.7) |
| Left sided colitis | 25 (31.6) |
| Pancolitis | 10 (12.7) |
| Mayo endoscopic score, n (%) | |
| Normal or inactive | 3 (3.8) |
| Mild disease | 23 (29.1) |
| Moderate disease | 44 (55.7) |
| Severe disease | 9 (11.4) |
| Clinical type, n (%) | |
| Only one episode | 39 (49.4) |
| Chronic relapsing | 35 (44.3) |
| Chronic continuous | 5 (6.3) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, K.; Bae, J.-H.; Han, K.; Kim, E.S.; Kim, T.-O.; Yi, J.M. A Genome-Wide Methylation Approach Identifies a New Hypermethylated Gene Panel in Ulcerative Colitis. Int. J. Mol. Sci. 2016, 17, 1291. https://doi.org/10.3390/ijms17081291
Kang K, Bae J-H, Han K, Kim ES, Kim T-O, Yi JM. A Genome-Wide Methylation Approach Identifies a New Hypermethylated Gene Panel in Ulcerative Colitis. International Journal of Molecular Sciences. 2016; 17(8):1291. https://doi.org/10.3390/ijms17081291
Chicago/Turabian StyleKang, Keunsoo, Jin-Han Bae, Kyudong Han, Eun Soo Kim, Tae-Oh Kim, and Joo Mi Yi. 2016. "A Genome-Wide Methylation Approach Identifies a New Hypermethylated Gene Panel in Ulcerative Colitis" International Journal of Molecular Sciences 17, no. 8: 1291. https://doi.org/10.3390/ijms17081291