
How the Proximal Pocket May Influence the Enantiospecificities of Chloroperoxidase-Catalyzed Epoxidations of Olefins

Abstract
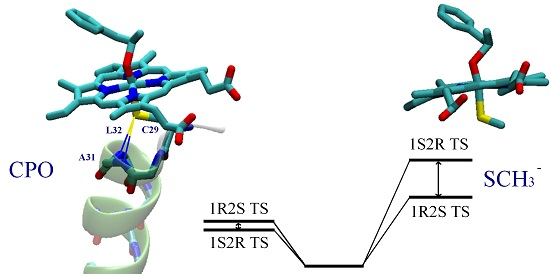
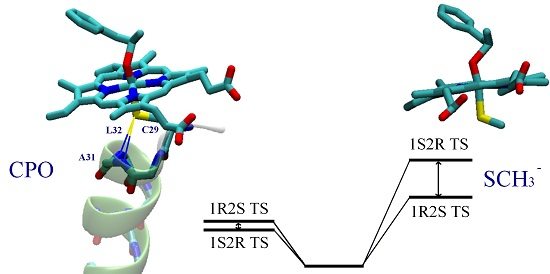
:
1. Introduction
2. Computational Details
2.1. Methods
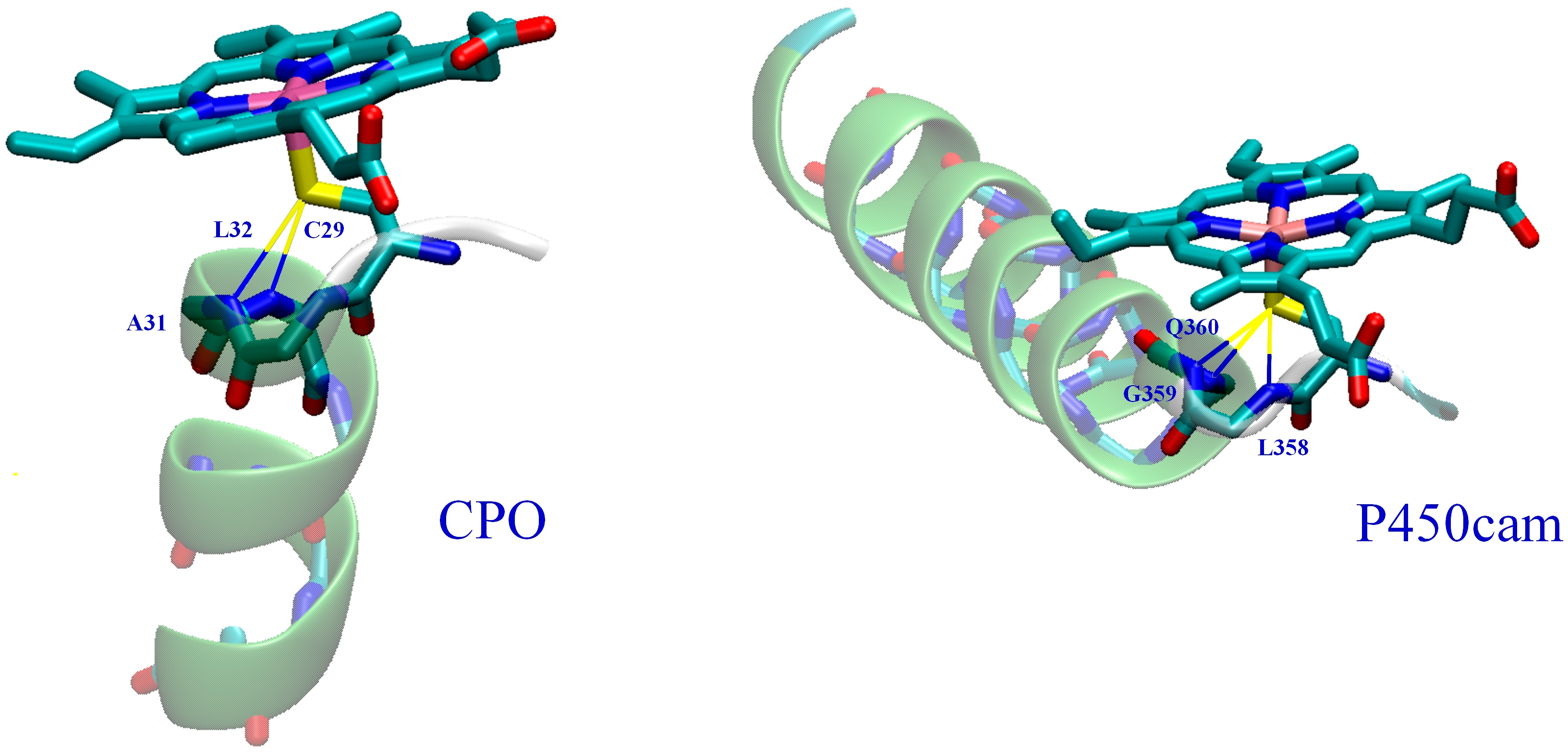
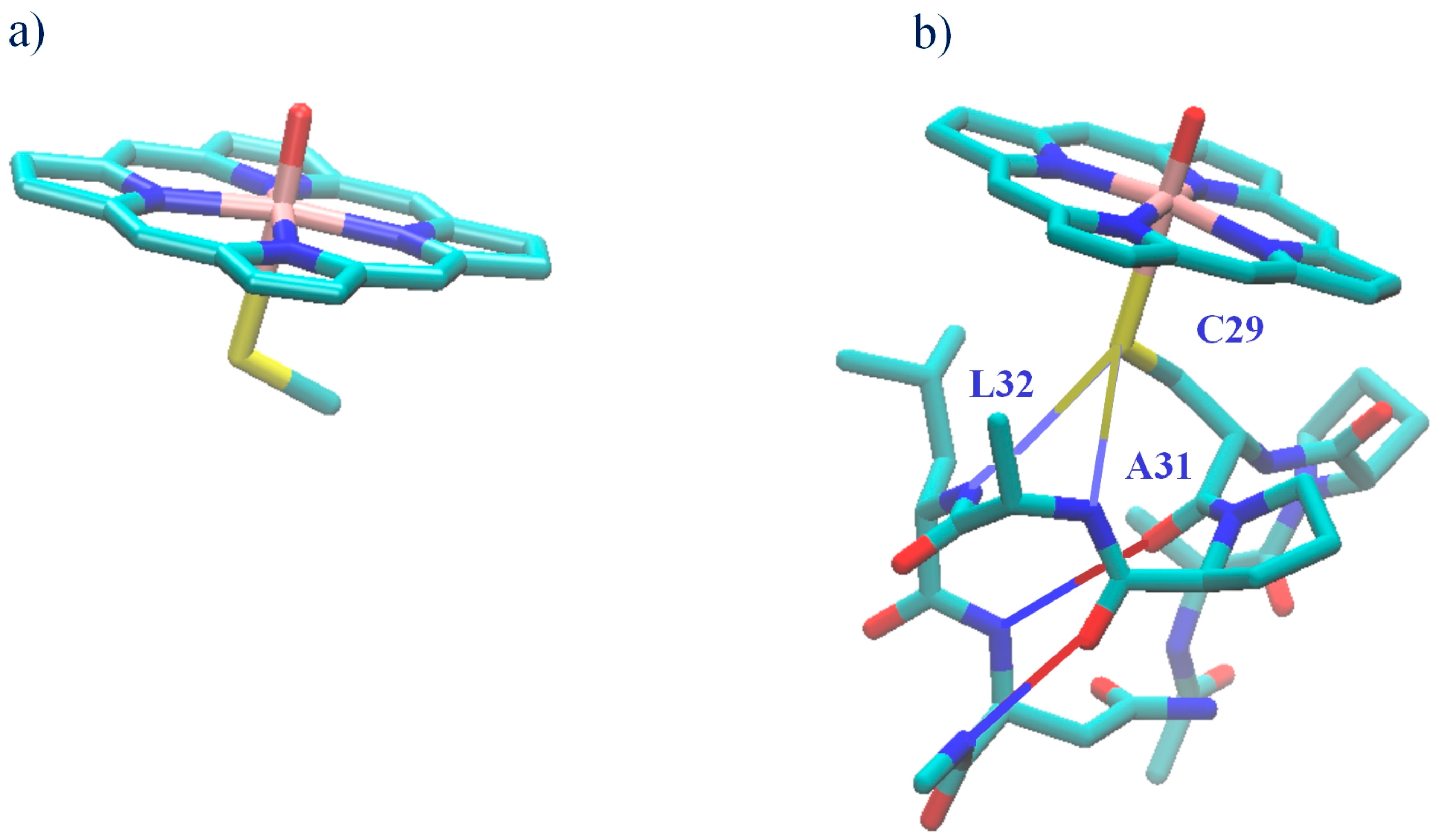
2.2. CPO-I Models
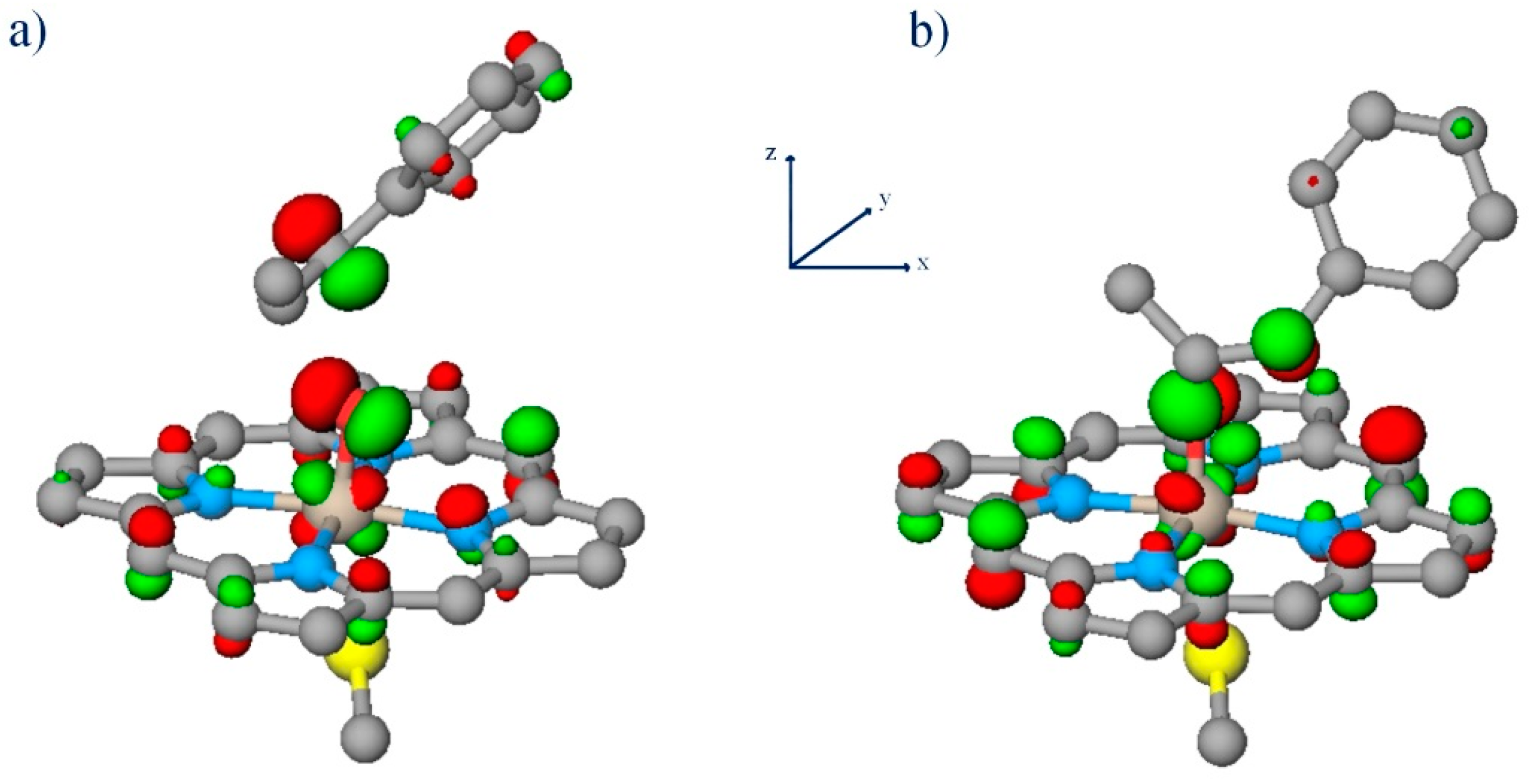
2.3. Initial Structures for 1R2S and 1S2R CPO-I/CBMS Transition State Complexes
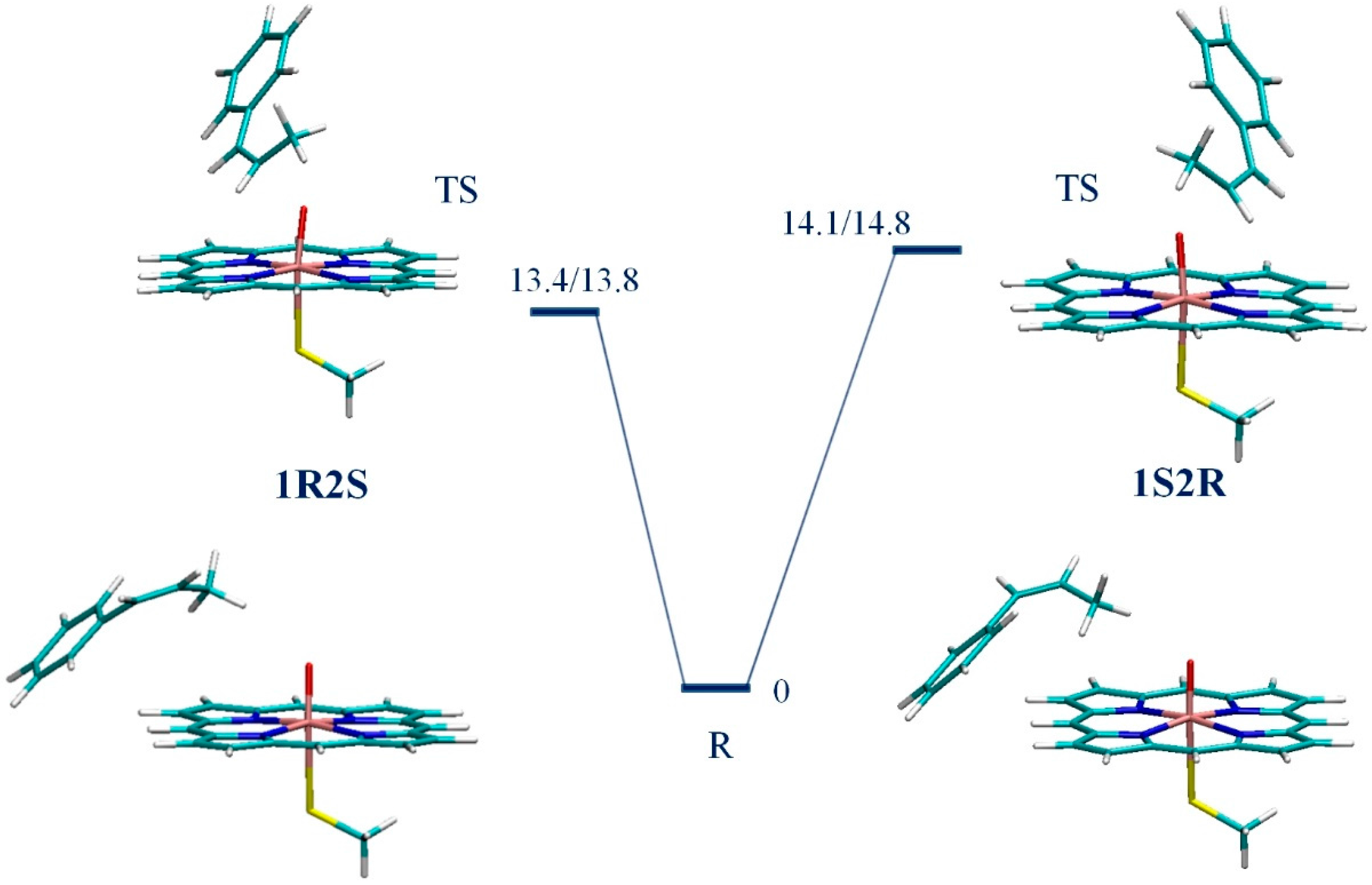
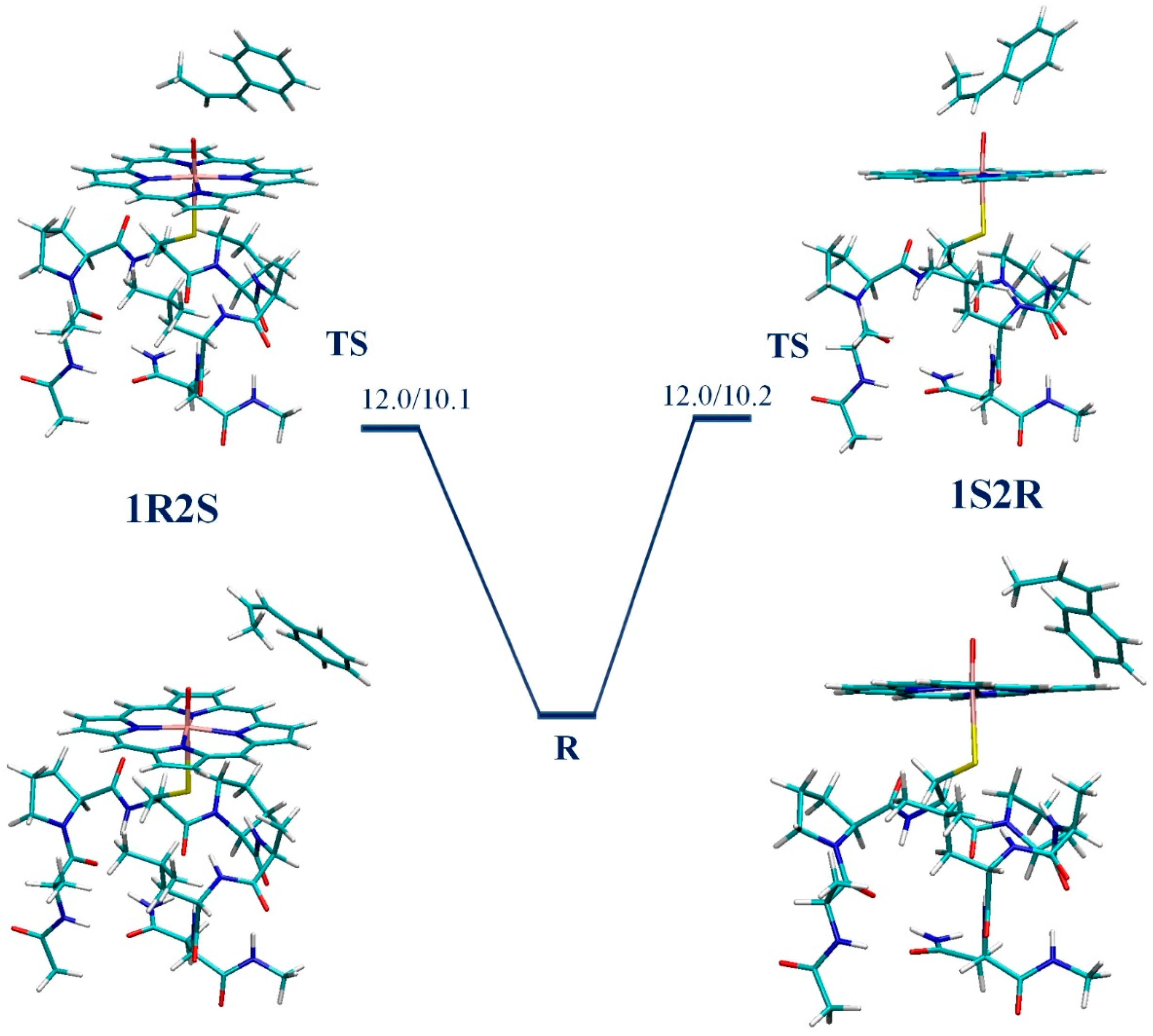
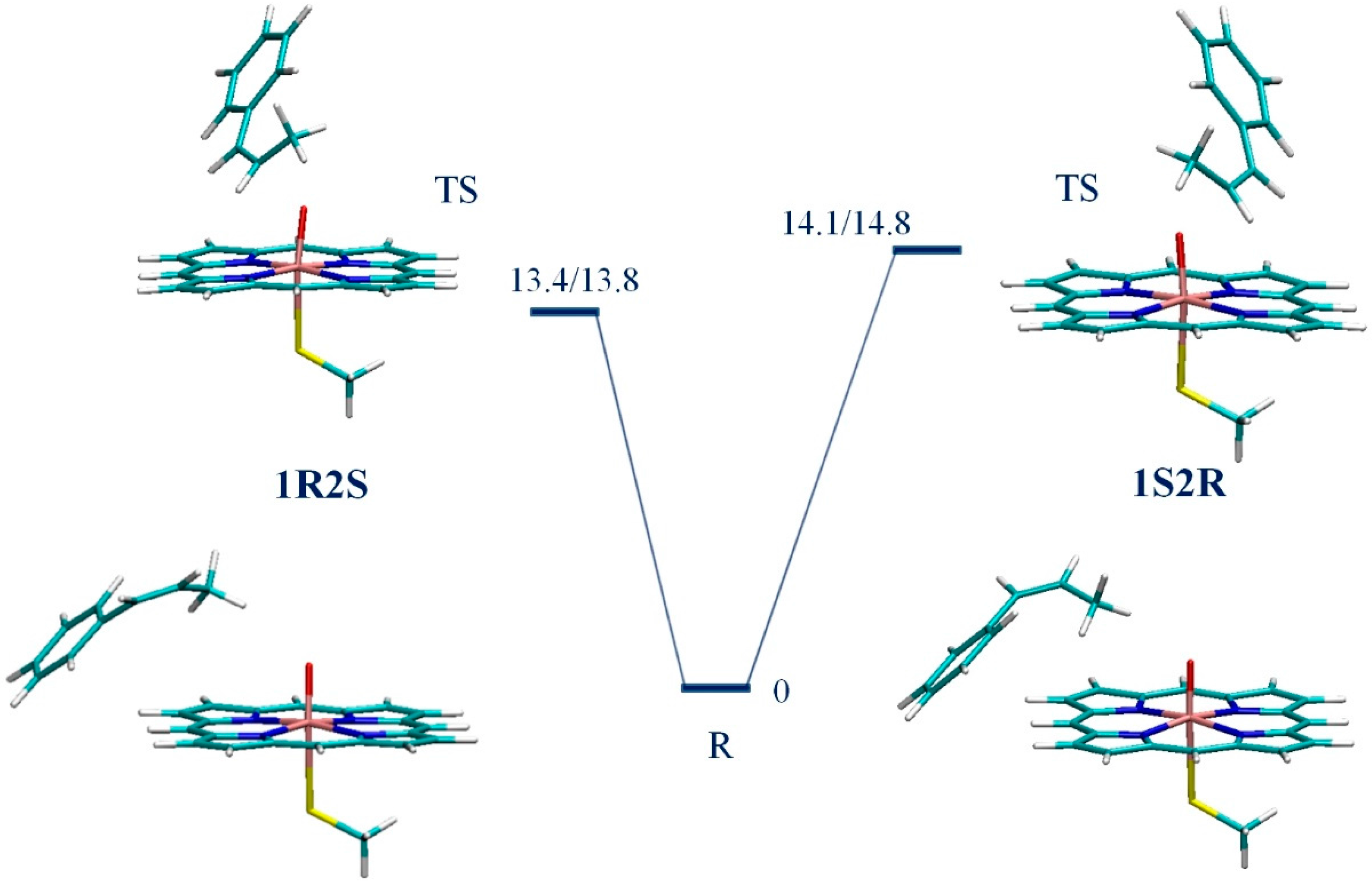
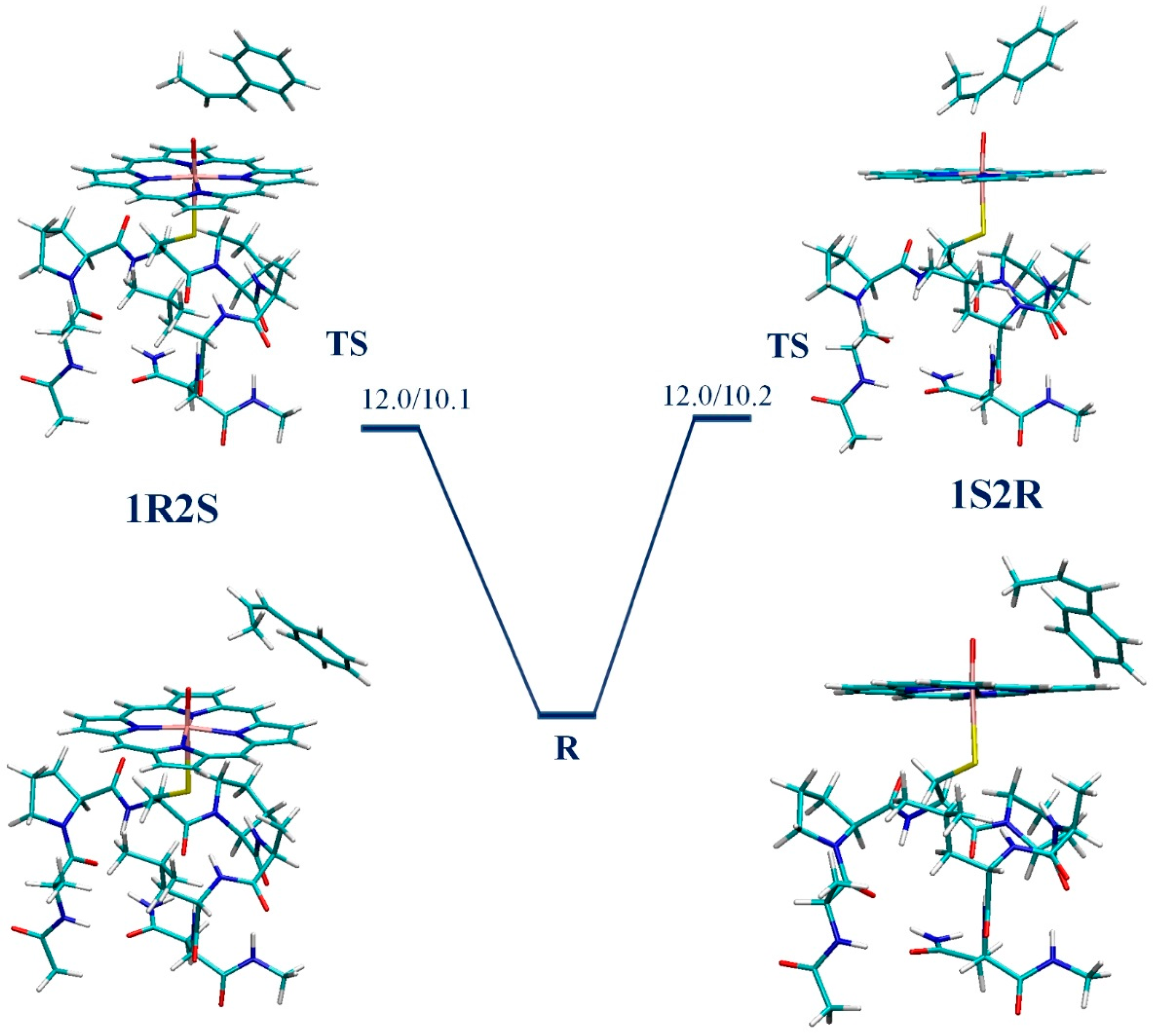
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.D.; Hager, L.P. Biological chlorination. IV. Peroxidative nature of enzymatic chlorination. J. Am. Chem. Soc. 1959, 81, 1011–1012. [Google Scholar] [CrossRef]
- Shaw, P.D.; Hager, L.P. Biological Chlorination. J. Biol. Chem. 1961, 236, 1626–1630. [Google Scholar]
- Hager, L.P.; Morris, D.R.; Brown, F.S.; Eberwein, H. Chloroperoxidase II: Utilization of halogen ions. J. Biol. Chem. 1966, 241, 1769–1777. [Google Scholar] [PubMed]
- Hofrichter, M.; Ullrich, R. Heme-thiolate haloperoxidases: Versatile biocatalysts with biotechnological and environmental significance. Appl. Microbiol. Biotechnol. 2006, 71, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Osborne, R.L.; Coggins, M.K.; Terner, J.; Dawson, J.H. Caldariomyces fumago chloroperoxidase catalyzes the oxidative dehalogenation of chlorophenols by a mechanism involving two one-electron steps. J. Am. Chem. Soc. 2007, 129, 14838–14839. [Google Scholar] [CrossRef] [PubMed]
- Gruia, F.; Ionascu, D.; Kubo, M.; Ye, X.; Dawson, J.; Osborne, R.L.; Sligar, S.G.; Denisov, I.; Das, A.; Poulos, T.L.; et al. Low-frequency dynamics of caldariomyces fumago chloroperoxidase probed by femtosecond coherence spectroscopy. Biochem. 2008, 47, 5156–5167. [Google Scholar] [CrossRef] [PubMed]
- Tzialla, A.A.; Kalogeris, E.; Gournis, D.; Sanakis, Y.; Stamatis, H. Enhanced catalytic performance and stability of chloroperoxidase from Caldariomyces fumago in surfactant free ternary water-organic solvent systems. J. Mol. Catal. B Enzym. 2008, 51, 24–35. [Google Scholar] [CrossRef]
- Hager, L.P. A lifetime of playing with enzymes. J. Biol. Chem. 2010, 285, 14852–14860. [Google Scholar] [CrossRef] [PubMed]
- Ayala, M.; Hernandez-Lopez, E.L.; Perezgasga, L.; Vazquez-Duhalt, R. Reduced coke formation and aromaticity due to chloroperoxidase-catalyzed transformation of asphaltenes from Maya crude oil. Fuel 2012, 92, 245–249. [Google Scholar] [CrossRef]
- Zhang, R.; He, Q.H.; Chatfield, D.; Wang, X.T. Paramagnetic nuclear magnetic resonance relaxation and molecular mechanics studies of the chloroperoxidase-indole complex: Insights into the mechanism of chloroperoxidase-catalyzed regioselective oxidation of indole. Biochemistry 2013, 52, 3688–3701. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Jiang, Y.; Hu, M.; Li, S.; Zhai, Q. Biocatalytic synthesis of C3 chiral building blocks by chloroperoxidase-catalyzed enantioselective halo-hydroxylation and epoxidation in the presence of ionic liquids. Biotechnol. Prog. 2015, 31, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Guerrero, F.A.; Aguila, S.; Vazquez-Duhalt, R.; Campos, C.H.; Torres, C.C.; Alderete, J.B. Substrate ionization energy influences the epoxidation of m-substituted styrenes catalyzed by chloroperoxidase from Caldariomyces fumago. Catal. Commun. 2016, 77, 52–54. [Google Scholar] [CrossRef]
- Zhang, R.; He, Q.; Huang, Y.; Wang, X. Spectroscopic and QM/MM investigations of chloroperoxidase catalyzed degradation of orange G. Arch. Biochem. Biophys. 2016, 596, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hirao, H.; Derat, E.; Schlichting, I.; Shaik, S. Quantum mechanical/molecular mechanical study on the mechanisms of Compound I formation in the catalytic cycle of chloroperoxidase: An overview on heme enzymes. J. Phys. Chem. B 2008, 112, 9490–9500. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.; Chen, H.; Cho, K.B.; Shaik, S. Effects of substrate, protein environment, and proximal ligand mutation on Compound I and compound 0 of chloroperoxidase. J. Phys. Chem. A 2009, 113, 11763–11771. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.N.; D’Cunha, C.; Alvarez, C.A.; Chatfield, D.C. Enantiospecificity of chloroperoxidase-catalyzed epoxidation: Biased molecular dynamics study of a cis-β-methylstyrene/chloroperoxidase-Compound I complex. Biophys. J. 2011, 100, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.N.; Chatfield, D.C. Chloroperoxidase-catalyzed epoxidation of cis-β-methylstyrene: Distal pocket flexibility tunes catalytic reactivity. J. Phys. Chem. B 2012, 116, 12905–12914. [Google Scholar] [CrossRef] [PubMed]
- D’Cunha, C.; Morozov, A.N.; Chatfield, D.C. Theoretical study of HOCl-catalyzed keto-enol tautomerization of cyclopentanedione in an explicit water environment. J. Phys. Chem. A 2013, 117, 8437–8448. [Google Scholar] [CrossRef] [PubMed]
- Pardillo, A.D.; Morozov, A.N.; Chatfield, D.C. Proximal pocket hydrogen bonds significantly influence the mechanism of chloroperoxidase Compound I formation. J. Phys. Chem. B 2015, 119, 12590–12602. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.N.; Pardillo, A.D.; Chatfield, D.C. Chloroperoxidase-catalyzed epoxidation of cis-β-methylstyrene: NH–S hydrogen bonds and proximal helix dipole change the catalytic mechanism and significantly lower the reaction barrier. J. Phys. Chem. B 2015, 119, 14350–14363. [Google Scholar] [CrossRef] [PubMed]
- Rutter, R.; Hager, L.P. The detection of two electron paramagnetic resonance radical signals associated with chloroperoxidase Compound I. J. Biol. Chem. 1982, 257, 7958–7961. [Google Scholar] [PubMed]
- Kim, S.H.; Perera, R.; Hager, L.P.; Dawson, J.H.; Hoffman, B.M. Rapid freeze-quench ENDOR study of chloroperoxidase Compound I: The site of the radical. J. Am. Chem. Soc. 2006, 128, 5598–5599. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, M.; Terner, J.; Poulos, T.L. The crystal structure of chloroperoxidase: A heme peroxidase-cytochrome P450 functional hybrid. Structure 1995, 3, 1367–1378. [Google Scholar] [CrossRef]
- Sundaramoorthy, M.; Kishi, K.; Gold, M.H.; Poulos, T.L. The crystal-structure of manganese peroxidase from phanerochaete-chrysosporium at 2.06-angstrom resolution. J. Biol. Chem. 1994, 269, 32759–32767. [Google Scholar] [PubMed]
- Poulos, T.L.; Finzel, B.C.; Howard, A.J. High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987, 195, 687–700. [Google Scholar] [CrossRef]
- Dawson, J.H. Probing structure-function relations in heme-containing oxygenases and peroxidases. Science 1988, 240, 433–439. [Google Scholar] [CrossRef] [PubMed]
- McQuarters, A.B.; Wolf, M.W.; Hunt, A.P.; Lehnert, N. 1958–2014: After 56 years of research, cytochrome P450 reactivity is finally explained. Angew. Chem. Int. Ed. 2014, 53, 4750–4752. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T. Enzymatic C–H bond activation: Using push to get pull. Nat. Chem. 2014, 6, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.L.; Loew, G.H. Theoretical investigation of the proton assisted pathway to formation of cytochrome P450 Compound I. J. Am. Chem. Soc. 1998, 120, 8941–8948. [Google Scholar] [CrossRef]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants? J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Sigfridsson, E.; Ryde, U. On the role of the axial ligand in heme proteins: A theoretical strudy. J. Biol. Inorg. Chem. 2004, 9, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. The role of the proximal ligand in heme enzymes. J. Biol. Inorg. Chem. 1996, 1, 356–359. [Google Scholar] [CrossRef]
- Ueno, T.; Nishikawa, N.; Moriyama, S.; Adachi, S.; Lee, K.; Okamura, T.; Ueyama, N.; Nakamura, A. Role of the invariant peptide fragment forming nhs hydrogen bonds in the active site of cytochrome P450 and chloroperoxidase: Synthesis and properties of cys-containing peptide Fe(III) and Ga(III) (octaethylporphinato) complexes as models. Inorg. Chem. 1999, 38, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Tosha, T.; Takahashi, S.; Ishimori, K.; Hori, H.; Morishima, I. Roles of the proximal hydrogen bonding network in cytochrome P450cam-catalyzed oxygenation. J. Am. Chem. Soc. 2002, 124, 14571–14579. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Takahashi, S.; Ishimori, K.; Morishima, I. Roles of the axial push effect in cytochrome P450cam studied with the site-directed mutagenesis at the heme proximal site. J. Inorg. Biochem. 2000, 81, 141–151. [Google Scholar] [CrossRef]
- Dey, A.; Jiang, Y.; Ortiz de Montellano, P.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. S K-edge XAS and DFT calculations on cytochrome P450: Covalent and ionic contributions to the cysteine-Fe bond and their contribution to reactivity. J. Am. Chem. Soc. 2009, 131, 7869–7878. [Google Scholar] [CrossRef] [PubMed]
- Galinato, M.G.I.; Spolitak, T.; Ballou, D.P.; Lehnert, N. Elucidating the role of the proximal cysteine hydrogen-bonding network in ferric cytochrome P450cam and corresponding mutants using magnetic circular dichroism spectroscopy. Biochemistry 2011, 50, 1053–1069. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. Hydrogen bonding modulates the selectivity of enzymatic oxidation by P450: Chameleon oxidant behavior by Compound I. Angew. Chem. Int. Ed. 2002, 124, 11809–11826. [Google Scholar]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 130, 12961–12974. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Tan, L.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Sainna, M.A.; Rybak-Akimova, E.V.; de Visser, S.P. Does hydrogen-bonding donation to manganese(IV)-oxo and iron(IV)-oxo oxidants affect the oxygen-atom transfer ability? a computational study. Chem. Eur. J. 2013, 19, 4058–4068. [Google Scholar] [CrossRef] [PubMed]
- Hol, W.G.J.; van Duijnen, P.T.; Berendsen, H.J.C. The α-Helix dipole and the properties of proteins. Nature 1978, 273, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Goddard, W.A. Stabilization of α-helices by dipole-dipole interactions within α-helices. J. Phys. Chem. B 2000, 104, 7784–7789. [Google Scholar] [CrossRef]
- Hol, W.G.J.; Halie, L.M.; Sander, C. Dipoles of the α-helix and β-sheet: Their role in protein folding. Nature 1981, 294, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.N.; Shiu, Y.J.; Liang, C.T.; Tsai, M.Y.; Lin, S.H. Nonadditive interactions in protein folding: The zipper model of cytochrome C. J. Biol. Phys. 2007, 33, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Morozov, A.N.; Chu, K.Y.; Lin, S.H. Molecular dynamics insight into the role of tertiary (foldon) interactions on unfolding in cytochrome C. Chem. Phys. Lett. 2009, 475, 111–115. [Google Scholar] [CrossRef]
- Morozov, A.N.; Roach, J.P.; Kotzer, M.; Chatfield, D.C. A possible mechanism for redox control of human neuroglobin activity. J. Chem. Inf. Model. 2014, 54, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, D.J.; Kim, P.S. Internal stark effect measurement of the electric field at the amino terminus of an α-helix. Science 1992, 257, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Kousumi, Y.; Yoshizawa-Kumagaye, K.; Nakajima, K.; Ueyama, N.; Okamura, T.; Nakamura, A. Role of α-helix conformation cooperating with NH···S hydrogen bond in the active site of cytochrome P-450 and chloroperoxidase: Synthesis and Properties of [MIII(OEP)(Cys-Helical Peptide)] (M = Fe and Ga). J. Am. Chem. Soc. 1998, 120, 12264–12273. [Google Scholar] [CrossRef]
- Allain, E.J.; Hager, L.P.; Deng, L.; Jacobsen, E.N. Highly enantioselective epoxidation of disubstituted alkenes with hydrogen peroxide catalyzed by chloroperoxidase. J. Am. Chem. Soc. 1993, 115, 4415–4416. [Google Scholar] [CrossRef]
- Hu, S.; Hager, L.P. Asymmetric epoxidation of functionalized cis-olefins catalyzed by chloroperoxidase. Tetrahedron Lett. 1999, 40, 1641–1644. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their structure, reactivity, and selectivity modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural Bond Orbital Analysis Program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Norrby, P.O. Selectivity in asymmetric synthesis from QM-guided molecular mechanics. J. Mol. Struct. Theochem. 2000, 506, 9–16. [Google Scholar] [CrossRef]
- Fruetel, J.A.; Collins, J.R.; Camper, D.L.; Loew, G.H.; Ortiz de Montellano, P.R. Calculated and experimental absolute stereochemistry of the styrene and β-methylstyrene epoxides formed by cytochrome P450cam. J. Am. Chem. Soc. 1992, 114, 6987–6993. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Spin Densities/Natural Atomic Charges | Bond Lengths | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S–R | Por | Fe | O | CβH | CαH | R1+ | R2+ | S–Fe | Fe–O | O–Cβ | ||
| 1S2R | ||||||||||||
| R | A | −0.75/−0.05 | −0.30/−0.49 | 1.10/0.91 | 0.95/−0.37 | 0.00/0.06 | 0.00/−0.03 | 0.00/−0.05 | 0.00/0.02 | 2.619 | 1.623 | – |
| B | −0.60/−0.27 | −0.50/−0.32 | 1.15/0.95 | 0.95/−0.36 | 0.00/0.06 | 0.00/−0.03 | 0.00/−0.05 | 0.00/0.02 | 2.776 | 1.619 | – | |
| TS | A | −0.70/−0.08 | −0.30/−0.58 | 0.95/0.90 | 0.75/−0.44 | −0.10/0.15 | 0.30/0.04 | 0.10/−0.02 | 0.00/0.03 | 2.554 | 1.705 | 1.985 |
| B | −0.30/−0.40 | −0.25/−0.51 | 1.40/0.96 | 0.50/−0.42 | −0.05/0.18 | −0.20/0.08 | −0.10/0.07 | 0.00/0.04 | 2.570 | 1.658 | 2.099 | |
| 1R2S | ||||||||||||
| R | A | −0.76/−0.05 | −0.30/−0.49 | 1.10/0.91 | 0.96/−0.37 | 0.00/0.06 | 0.00/−0.03 | 0.00/−0.05 | 0.00/0.02 | 2.624 | 1.623 | – |
| B | −0.59/−0.27 | −0.48/−0.32 | 1.15/0.95 | 0.95/−0.36 | 0.00/0.06 | 0.00/−0.03 | 0.00/−0.05 | 0.00/0.02 | 2.776 | 1.619 | – | |
| TS | A | −0.65/−0.08 | −0.28/−0.59 | 0.91/0.89 | 0.76/−0.43 | −0.11/0.15 | 0.27/0.05 | 0.10/−0.01 | 0.00/0.03 | 2.512 | 1.702 | 1.996 |
| B | −0.30/−0.41 | −0.26/−0.50 | 1.38/0.96 | 0.52/−0.42 | −0.06/0.18 | −0.17/0.08 | −0.11/0.07 | 0.00/0.04 | 2.576 | 1.658 | 2.116 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morozov, A.N.; Chatfield, D.C. How the Proximal Pocket May Influence the Enantiospecificities of Chloroperoxidase-Catalyzed Epoxidations of Olefins. Int. J. Mol. Sci. 2016, 17, 1297. https://doi.org/10.3390/ijms17081297
Morozov AN, Chatfield DC. How the Proximal Pocket May Influence the Enantiospecificities of Chloroperoxidase-Catalyzed Epoxidations of Olefins. International Journal of Molecular Sciences. 2016; 17(8):1297. https://doi.org/10.3390/ijms17081297
Chicago/Turabian StyleMorozov, Alexander N., and David C. Chatfield. 2016. "How the Proximal Pocket May Influence the Enantiospecificities of Chloroperoxidase-Catalyzed Epoxidations of Olefins" International Journal of Molecular Sciences 17, no. 8: 1297. https://doi.org/10.3390/ijms17081297