
De Novo Assembly and Characterization of the Transcriptome of Grasshopper Shirakiacris shirakii

Abstract
:1. Introduction
2. Results
2.1. Transcriptome Assembly
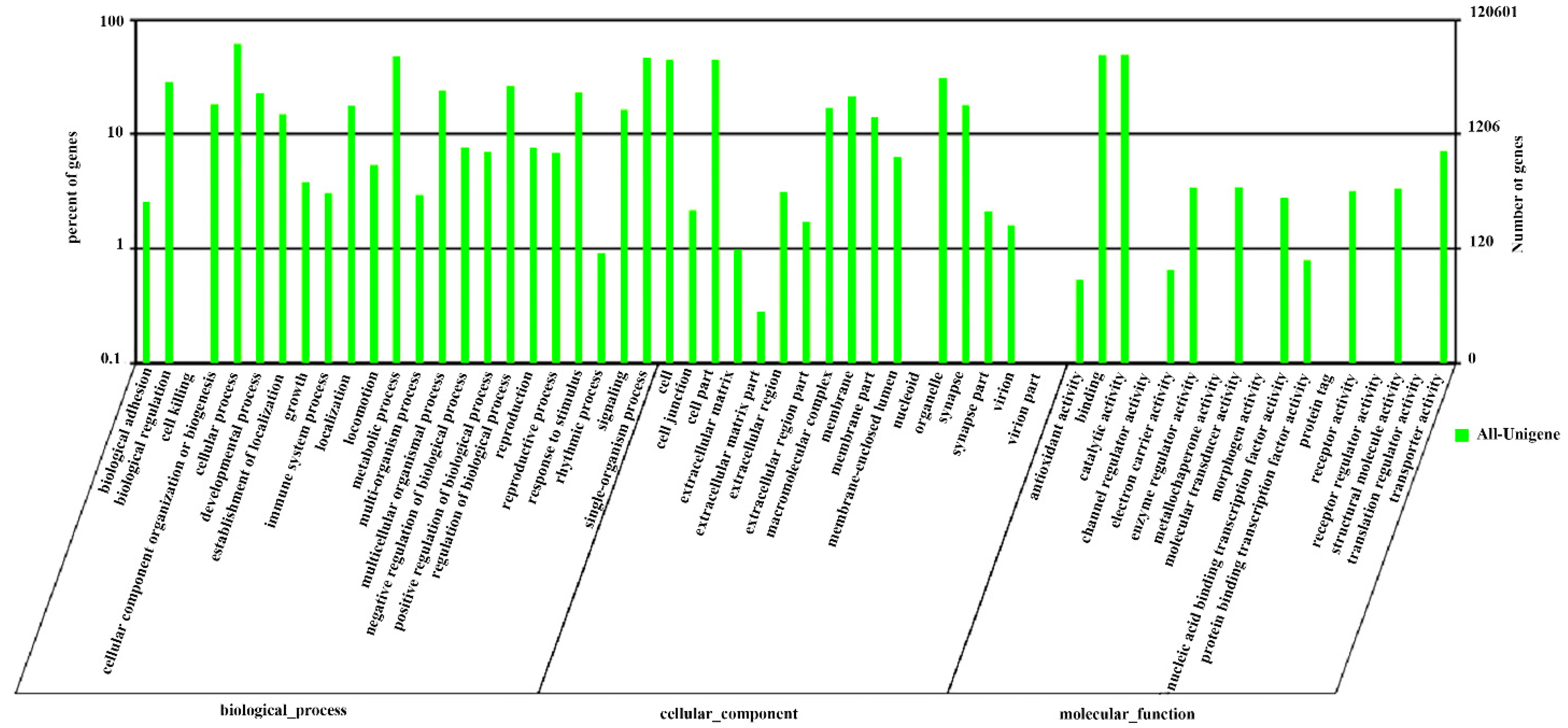
2.2. Functional Annotation
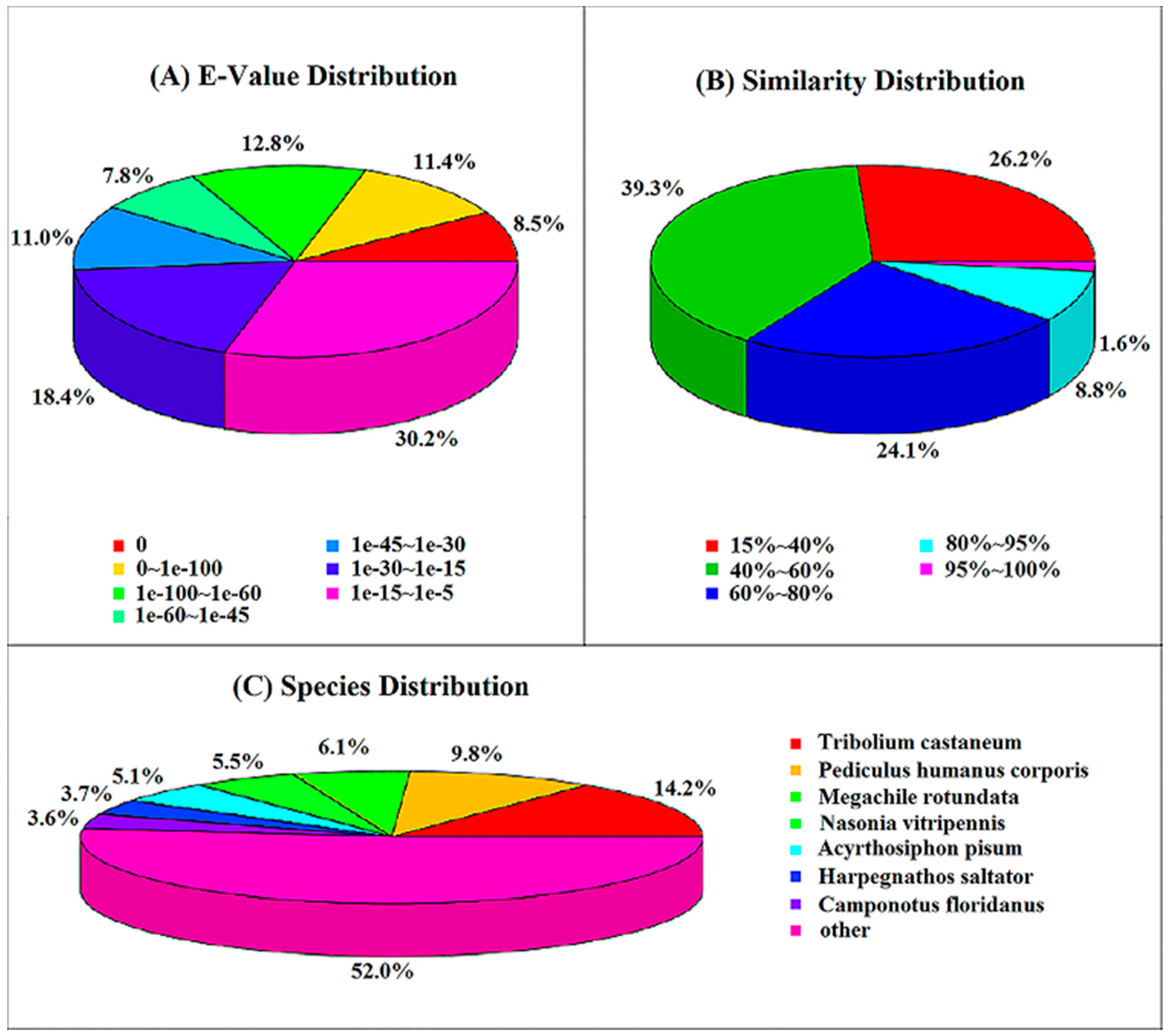
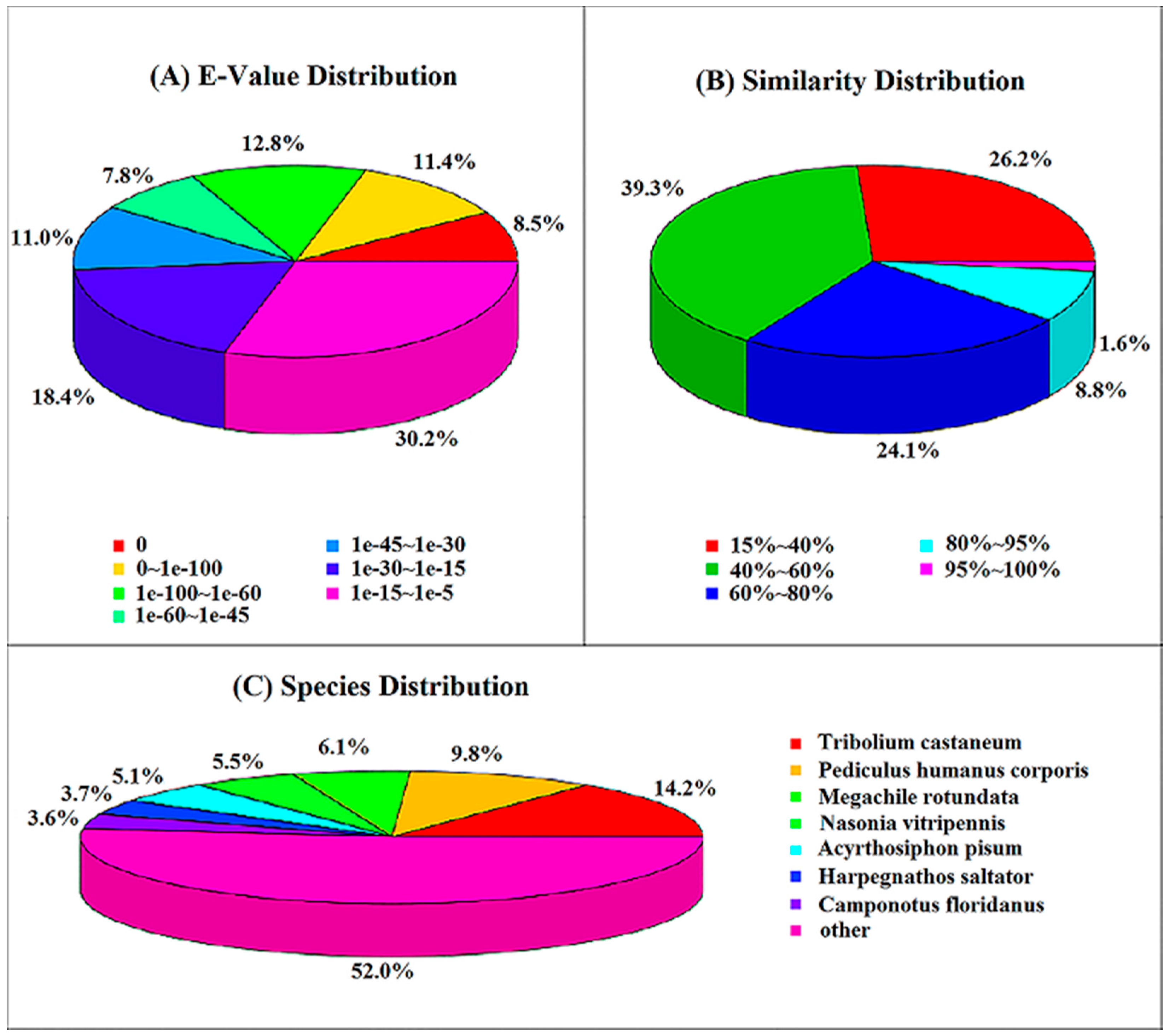
2.3. Nr Annotation
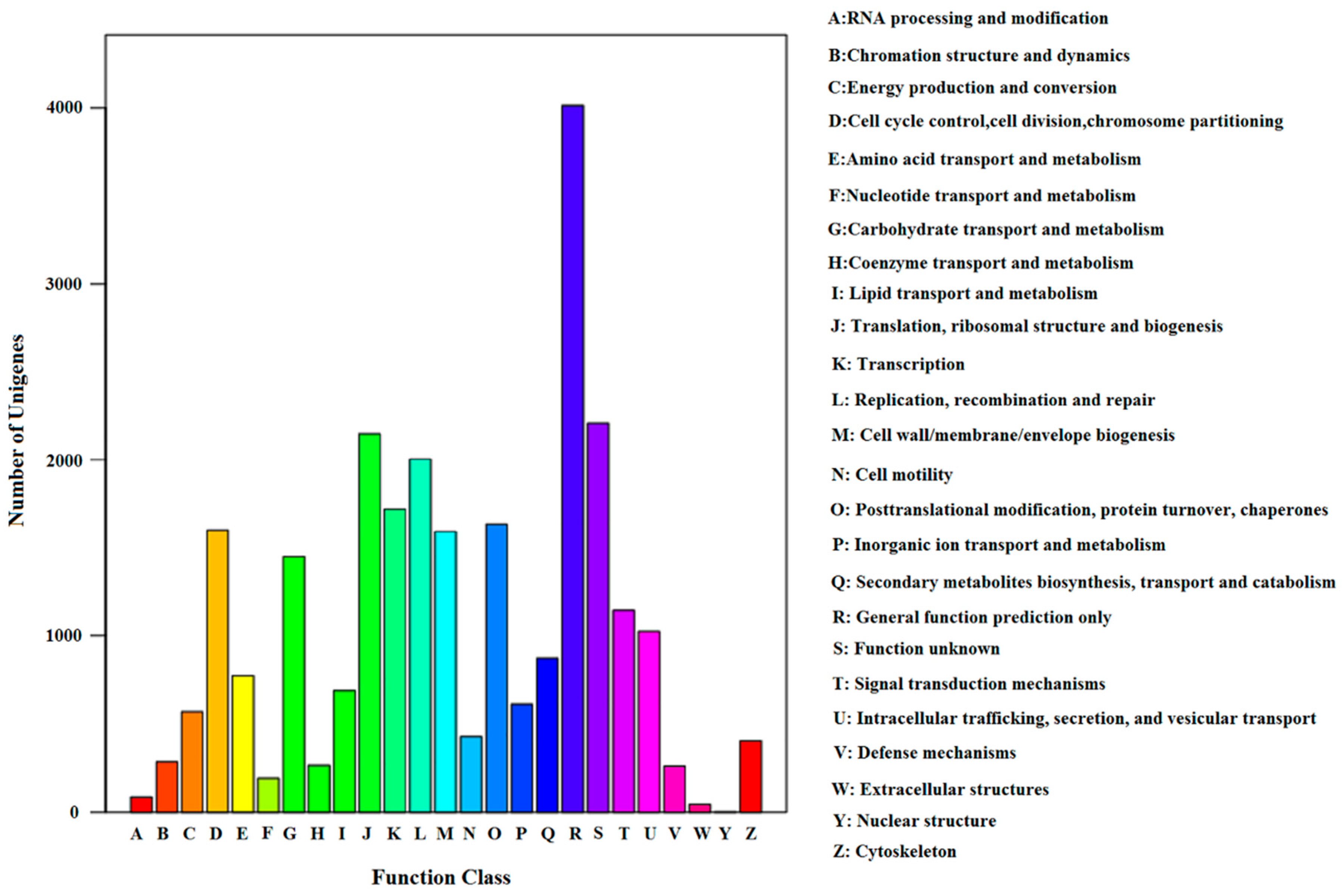
2.4. GO, COG Classification, Swiss-Prot and KEGG Pathway
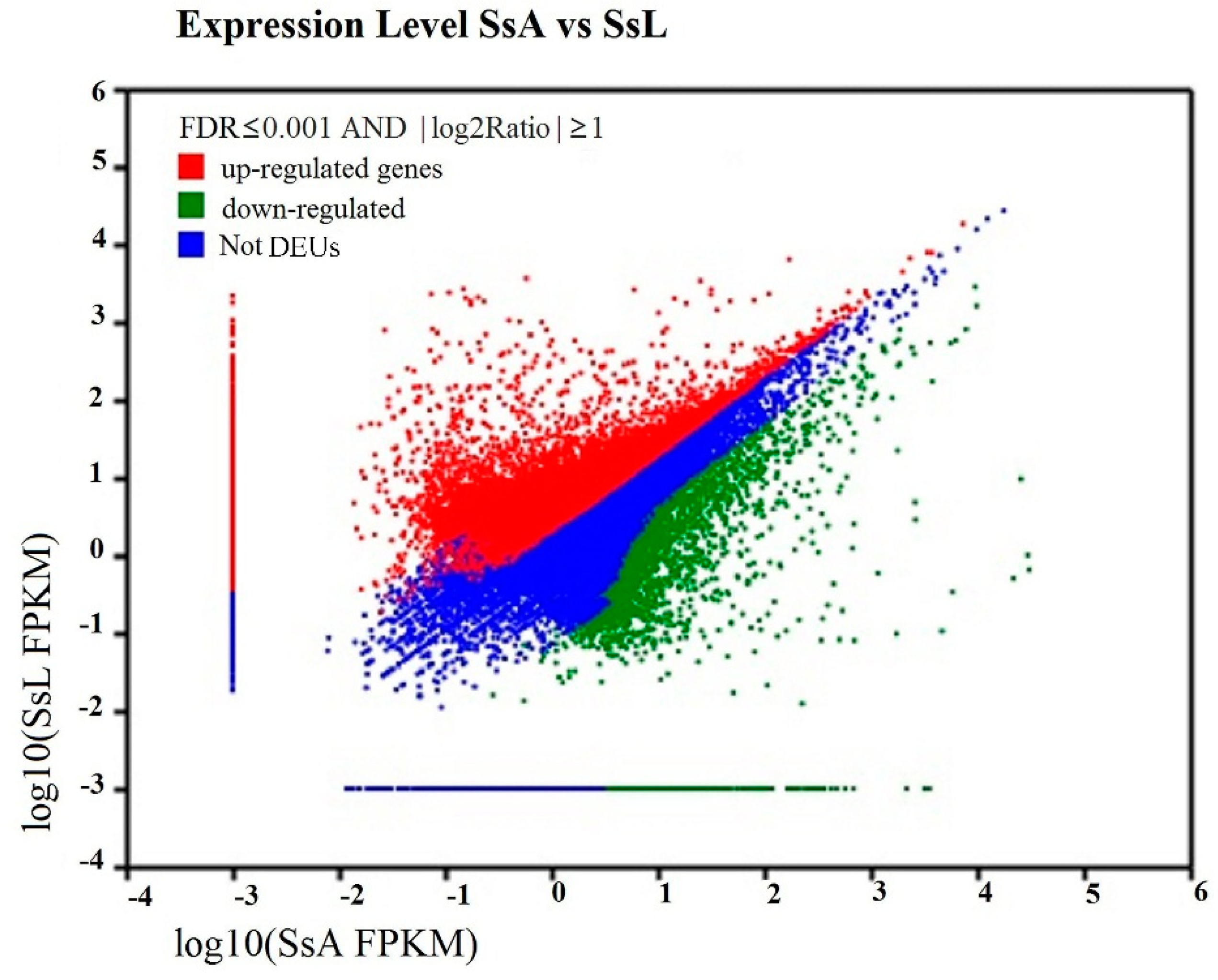
2.5. Differentially-Expressed Unigenes’ Analysis
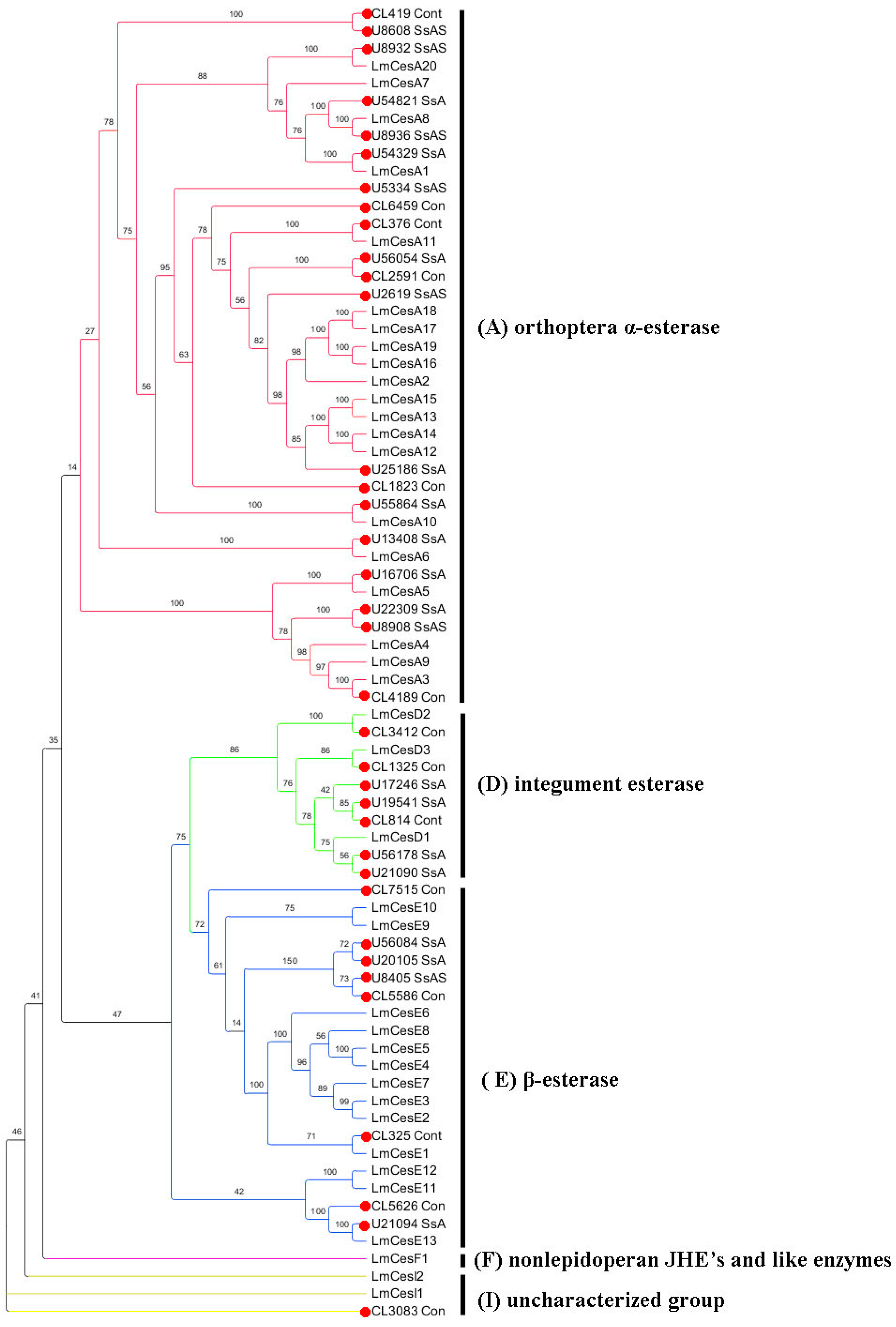
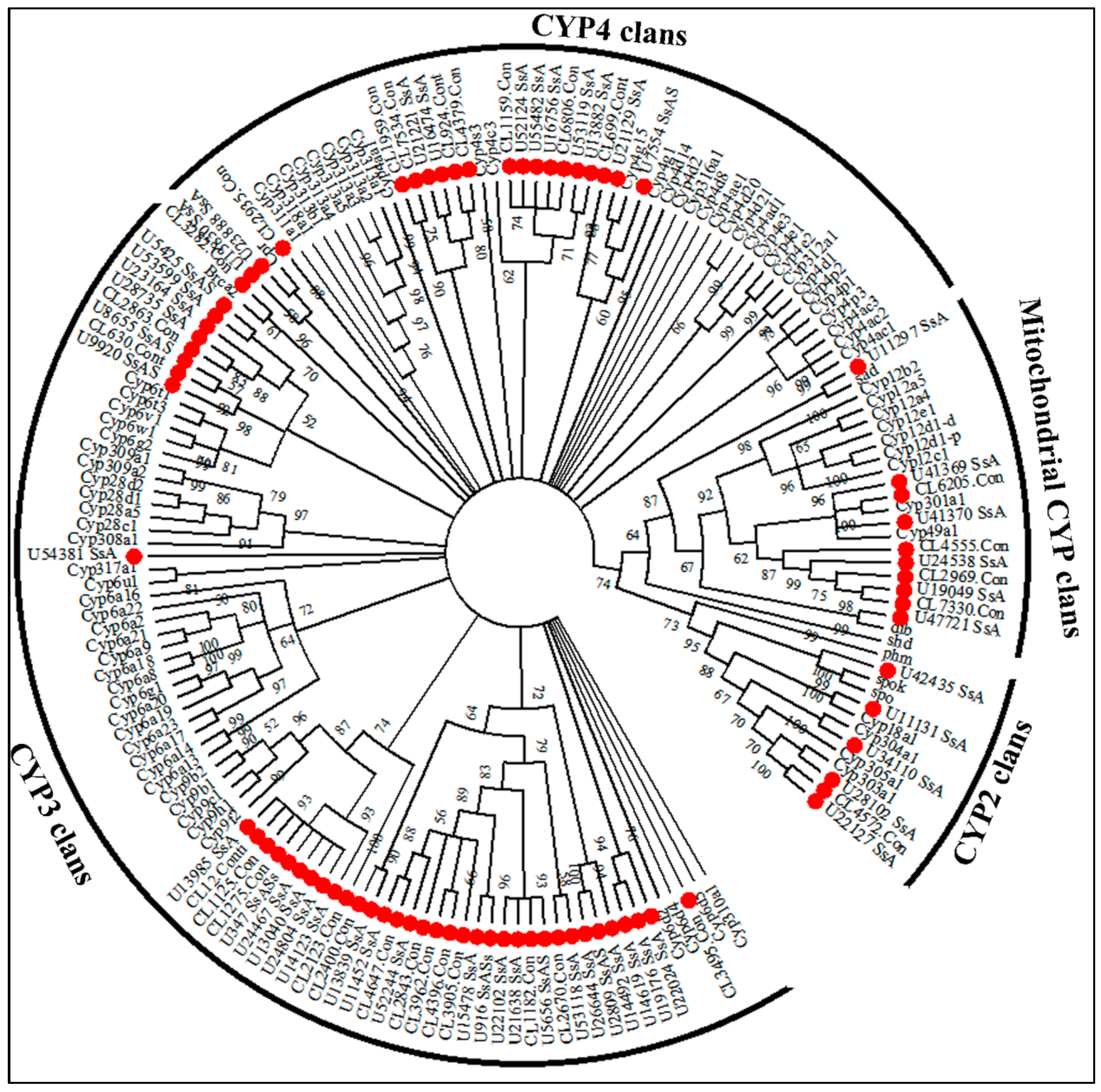
2.6. Insecticide Resistance
2.7. Core RNAi Components
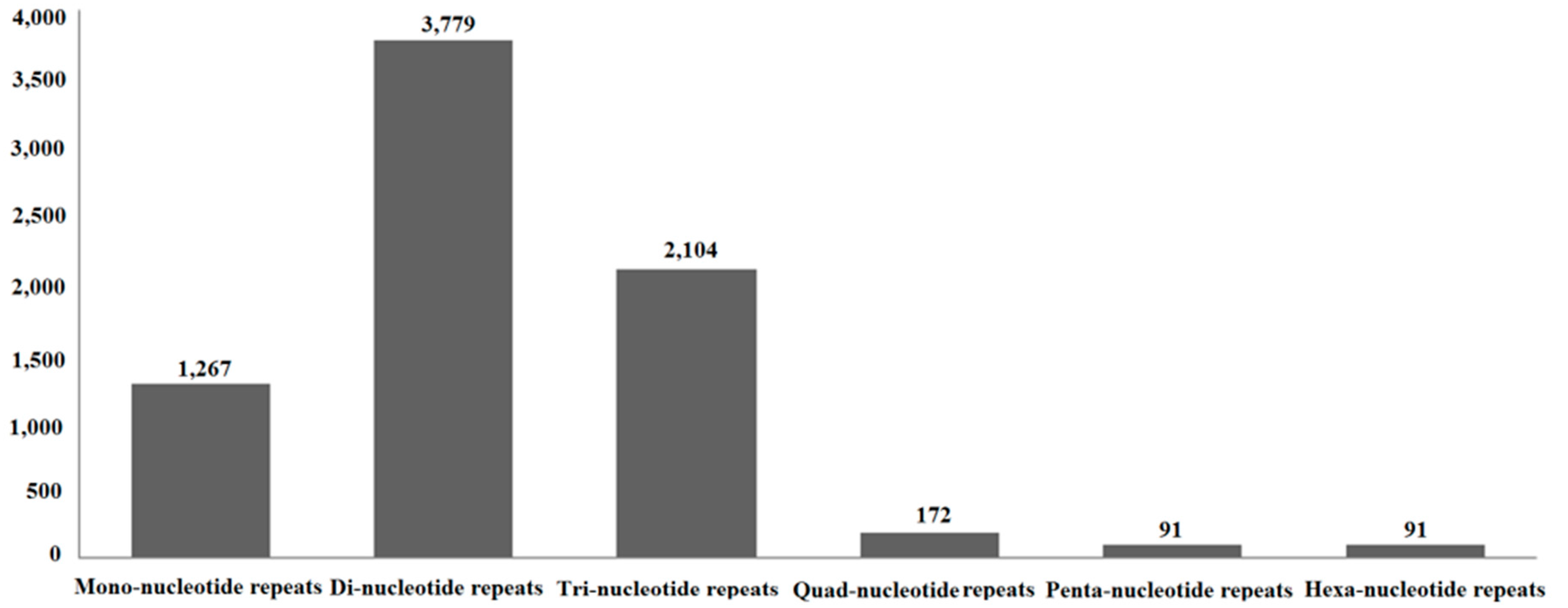
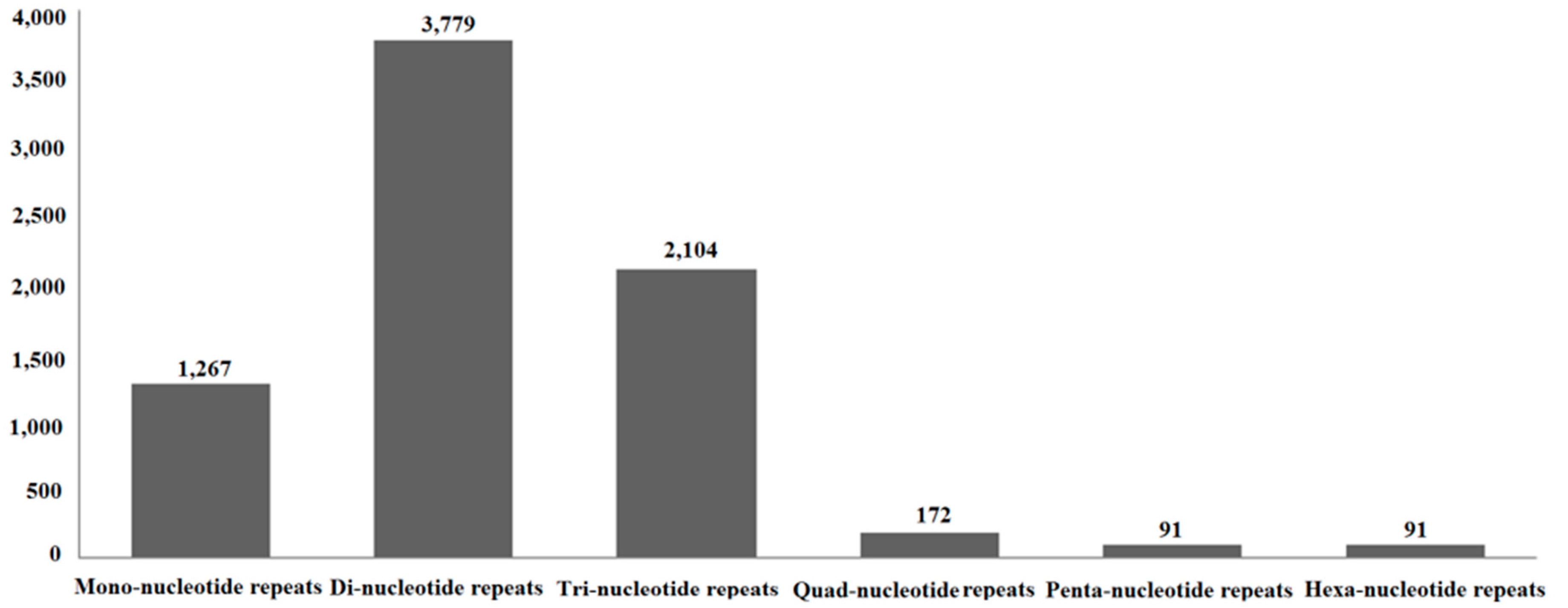
2.8. Simple Sequence Repeats (SSRs)
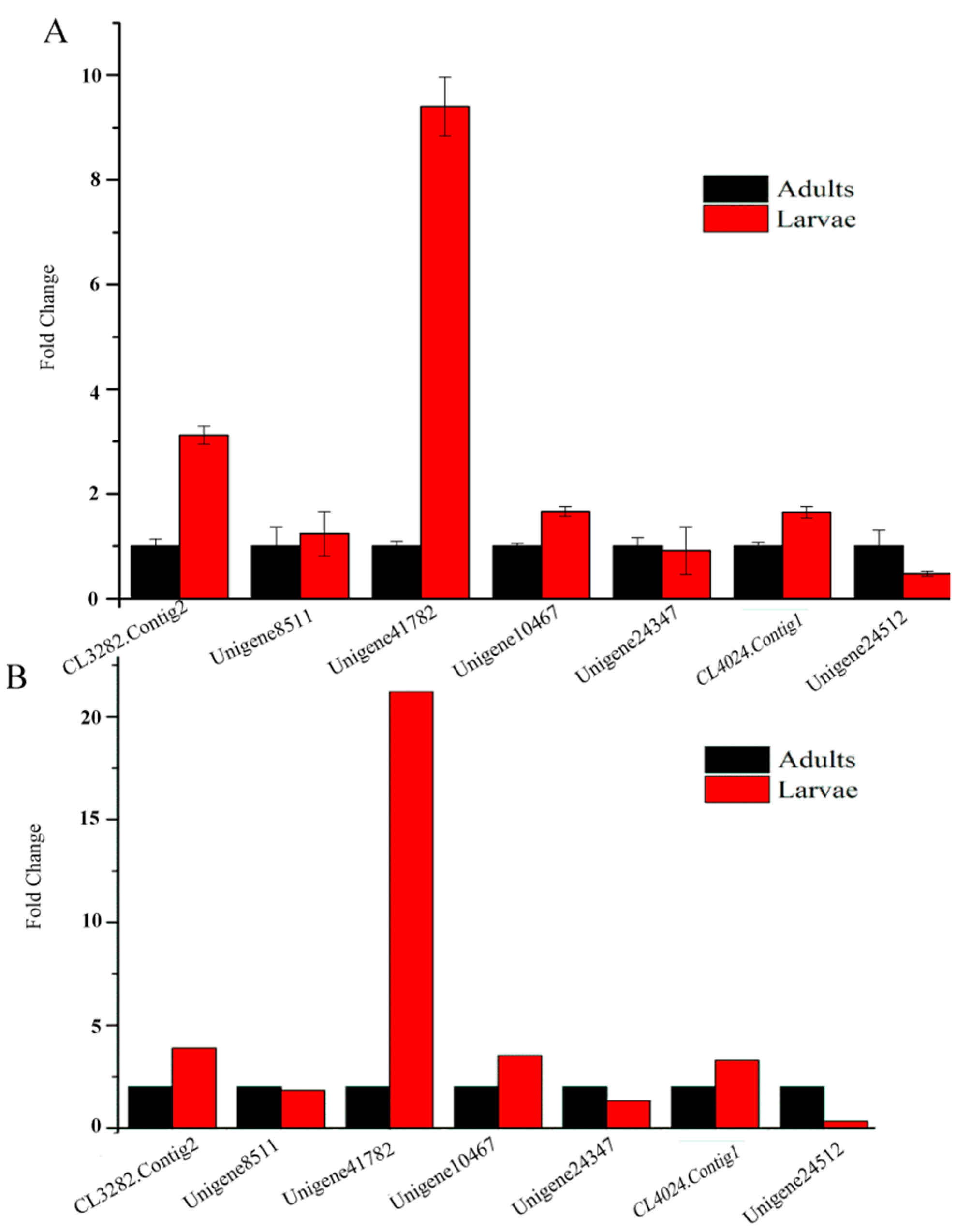
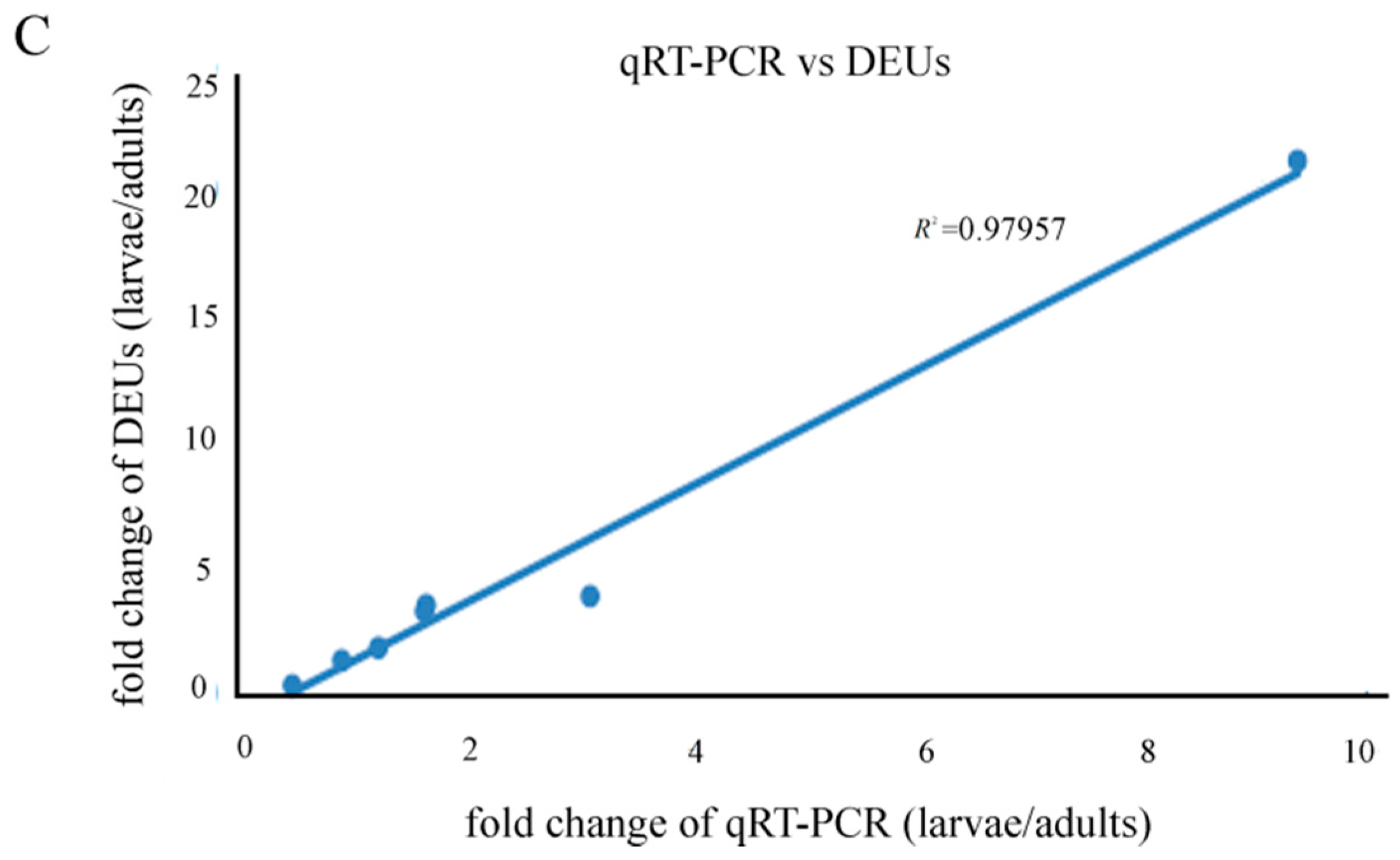
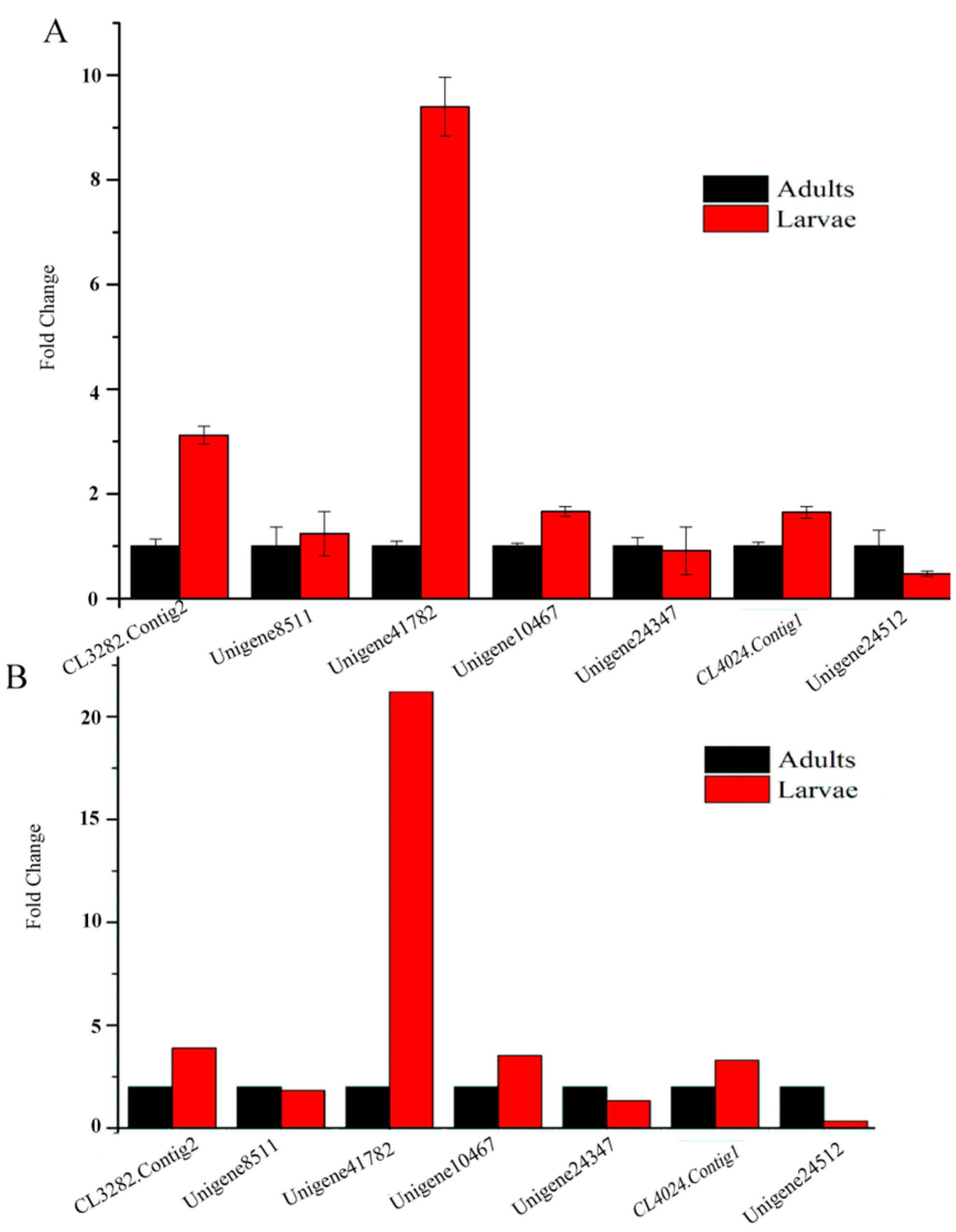
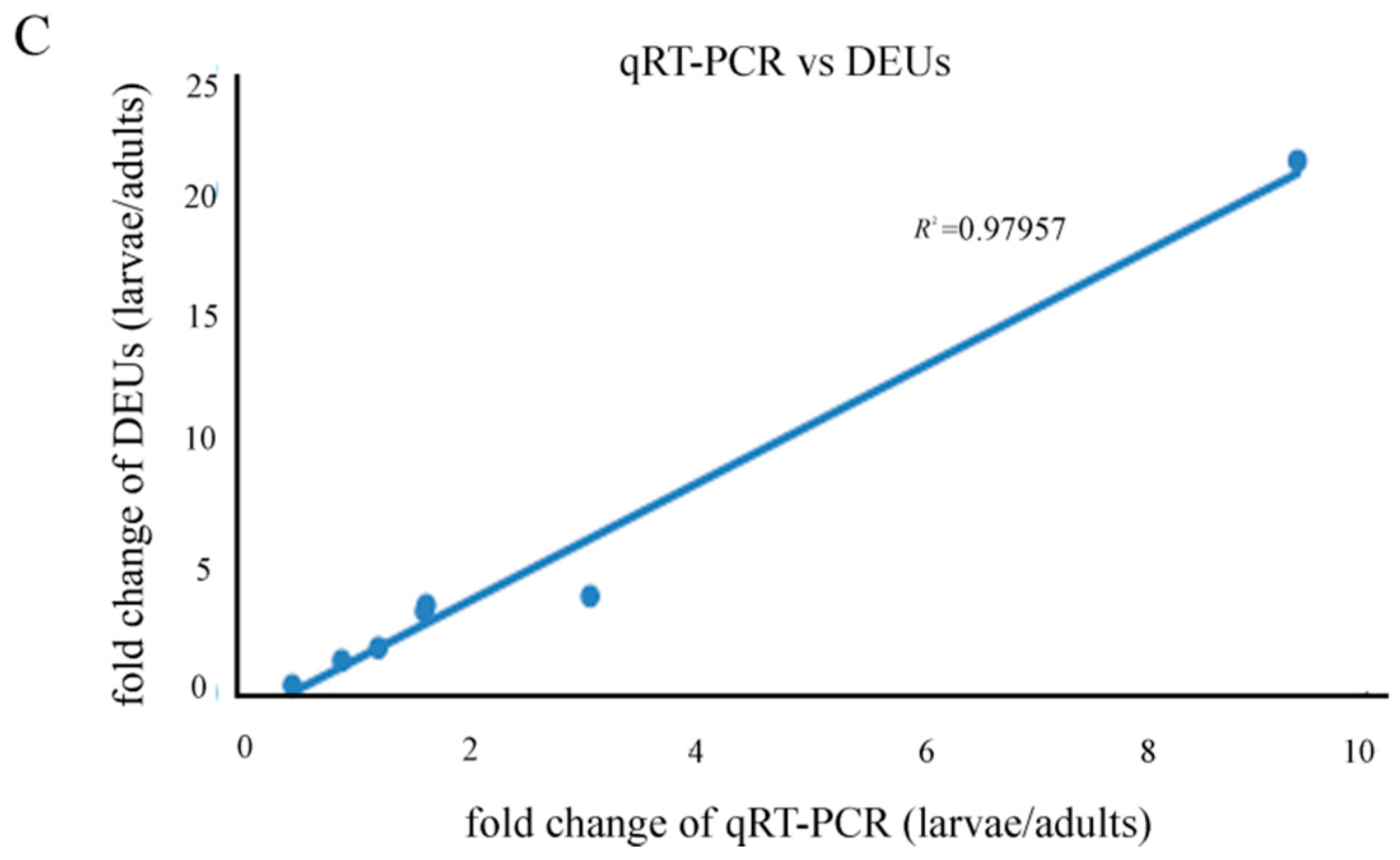
2.9. Quantitative Real-Time PCR Results
3. Discussion
4. Materials and Methods
4.1. Species Collection, RNA Extraction and RNA-Seq
4.2. De Novo Assembly and Functional Annotation
4.3. Differentially-Expressed Unigenes (DEUs)
4.4. Core RNAi Components Identification
4.5. Identification of Genes Related to Insecticide Resistance and Phylogenetic Analysis
4.6. qRT-PCR
4.7. SSRs Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cabrero, J.; Bugrov, A.; Warchalowska-Sliwa, E.; Lopez-Leon, M.D.; Perfectti, F.; Camacho, J.P. Comparative FISH analysis in five species of Eyprepocnemidine grasshoppers. Heredity 2003, 90, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Ma, E.B.; Zheng, X.Y. Genetic differentiation of different populations of four locust species in China. Yi Chuan Xue Bao 2003, 30, 234–244. [Google Scholar] [PubMed]
- Raymond-Delpech, V.; Matsuda, K.; Sattelle, B.M.; Rauh, J.J.; Sattelle, D.B. Ion channels: Molecular targets of neuroactive insecticides. Invertebr. Neurosci. 2005, 5, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, M.; Jia, Q.; Guo, Y.; Ma, E.; Zhu, K.Y. Genomics-based approaches to screening carboxylesterase-like genes potentially involved in malathion resistance in oriental migratory locust (Locusta migratoria manilensis). Pest Manag. Sci. 2011, 67, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Zhang, M.; Qin, G.; Li, D.; Zhu, K.Y.; Ma, E.; Zhang, J. Molecular cloning, characterization and positively selected sites of the glutathione S-transferase family from Locusta migratoria. PLoS ONE 2014, 9, e114776. [Google Scholar] [CrossRef] [PubMed]
- Ketting, R.F. The many faces of RNAi. Dev. Cell 2011, 20, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.C.; Miao, X.X. Feasibility, limitation and possible solutions of RNAi-based technology for insect pest control. Insect Sci. 2013, 20, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Huvenne, H.; Smagghe, G. Mechanisms of dsRNA uptake in insects and potential of RNAi for pest control: A review. J. Insect Physiol. 2010, 56, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, X.; Yu, D.; Kang, L. The SID-1 double-stranded RNA transporter is not required for systemic RNAi in the migratory locust. RNA Biol. 2012, 9, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Wynant, N.; Verlinden, H.; Breugelmans, B.; Simonet, G.; Vanden Broeck, J. Tissue-dependence and sensitivity of the systemic RNA interference response in the desert locust, Schistocerca gregaria. Insect Biochem. Mol. Biol. 2012, 42, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Wynant, N.; Santos, D.; Verdonck, R.; Spit, J.; van Wielendaele, P.; Vanden Broeck, J. Identification, functional characterization and phylogenetic analysis of double stranded RNA degrading enzymes present in the gut of the desert locust, Schistocerca gregaria. Insect Biochem. Mol. Biol. 2014, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tomoyasu, Y.; Miller, S.C.; Tomita, S.; Schoppmeier, M.; Grossmann, D.; Bucher, G. Exploring systemic RNA interference in insects: A genome-wide survey for RNAi genes in Tribolium. Genome Biol. 2008, 9, R10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Allan, A.C.; Li, C.; Wang, Y.; Yao, Q. De Novo Assembly and Characterization of the Transcriptome of the Chinese Medicinal Herb, Gentiana rigescens. Int. J. Mol. Sci. 2015, 16, 11550–11573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hao, Y.; Si, F.; Ren, S.; Hu, G.; Shen, L.; Chen, B. The De Novo Transcriptome and Its Analysis of the Worldwide Vegetable Pest, Delia antiqua (Diptera: Anthomyiidae). G3 (Bethesda) 2014, 4, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Silva, I.; Whitney, J.; Wainwright, B.; Andrews, K.R.; Ylitalo-Ward, H.; Bowen, B.W.; Toonen, R.J.; Goetze, E.; Karl, S.A. Microsatellites for next-generation ecologists: A post-sequencing bioinformatics pipeline. PLoS ONE 2013, 8, e55990. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Tijet, N.; Helvig, C.; Feyereisen, R. The cytochrome P450 gene superfamily in Drosophila melanogaster: Annotation, intron-exon organization and phylogeny. Gene 2001, 262, 189–198. [Google Scholar] [CrossRef]
- Wynant, N.; Santos, D.; Vanden Broeck, J. Biological mechanisms determining the success of RNA interference in insects. Int. Rev. Cell Mol. Biol. 2014, 312, 139–167. [Google Scholar] [PubMed]
- Lv, J.; Liu, P.; Gao, B.; Wang, Y.; Wang, Z.; Chen, P.; Li, J. Transcriptome analysis of the Portunus trituberculatus: De novo assembly, growth-related gene identification and marker discovery. PLoS ONE 2014, 9, e94055. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, O.; Bai, X.; Mamidala, P.; Rajarapu, S.P.; Bonello, P.; Herms, D.A. Tissue-specific transcriptomics of the exotic invasive insect pest emerald ash borer (Agrilus planipennis). PLoS ONE 2010, 5, e13708. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.D.; Chen, E.H.; Ding, T.B.; Chen, S.C.; Dou, W.; Wang, J.J. De novo assembly, gene annotation, and marker discovery in stored-product pest Liposcelis entomophila (Enderlein) using transcriptome sequences. PLoS ONE 2013, 8, e80046. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Singh, H.; Dixit, S.; Mendu, V.; Verma, P.C. Molecular Characterization of Vitellogenin and Vitellogenin Receptor of Bemisia tabaci. PLoS ONE 2016, 11, e0155306. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Luan, J.B.; Li, J.M.; Bao, Y.Y.; Zhang, C.X.; Liu, S.S. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genom. 2010, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Feyereisen, R. Evolution of insect P450. Biochem. Soc. Trans. 2006, 34, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Claudianos, C.; Ranson, H.; Johnson, R.M.; Biswas, S.; Schuler, M.A.; Berenbaum, M.R.; Feyereisen, R.; Oakeshott, J.G. A deficit of detoxification enzymes: Pesticide sensitivity and environmental response in the honeybee. Insect Mol. Biol. 2006, 15, 615–636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, D.; Ge, P.; Guo, Y.; Zhu, K.Y.; Ma, E.; Zhang, J. Molecular and functional characterization of cDNAs putatively encoding carboxylesterases from the migratory locust, Locusta migratoria. PLoS ONE 2014, 9, e94809. [Google Scholar] [CrossRef] [PubMed]
- Dabour, N.; Bando, T.; Nakamura, T.; Miyawaki, K.; Mito, T.; Ohuchi, H.; Noji, S. Cricket body size is altered by systemic RNAi against insulin signaling components and epidermal growth factor receptor. Dev. Growth Differ. 2011, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Belles, X. Beyond Drosophila: RNAi in vivo and functional genomics in insects. Annu. Rev. Entomol. 2010, 55, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Karatolos, N.; Williamson, M.S.; Denholm, I.; Gorman, K.; Ffrench-Constant, R.H.; Bass, C. Over-expression of a cytochrome P450 is associated with resistance to pyriproxyfen in the greenhouse whitefly Trialeurodes vaporariorum. PLoS ONE 2012, 7, e31077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edi, C.V.; Djogbenou, L.; Jenkins, A.M.; Regna, K.; Muskavitch, M.A.; Poupardin, R.; Jones, C.M.; Essandoh, J.; Ketoh, G.K.; Paine, M.J.; et al. CYP6 P450 enzymes and ACE-1 duplication produce extreme and multiple insecticide resistance in the malaria mosquito Anopheles gambiae. PLoS Genet. 2014, 10, e1004236. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wu, H.; Zhang, X.; Ma, E.; Guo, Y.; Zhu, K.Y.; Zhang, J. RNA interference of cytochrome P450 CYP6F subfamily genes affects susceptibility to different insecticides in Locusta migratoria. Pest Manag. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Grubor, V.D.; Heckel, D.G. Evaluation of the role of CYP6B cytochrome P450s in pyrethroid resistant Australian Helicoverpa armigera. Insect Mol. Biol. 2007, 16, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Ma, J.; Zhao, J. Silencing of cytochrome P450 CYP6B6 gene of cotton bollworm (Helicoverpa armigera) by RNAi. Bull. Entomol. Res. 2013, 103, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Zhao, C.; Feng, X.; Liu, X.; Qiu, L. Knockdown of several components of cytochrome P450 enzyme systems by RNA interference enhances the susceptibility of Helicoverpa armigera to fenvalerate. Pest Manag. Sci. 2012, 68, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, D.; Ge, P.; Yang, M.; Guo, Y.; Zhu, K.Y.; Ma, E. RNA interference revealed the roles of two carboxylesterase genes in insecticide detoxification in Locusta migratoria. Chemosphere 2013, 93, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Duan, X.; Lv, Y.; Zhang, X.; Nie, Z.; Xie, C.; Ni, Z.; Liang, R. Silencing of an aphid carboxylesterase gene by use of plant-mediated RNAi impairs Sitobion avenae tolerance of Phoxim insecticides. Transgenic Res. 2014, 23, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Kola, V.S.; Renuka, P.; Madhav, M.S.; Mangrauthia, S.K. Key enzymes and proteins of crop insects as candidate for RNAi based gene silencing. Front. Physiol. 2015, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Winston, W.M.; Molodowitch, C.; Hunter, C.P. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science 2002, 295, 2456–2459. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.J.; Chen, T.; Ma, X.F.; Xue, J.; Pan, P.L.; Zhang, X.C.; Cheng, J.A.; Zhang, C.X. Genome-wide screening for components of small interfering RNA (siRNA) and micro-RNA (miRNA) pathways in the brown planthopper, Nilaparvata lugens (Hemiptera: Delphacidae). Insect Mol. Biol. 2013, 22, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Tribolium Genome Sequencing, C.; Richards, S.; Gibbs, R.A.; Weinstock, G.M.; Brown, S.J.; Denell, R.; Beeman, R.W.; Gibbs, R.; Beeman, R.W.; Brown, S.J.; et al. The genome of the model beetle and pest Tribolium castaneum. Nature 2008, 452, 949–955. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B. J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. ISMB 1999, 99, 138–148. [Google Scholar]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011, 27, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Adult | Larvae | All |
|---|---|---|---|
| Raw reads | 58,063,258 | 62,093,046 | 120,156,304 |
| Total clean reads | 51,299,516 | 54,109,362 | 105,408,878 |
| Total Nucleotides (bp) | 4,616,956,440 | 4,869,842,580 | - |
| Q20 1 percentage (%) | 97.45% | 97.42% | - |
| N percentage (%) | 0 | 0 | - |
| Total length of contigs (nt) | 39,306,387 | 54,595,446 | - |
| Total length of unigenes (nt) | 35,186,305 | 49,464,707 | - |
| Number of unigenes | 69,189 | 93,948 | 74,657 |
| Mean size of contigs (nt) | 290 | 292 | - |
| Mean size of unigenes (nt) | 509 | 527 | 680 |
| N50 2 of contigs (nt) | 428 | 445 | |
| N50 2 of unigenes (nt) | 722 | 800 | 1057 |
| GC content (%) | 47.93% | 45.31% | - |
| Database Name | Nr | Nt | Swiss-Prot | KEGG | COG | GO | All |
|---|---|---|---|---|---|---|---|
| Numbers | 25,652 | 12,671 | 20,438 | 18,053 | 9558 | 12,061 | 28,173 |
| KEGG Pathways | Sub-Pathways | Numbers of Unigenes |
|---|---|---|
| Metabolism | Metabolic pathways | 2520 |
| Carbohydrate metabolism | 978 | |
| Energy metabolism | 204 | |
| Lipid metabolism | 745 | |
| Nucleotide metabolism | 575 | |
| Amino acid metabolism | 895 | |
| Metabolism of other amino acids | 288 | |
| Glycan biosynthesis and metabolism | 493 | |
| Metabolism of cofactors and vitamins | 422 | |
| Metabolism of terpenoids and polyketides | 184 | |
| Biosynthesis of other secondary metabolites | 22 | |
| Xenobiotics biodegradation and metabolism | 345 | |
| Genetic Information Processing | Transcription | 945 |
| Translation | 1398 | |
| Folding, sorting and degradation | 1115 | |
| Replication and repair | 337 | |
| Environmental Information Processing | Membrane transport | 313 |
| Signal transduction | 1603 | |
| Signaling molecules and interaction | 884 | |
| Cellular Processes | Transport and catabolism | 1262 |
| Cell motility | 804 | |
| Cell growth and death | 597 | |
| Cellular community | 1218 | |
| Organismal Systems | Immune system | 1307 |
| Endocrine system | 907 | |
| Circulatory system | 475 | |
| Digestive system | 1399 | |
| Excretory system | 454 | |
| Nervous system | 806 | |
| Sensory system | 165 | |
| Development | 538 | |
| Environmental adaptation | 89 |
| Gene | Unigene | Length (bp) | E-Value | Identity | Species Homologue | ACCESSION |
|---|---|---|---|---|---|---|
| mirRNA | ||||||
| Ago-1 | Unigene6375_SsASsL | 1122 | 0 | 100% | L. migratoria | AGO85968 |
| Unigene24483_SsASsL | 612 | 7.00 × 10−122 | 100% | L .migratoria | AGO85968 | |
| Unigene10467_SsASsL | 444 | 3.00 × 10−82 | 100% | L. migratoria | AGO85968 | |
| Loqs | Unigene5566_SsASsL | 345 | 3.00 × 10−121 | 65% | T. castaneum | XP_966668 |
| Drosha | Unigene22829_SsASsL | 339 | 2.00 × 10−47 | 78% | T. castaneum | XP_967454 |
| Unigene718_SsASsL | 300 | 3.00 × 10−67 | 74% | T. castaneum | XP_967454 | |
| Unigene18278_SsASsL | 312 | 3.00 × 10−51 | 78% | T. castaneum | XP_967454 | |
| Pasha | Unigene1479_SsASsL | 672 | 5.00 × 10−27 | 59% | T. castaneum | XP_971282 |
| Exportin-5 | Unigene24927_SsASsL | 1098 | 2.00 × 10−105 | 60% | T. castaneum | XP_974696 |
| siRNA | ||||||
| Dicer-2 | Unigene11038_SsASsL | 531 | 4.00 × 10−88 | 87% | S. gregaria | AFY13245 |
| Unigene24754_SsASsL | 237 | 7.00 × 10−65 | 78% | S. gregaria | AFY13245 | |
| Ago-2 | Unigene19329_SsASsL | 1737 | 0 | 88% | S. gregaria | AFY13246 |
| 0 | 72% | L. migratoria | AGO85972 | |||
| Unigene24512_SsASsL | 2742 | 0 | 78% | L. migratoria | AGO85972 | |
| 0 | 68% | S. gregaria | AFY13246 | |||
| Sid-1 | Unigene8511_SsASsL | 234 | 4.00 × 10−142 | 96% | L. migratoria | AFQ00936 |
| Unigene18465_SsASsL | 624 | 4.00 × 10−99 | 92% | L. migratoria | AFQ00936 | |
| Unigene24347_SsASsL | 474 | 2.00 × 10−75 | 99% | L. migratoria | AFQ00936 | |
| Unigene10814_SsASsL | 351 | 8.00 × 10−47 | 94% | L. migratoria | AFQ00936 | |
| Unigene39691_SsASsL | 201 | 7.00 × 10−29 | 92% | L. migratoria | AFQ00936 | |
| piRNA | ||||||
| Ago-3 | Unigene11235_SsASsL | 1062 | 4.00 × 10−92 | 45% | T. castaneum | EFA02921 |
| Piwi3 | Unigene24609_SsASsL | 2439 | 0 | 49% | L. migratoria | AGO85971 |
| Aub | Unigene11344_SsASsL | 1212 | 8.00 × 10−135 | 54% | T. castaneum | XP_001811159 |
| Zucchini | Unigene5197_SsASsL | 684 | 1.00 × 10−20 | 32% | T. castaneum | EEZ99465 |
| Eri-1 | Unigene17265_SsASsL | 609 | 4.00 × 10−121 | 93% | S. gregaria | JX516790 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Z.; Liu, F.; Lu, H.; Yuan, H.; Zhang, Q.; Huang, Y. De Novo Assembly and Characterization of the Transcriptome of Grasshopper Shirakiacris shirakii. Int. J. Mol. Sci. 2016, 17, 1110. https://doi.org/10.3390/ijms17071110
Qiu Z, Liu F, Lu H, Yuan H, Zhang Q, Huang Y. De Novo Assembly and Characterization of the Transcriptome of Grasshopper Shirakiacris shirakii. International Journal of Molecular Sciences. 2016; 17(7):1110. https://doi.org/10.3390/ijms17071110
Chicago/Turabian StyleQiu, Zhongying, Fei Liu, Huimeng Lu, Hao Yuan, Qin Zhang, and Yuan Huang. 2016. "De Novo Assembly and Characterization of the Transcriptome of Grasshopper Shirakiacris shirakii" International Journal of Molecular Sciences 17, no. 7: 1110. https://doi.org/10.3390/ijms17071110