
Kynurenine Aminotransferase Isozyme Inhibitors: A Review

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
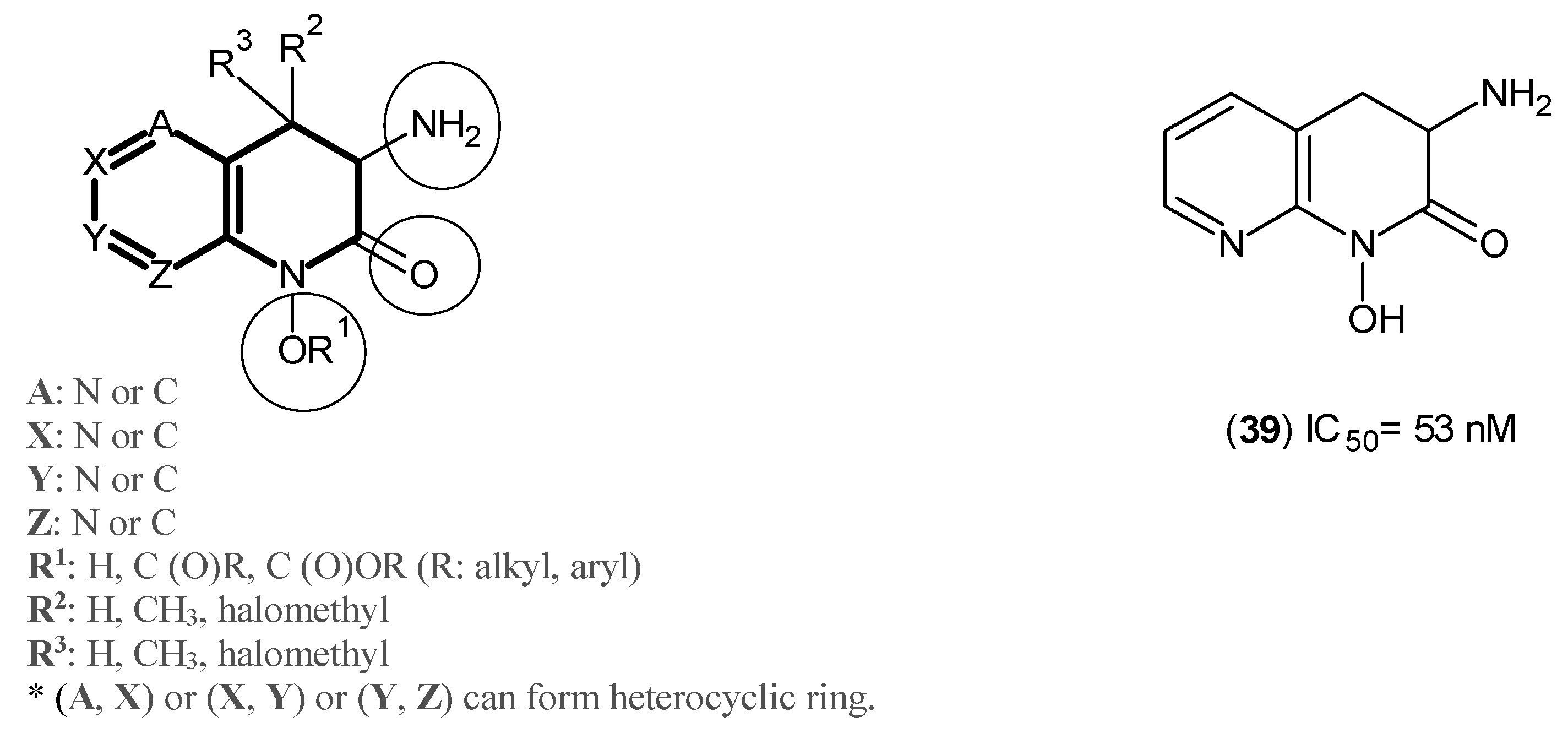{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
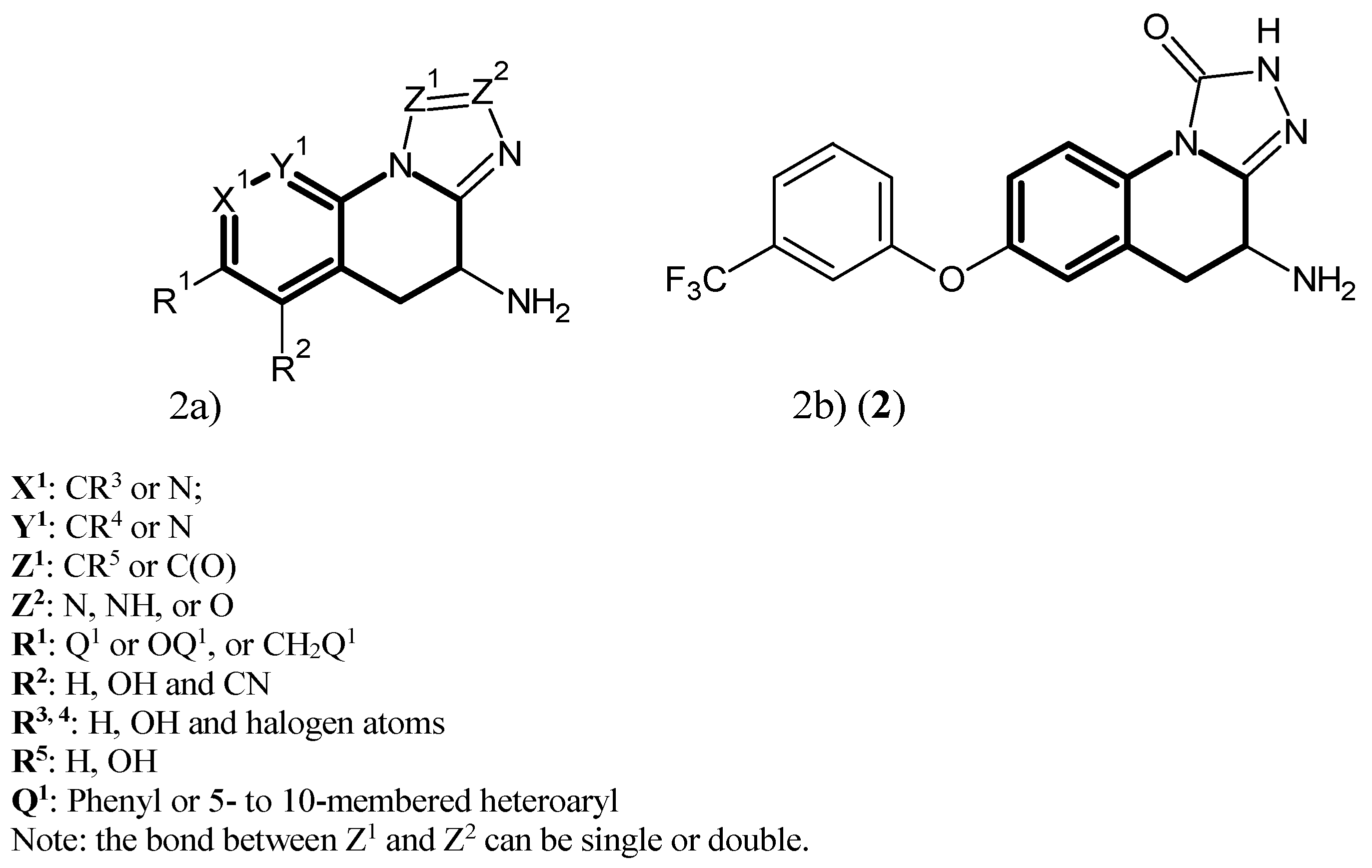
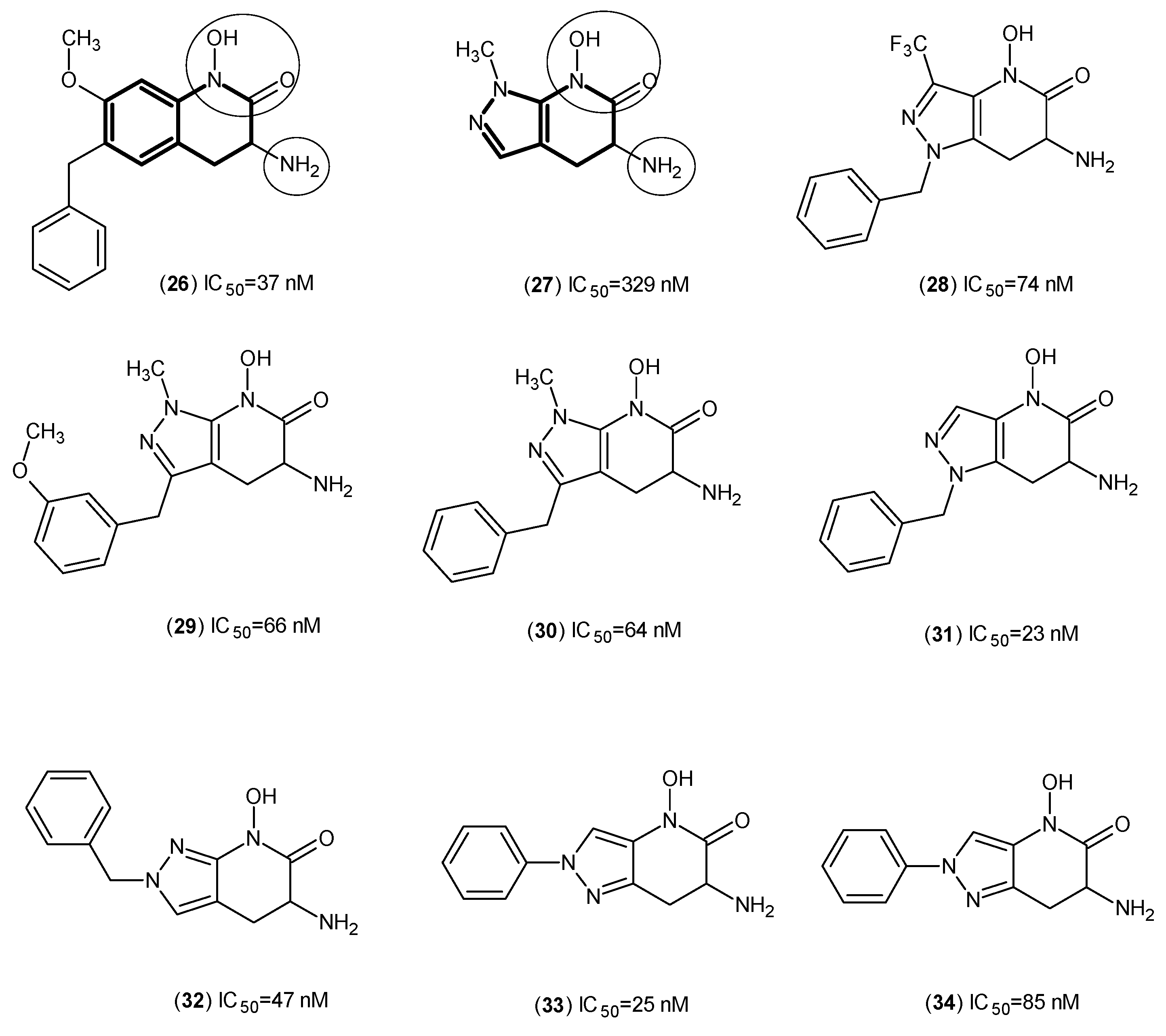
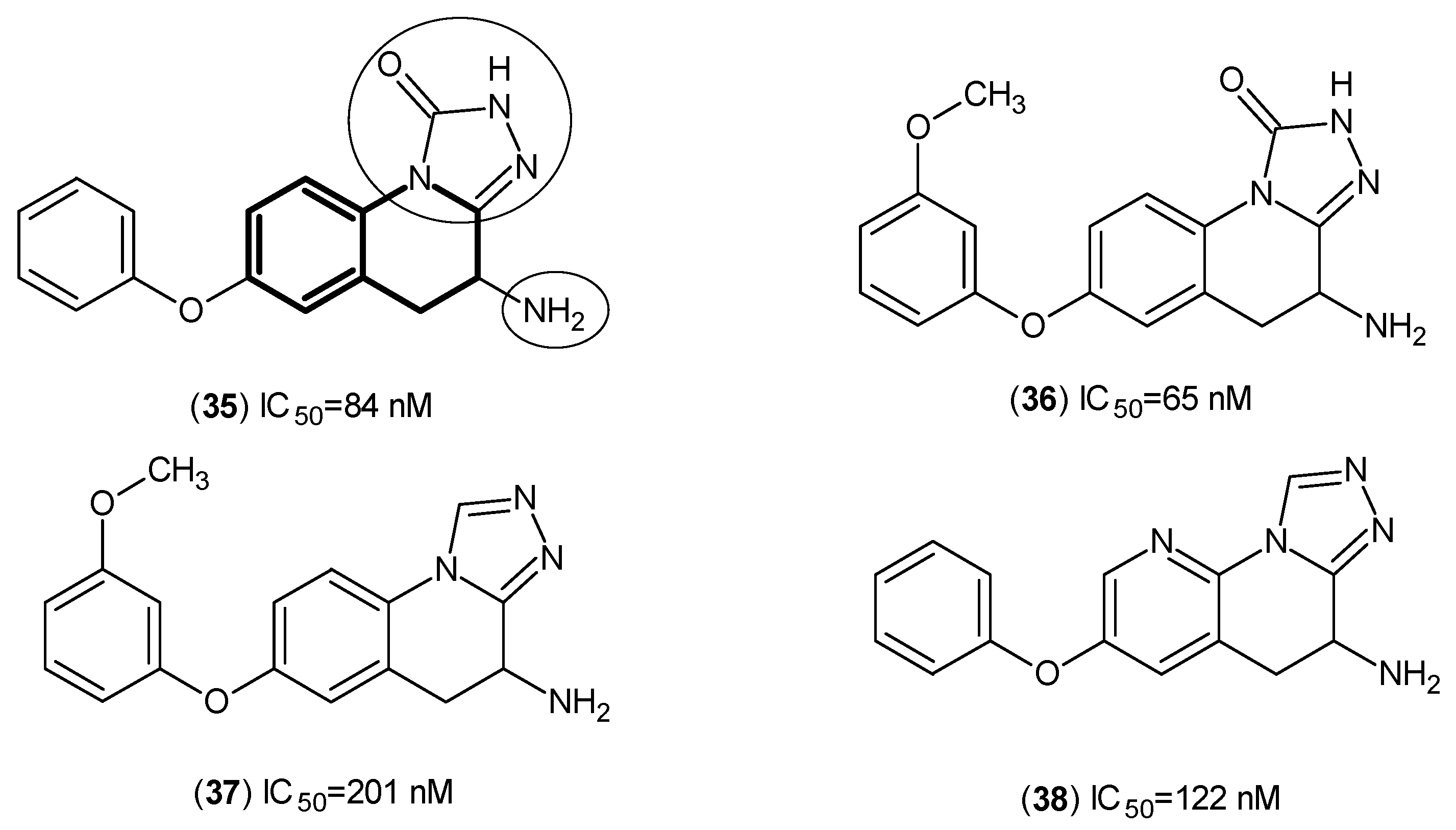
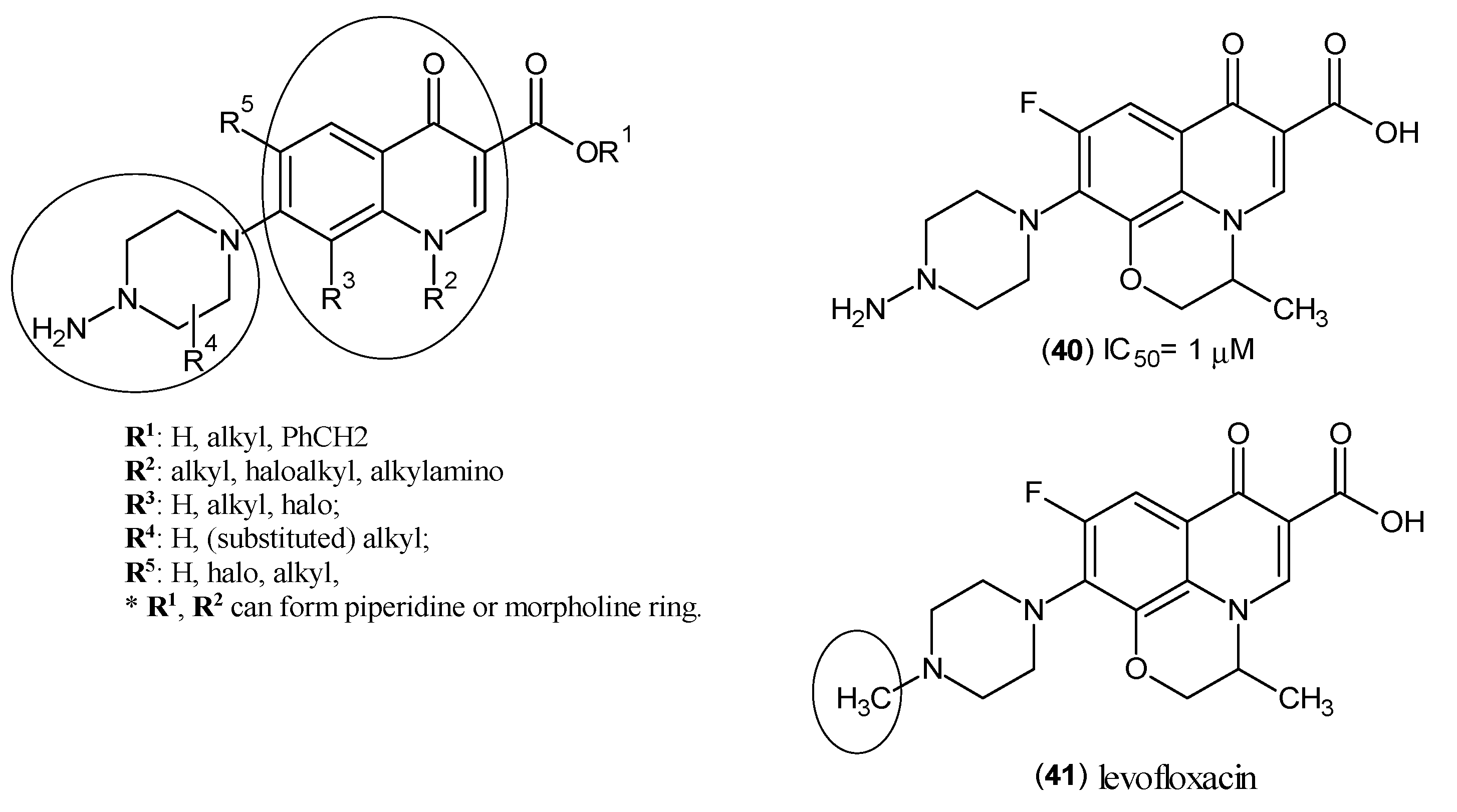
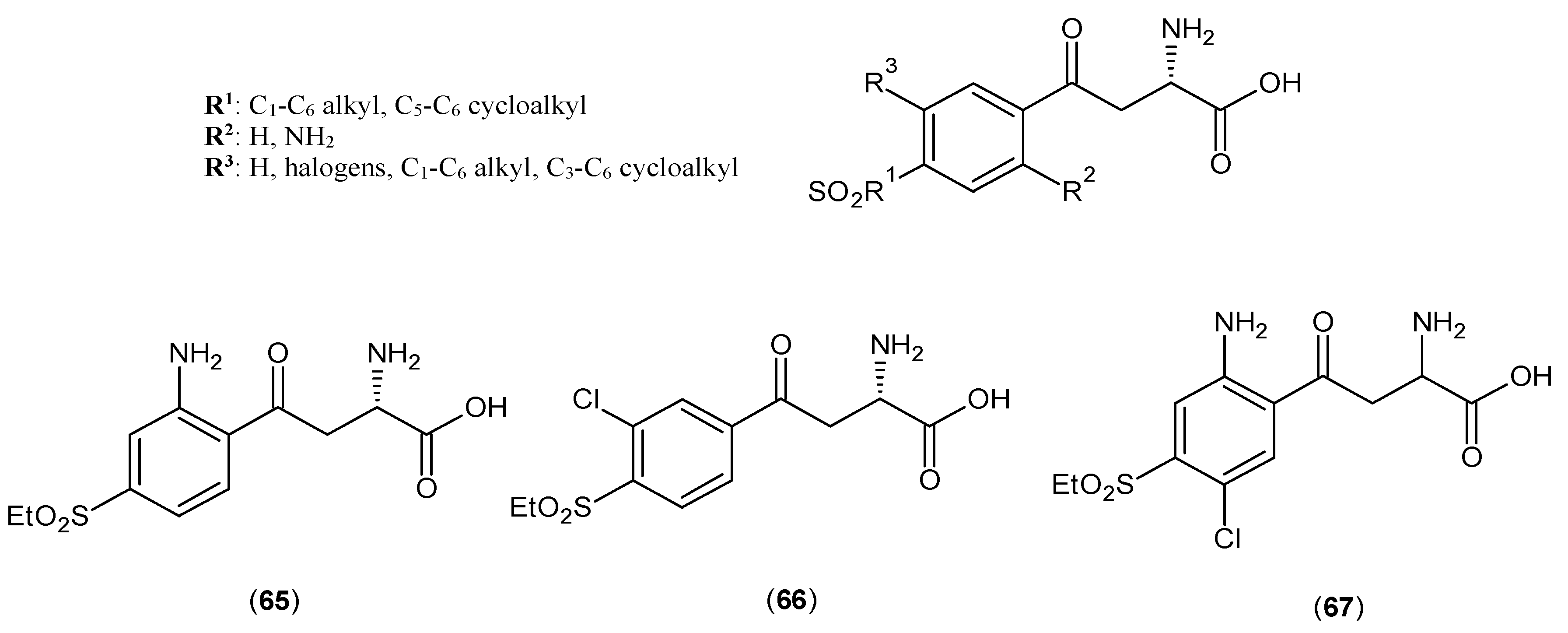
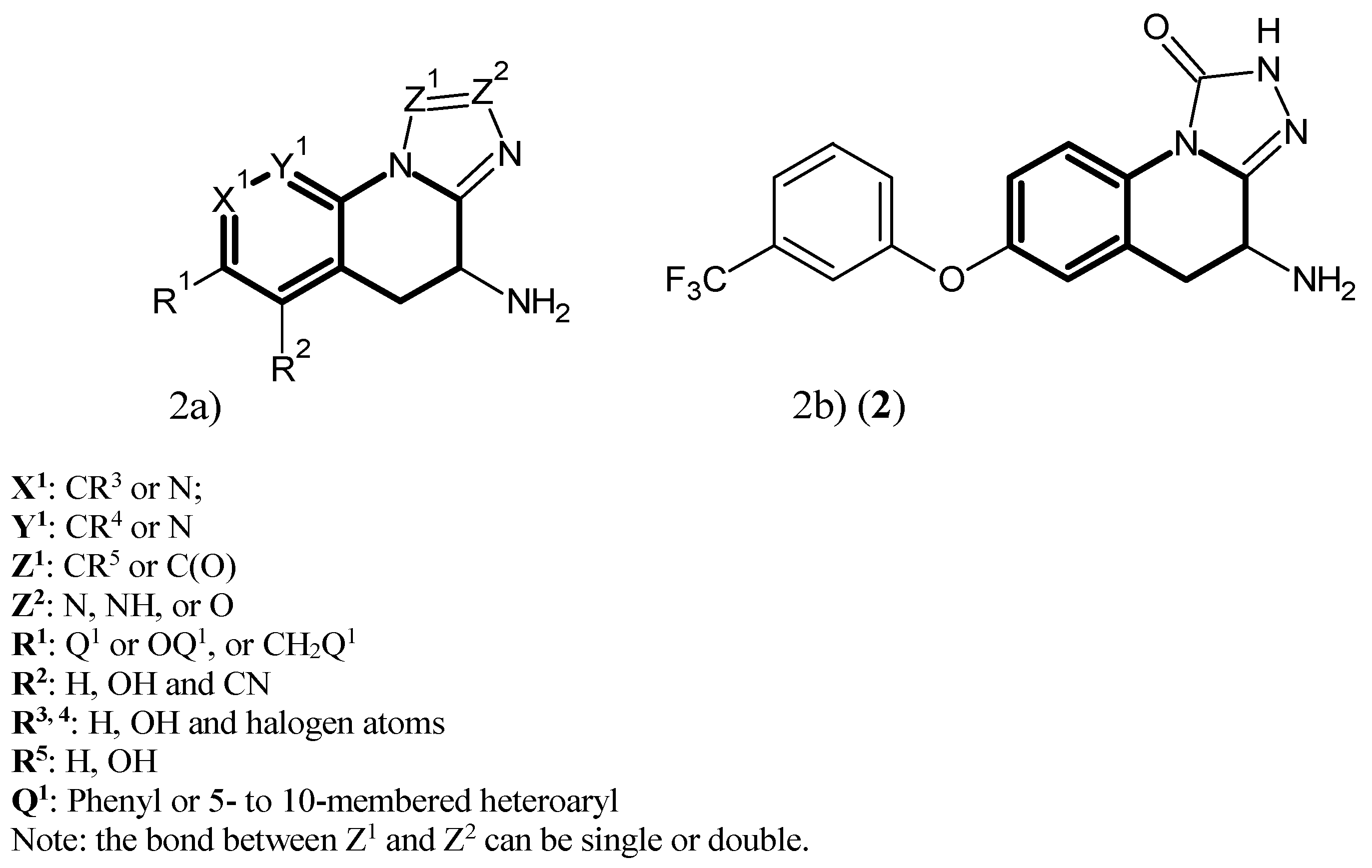
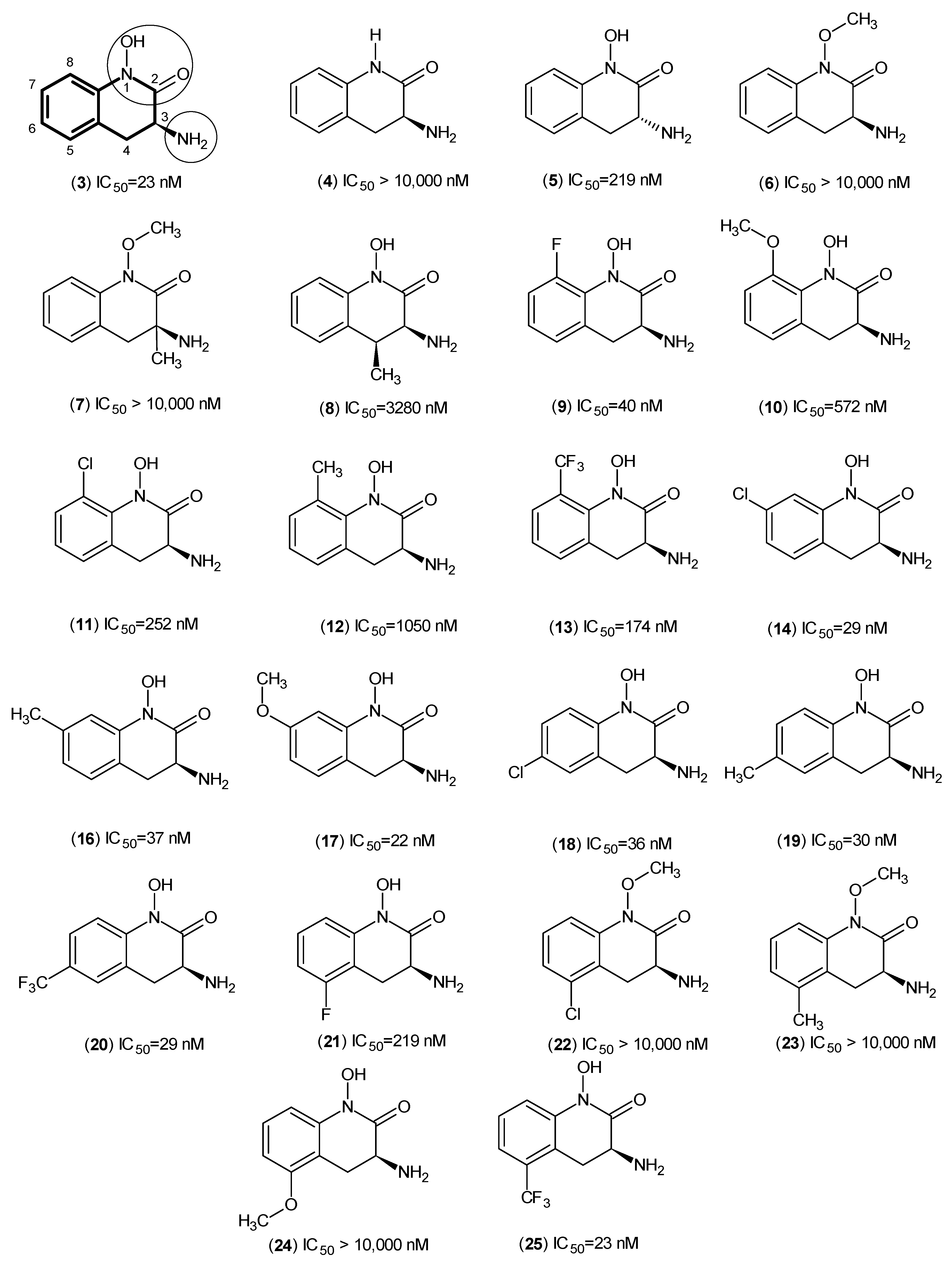
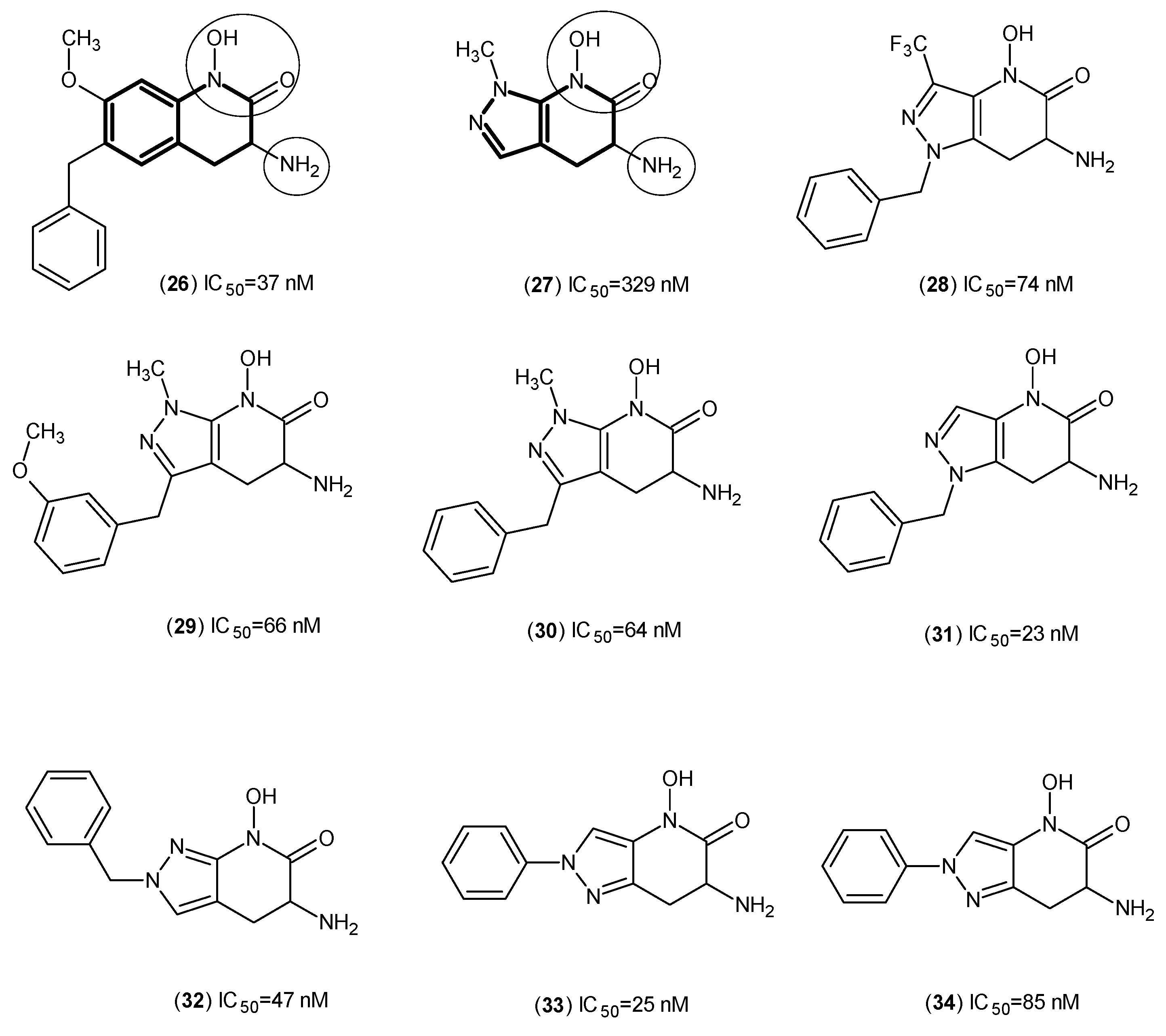
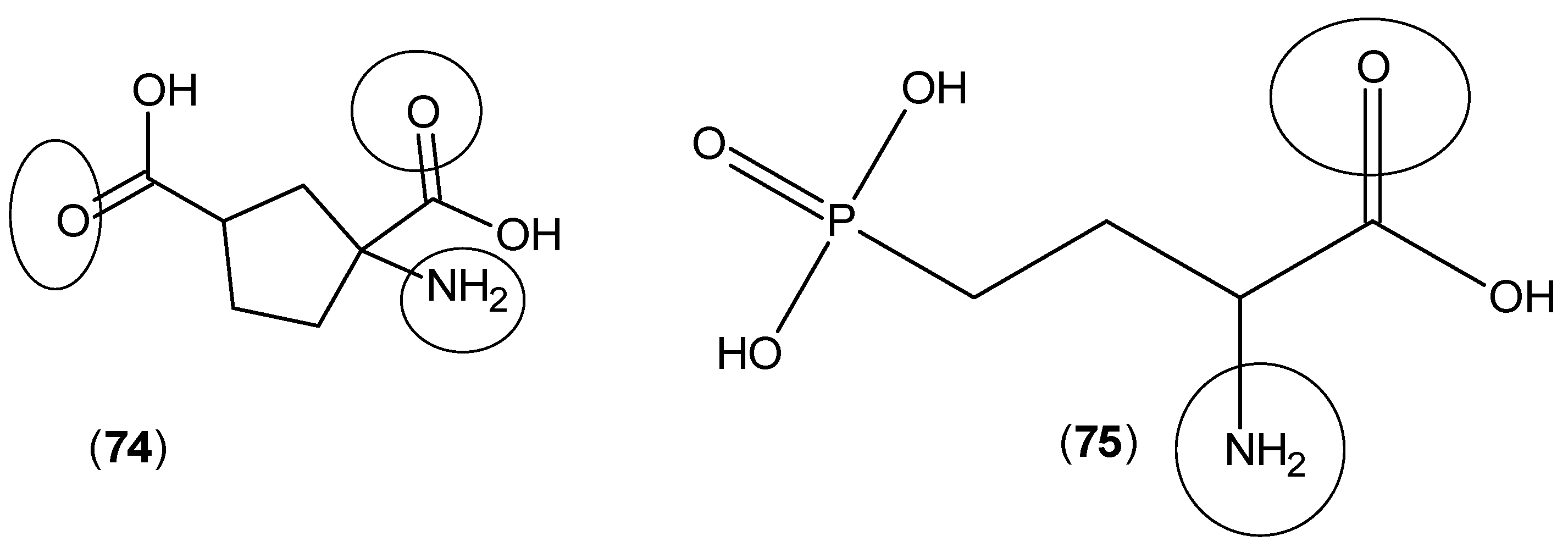
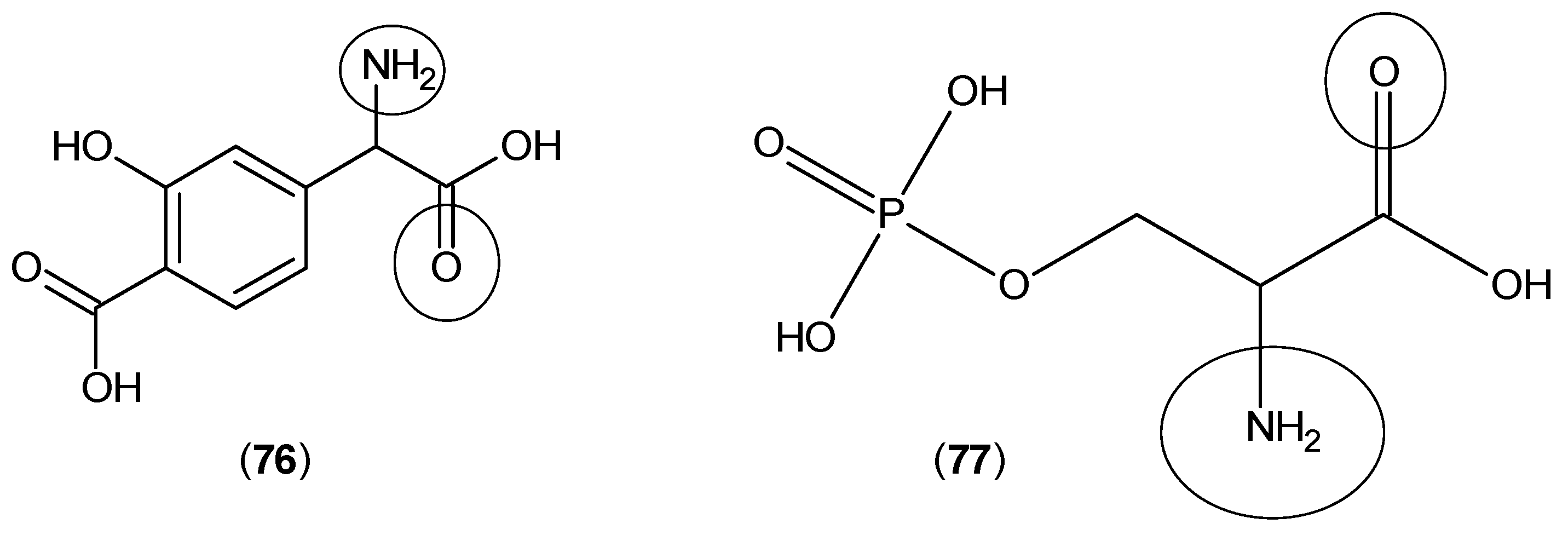
2. Kynurenine Aminotransferases Inhibitors
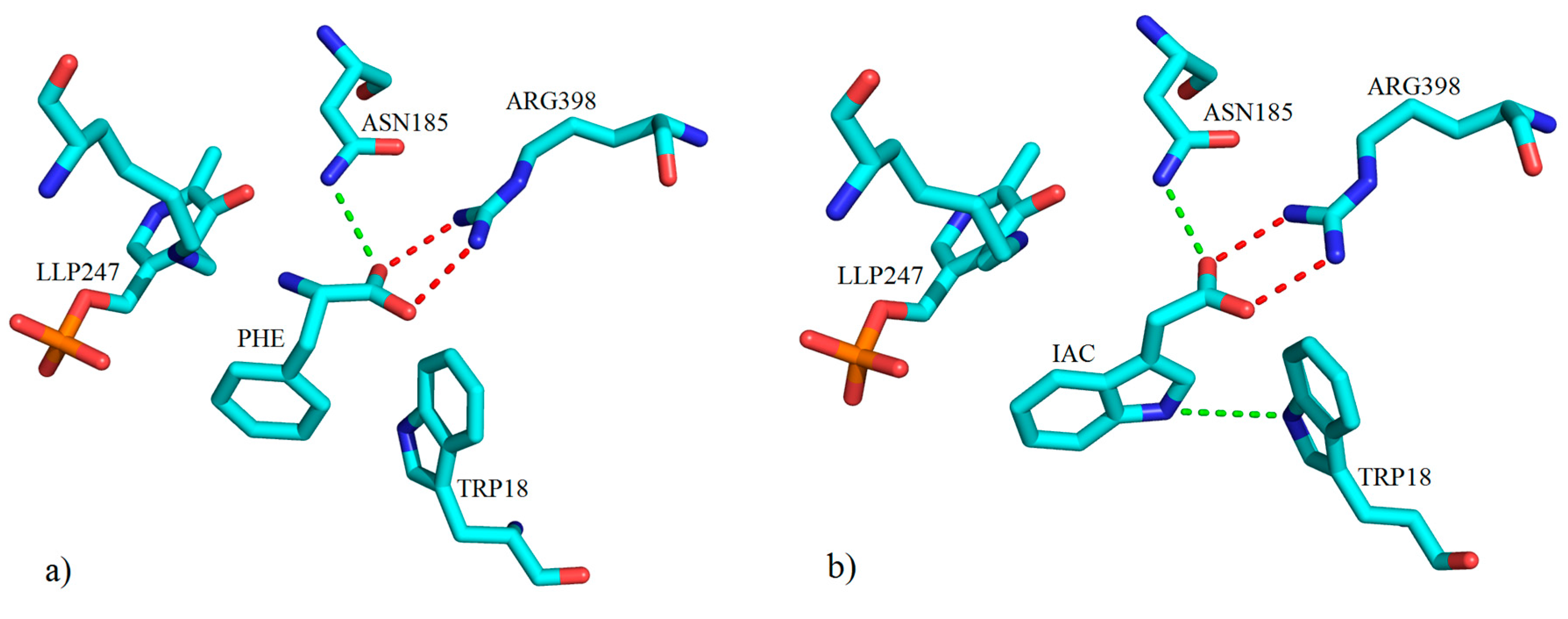
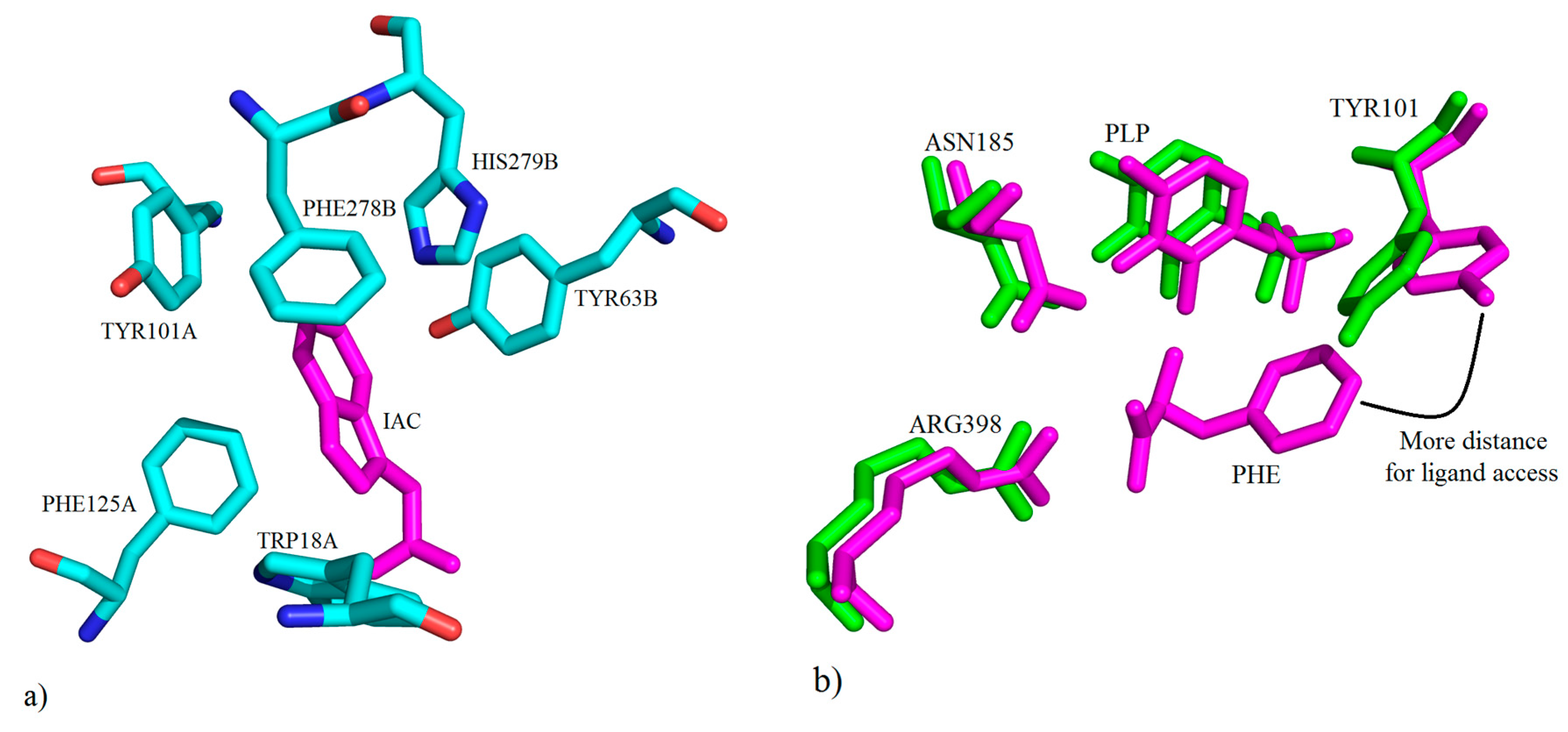
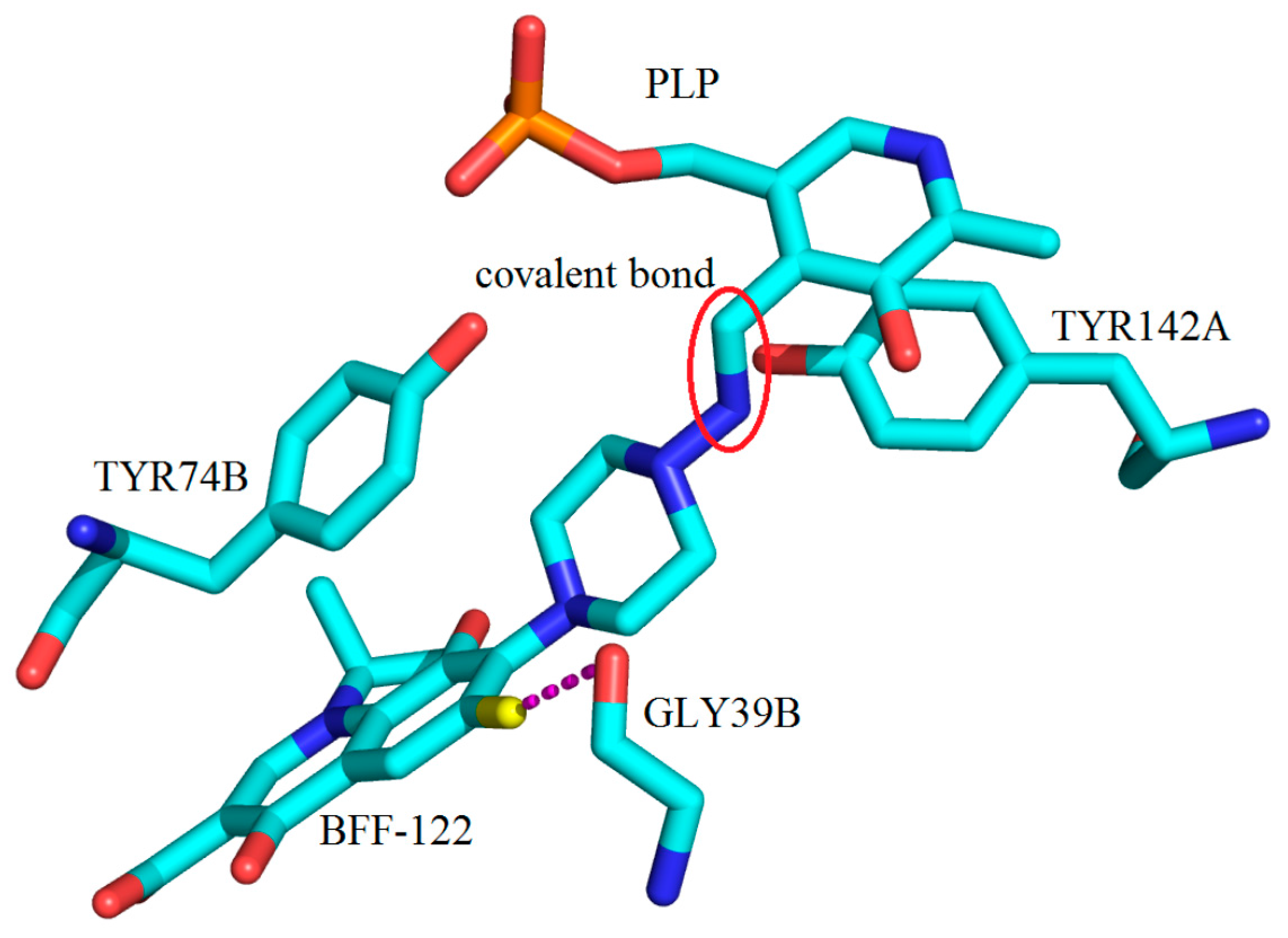
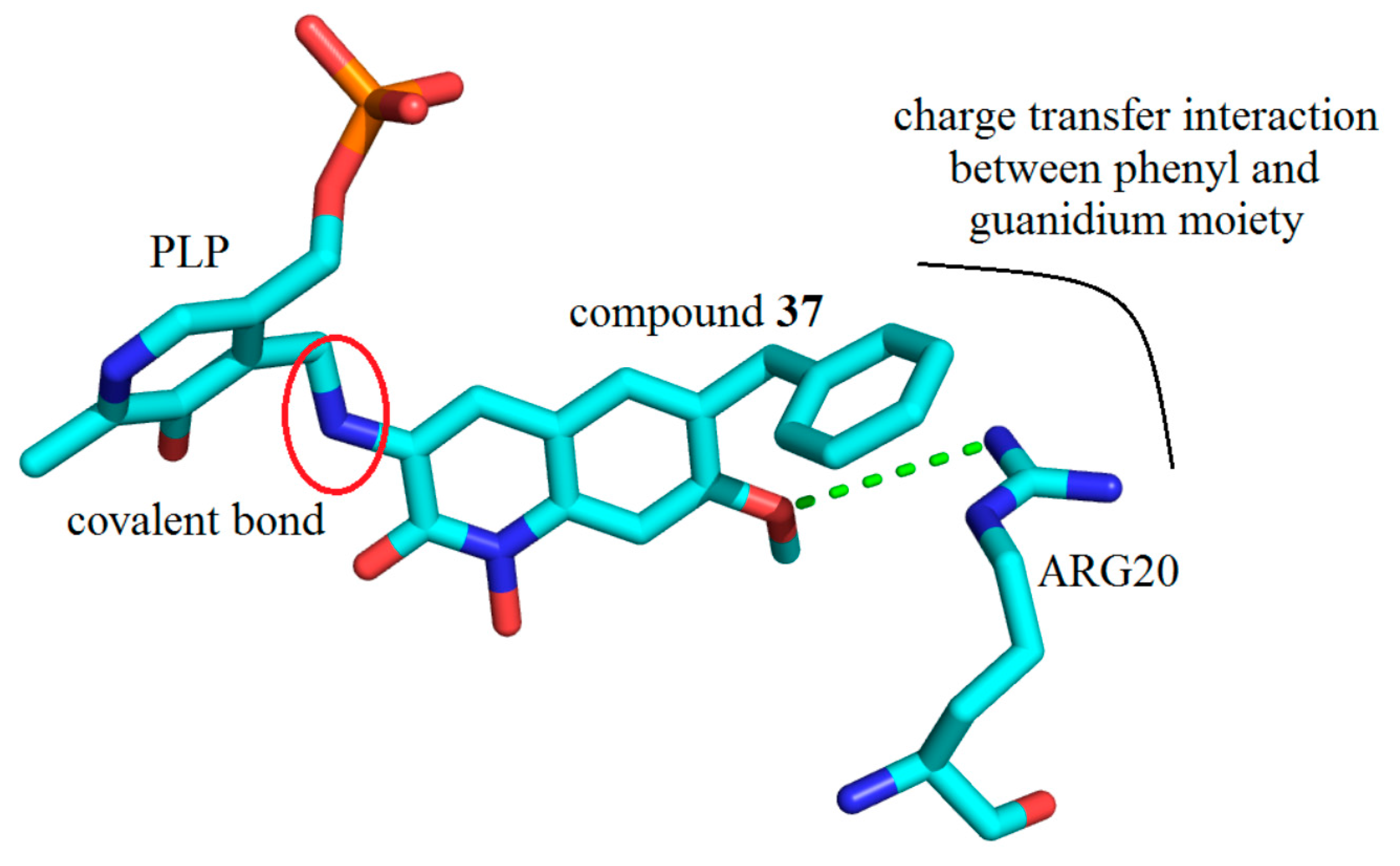
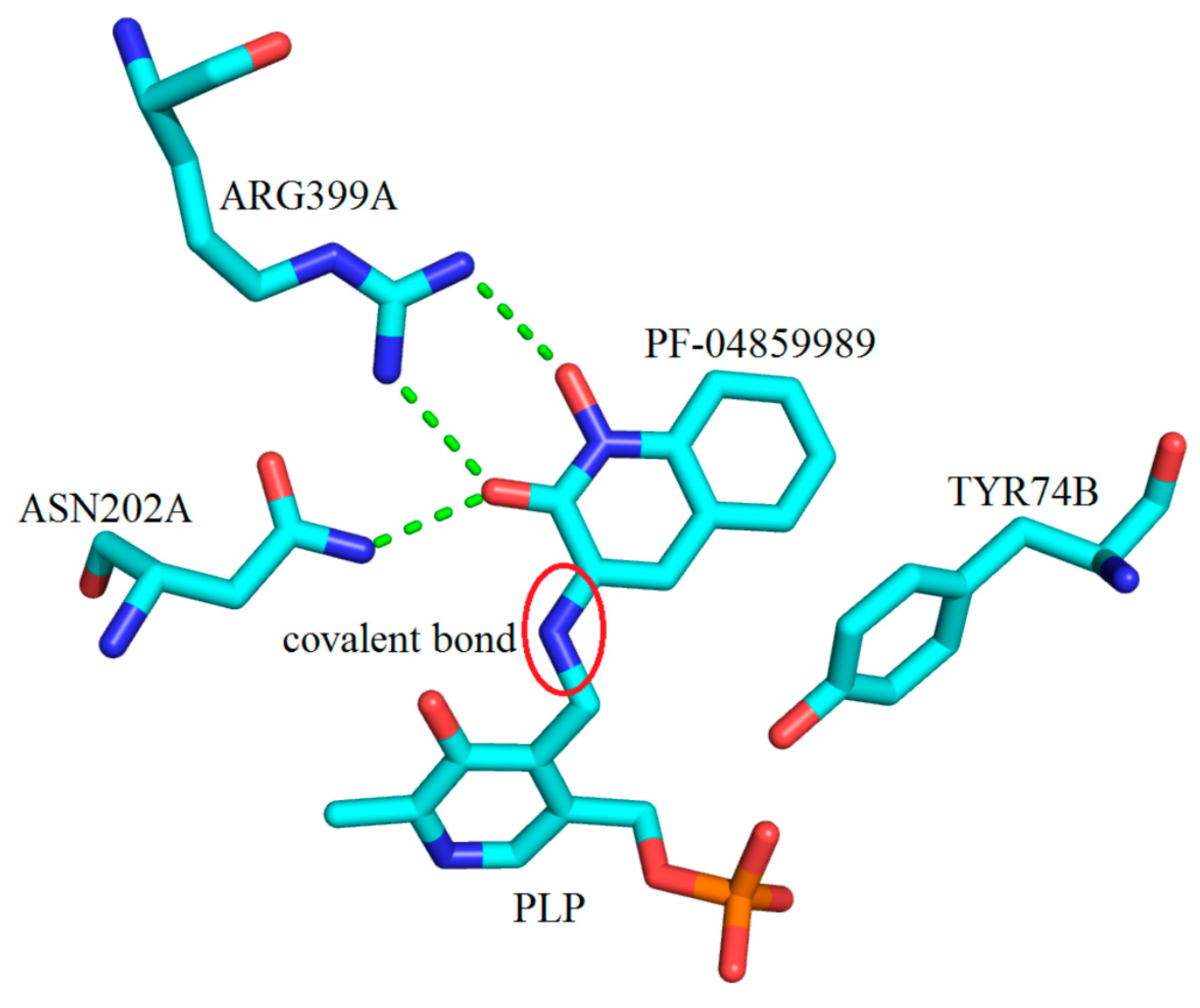
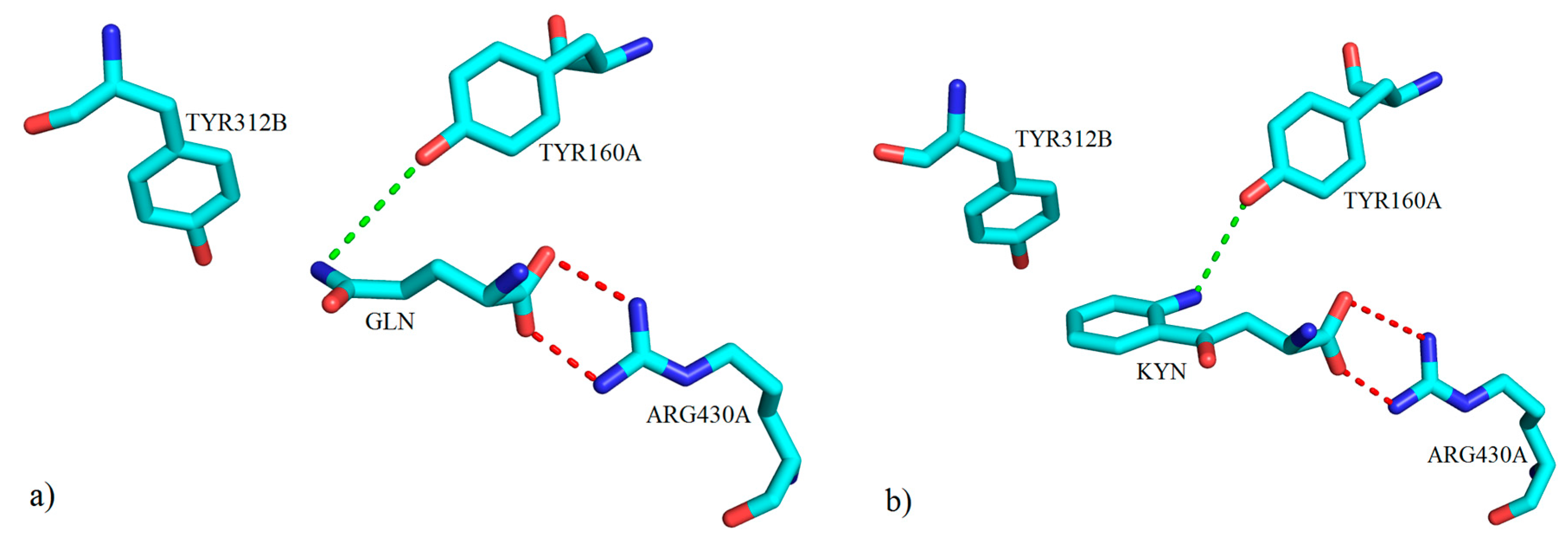
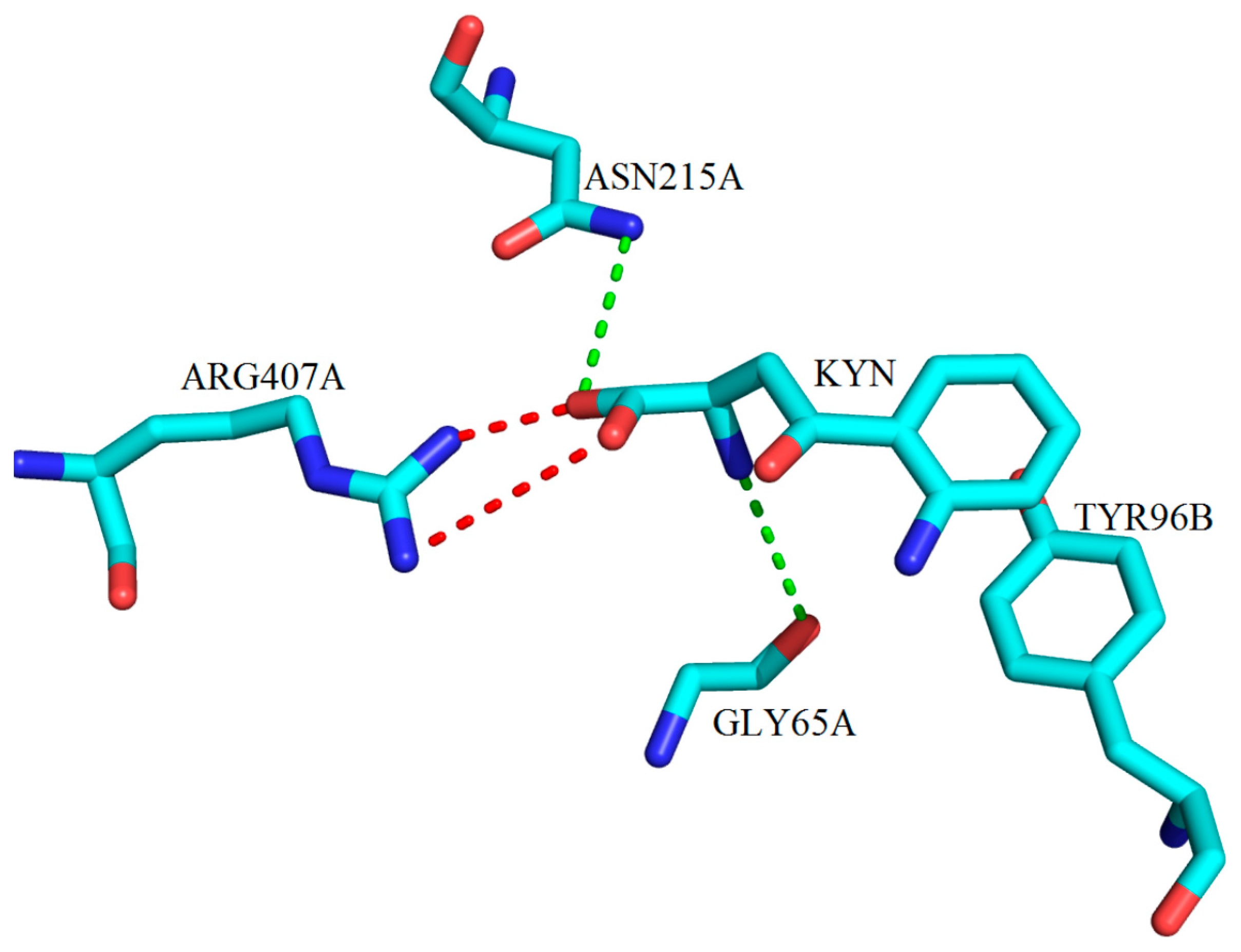
3. Interactions of Inhibitors and KATs
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jayawickrama, G.S.; Sadig, R.R.; Sun, G.; Nematollahi, A.; Nadvi, N.A.; Hanrahan, J.R.; Gorrell, M.D.; Church, W.B. Kynurenine aminotransferases and the prospects of inhibitors for the treatment of schizophrenia. Curr. Med. Chem. 2015, 22, 2902–2918. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, A.; Church, W.B.; Nadvi, N.A.; Gorrell, M.D.; Sun, G. Homology modeling of human kynurenine aminotransferase III and observations on inhibitor binding using molecular docking. Cent. Nerv. Syst. Agents Med. Chem. 2014, 14, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Jansonius, J.N. Structure, evolution and action of vitamin B6-dependent enzymes. Curr. Opin. Struct. Biol. 1998, 8, 759–769. [Google Scholar] [CrossRef]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J.Y. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell. Mol. Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Garavaglia, S.; Montalbano, V.; Walsh, M.A.; Rizzi, M. Crystal structure of human kynurenine aminotransferase II, a drug target for the treatment of schizophrenia. J. Biol. Chem. 2008, 283, 3559–3566. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Robinson, H.; Cai, T.; Tagle, D.A.; Li, J.Y. Structural insight into the inhibition of human kynurenine aminotransferase I/glutamine transaminase K. J. Med. Chem. 2009, 52, 2786–2793. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Cai, T.; Tagle, D.A.; Robinson, H.; Li, J.Y. Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci. Rep. 2008, 28, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Valentina, C.; Garavaglia, S.; Sathyasaikumar, K.V.; Schwarcz, R.; Kojima, S.; Okuwaki, K.; Ono, S.; Kajii, Y.; Rizzi, M. Crystal structure-based selective targeting of the pyridoxal 5′-phosphate dependent enzyme kynurenine aminotransferase II for cognitive enhancement. J. Med. Chem. 2010, 53, 5684–5689. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, J.B.; Anderson, M.; Bechle, B.M.; Campbell, B.M.; Chang, C.; Dounay, A.B.; Evrard, E.; Fonseca, K.R.; Gan, X.M.; Ghosh, S.; et al. Structure-based design of irreversible human KAT II inhibitors: Discovery of new potency-enhancing interactions. ACS Med. Chem. Lett. 2013, 4, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Dounay, A.B.; Anderson, M.; Bechle, B.M.; Evrard, E.; Gan, X.M.; Kim, J.Y.; McAllister, L.A.; Pandit, J.; Rong, S.B.; Salafia, M.A.; et al. PF-04859989 as a template for structure-based drug design: Identification of new pyrazole series of irreversible KAT II inhibitors with improved lipophilic efficiency. Bioorg. Med. Chem. Lett. 2013, 23, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, A.; Sun, G.; Harrop, S.J.; Hanrahan, J.R.; Church, W.B. Structure of the PLP-form of the human kynurenine aminotransferase II in a novel spacegroup at 1.83 Å resolution. Int. J. Mol. Sci. 2016, 17, 446. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Chang, H.; Zhou, Y. Recombinant expression, purification and crystallographic studies of the mature form of human mitochondrial aspartate aminotransferase. Biosci. Trends 2016, 10, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Robinson, H.; Cai, T.; Tagle, D.A.; Li, J. Biochemical and structural properties of mouse kynurenine aminotransferase III. Mol. Cell. Biol. 2009, 29, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J. The role of glutamine transaminase K (GTK) in sulfur and alpha-keto acid metabolism in the brain, and in the possible bioactivation of neurotoxicants. Neurochem. Int. 2004, 44, 557–577. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.L.; Meister, A. Isolation and properties of highly purified glutamine transaminase. Biochemistry 1972, 11, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Shurubor, Y.I.; Dorai, T.; Pinto, J.T.; Isakova, E.P.; Deryabina, Y.I.; Denton, T.T.; Krasnikov, B.F. ω-Amidase: An underappreciated, but important enzyme in l-glutamine and l-asparagine metabolism; relevance to sulfur and nitrogen metabolism, tumor biology and hyperammonemic diseases. Amino Acids 2016, 48, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Li, J.; Li, J. pH dependence, substrate specificity and inhibition of human kynurenine aminotransferase I. Eur. J. Biochem. 2004, 271, 4804–4814. [Google Scholar] [CrossRef] [PubMed]
- Goh, D.L.; Patel, A.; Thomas, G.H.; Salomons, G.S.; Schor, D.S.; Jakobs, C.; Geraghty, M.T. Characterization of the human gene encoding α-aminoadipate aminotransferase (AADAT). Mol. Genet. Metab. 2002, 76, 172–180. [Google Scholar] [CrossRef]
- Sun, G.; Nematollahi, A.; Nadvi, N.A.; Kwan, A.H.; Jeffries, C.M.; Church, W.B. Expression, purification and crystallization of human kynurenine aminotransferase 2 exploiting a highly optimized codon set. Protein Expr. Purif. 2016, 121, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Li, Z.; Zhang, L.; Tagle, D.A.; Cai, T. Characterization of kynurenine aminotransferase III, a novel member of a phylogenetically conserved KAT family. Gene 2006, 365, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Sellgren, C.M.; Kegel, M.E.; Bergen, S.E.; Ekman, C.J.; Olsson, S.; Larsson, M.; Vawter, M.P.; Backlund, L.; Sullivan, P.F.; Sklar, P.; et al. A genome-wide association study of kynurenic acid in cerebrospinal fluid: Implications for psychosis and cognitive impairment in bipolar disorder. Mol. Psychiatry 2015. [Google Scholar] [CrossRef] [PubMed]
- Stazka, J.; Luchowski, P.; Urbanska, E.M. Homocysteine, a risk factor for atherosclerosis, biphasically changes the endothelial production of kynurenic acid. Eur. J. Pharmacol. 2005, 517, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Hoffman, G.E.; Melendez-Ferro, M.; Albuquerque, E.X.; Schwarcz, R. Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia 2007, 55, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.; Heredi, J.; Menyhart, A.; Czinege, Z.; Nagy, D.; Fuzik, J.; Kocsis, K.; Knapp, L.; Krucso, E.; Gellert, L.; et al. Unexpected effects of peripherally administered kynurenic acid on cortical spreading depression and related blood-brain barrier permeability. Drug Des. Dev. Ther. 2013, 7, 981–987. [Google Scholar]
- Erhardt, S.; Schwieler, L.; Nilsson, L.; Linderholm, K.; Engberg, G. The kynurenic acid hypothesis of schizophrenia. Physiol. Behav. 2007, 92, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, A.; Aminimoghadamfarouj, N.; Church, W.B. Essential structural features of novel antischizophrenic drugs: A review. Med. Chem. 2014, 10, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Amori, L.; Wu, H.Q.; Marinozzi, M.; Pellicciari, R.; Guidetti, P.; Schwarcz, R. Specific inhibition of kynurenate synthesis enhances extracellular dopamine levels in the rodent striatum. Neuroscience 2009, 159, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Hopper, A.T.; Jones, K.A.; Campbell, B.M.; Li, G. Neuroinflammation in mood disorders: Mechanisms and drug targets. In Annual Reports in Medicinal Chemistry; Manoj, C.D., Ed.; Academic Press: San Diego, CA, USA, 2013; Volume 48, pp. 317–331. [Google Scholar]
- Dounay, A.B.; Anderson, M.; Bechle, B.M.; Campbell, B.M.; Claffey, M.M.; Evdokimov, A.; Evrard, E.; Fonseca, K.R.; Gan, X.; Ghosh, S.; et al. Discovery of brain-penetrant, irreversible kynurenine aminotransferase ii inhibitors for schizophrenia. ACS Med. Chem. Lett. 2012, 3, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Dounay, A.B.; Tuttle, J.B.; Verhoest, P.R. Challenges and opportunities in the discovery of new therapeutics targeting the kynurenine pathway. J. Med. Chem. 2015, 58, 8762–8782. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L.; Szalardy, L.; Fulop, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug. Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: A web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, M.; Fukunaga, K.; Usui, K.; Hayashi, N.; Iijima, D.; Horiuchi, H.; Itagaki, N. Novel Bicyclic or Tricyclic Heterocyclic Compounds and Their Pharmaceutical Compositions for Prophylactic and Therapeutic Treatment of KAT (Kynurenine Aminotransferase) II-Associated Disorders. WIPO Patent WO2015163339A1, 29 October 2015. [Google Scholar]
- Dounay, A.B.; Tuttle, J.B.; Verhoest, P.R. Preparation of Tricyclic Compounds as KAT II Inhibitors for Treatment of Neurol. Disorders and Other Diseases. WIPO Patent WO2013186666A1, 19 December 2013. [Google Scholar]
- Henderson, J.L.; Sawant-Basak, A.; Tuttle, J.B.; Dounay, A.B.; McAllister, L.A.; Pandit, J.; Rong, S.B.; Hou, X.J.; Bechle, B.M.; Kim, J.Y.; et al. Discovery of hydroxamate bioisosteres as KAT II inhibitors with improved oral bioavailability and pharmacokinetics. Medchemcomm 2013, 4, 125–129. [Google Scholar] [CrossRef]
- Claffey, M.M.; Dounay, A.B.; Gan, X.; Hayward, M.M.; Rong, S.; Tuttle, J.B.; Verhoest, P.R. Preparation of Bicyclic and Tricyclic Compounds as KAT II Inhibitors for Treating Cognitive and Other Disorders. WIPO Patent WO2010146488A1, 23 December 2010. [Google Scholar]
- Schwarcz, R.; Kajii, Y.; Ono, S. Preparation of Aminopiperazinylquinolonecarboxylates as Kynurenine-Aminotransferase Inhibitors. WIPO Patent WO2009064836A2, 22 May 2009. [Google Scholar]
- Kim, G.K. The Risk of fluoroquinolone-induced tendinopathy and tendon rupture: What does the clinician need to know? J. Clin. Aesthet. Dermatol. 2010, 3, 49–54. [Google Scholar] [PubMed]
- Kushner, J.M.; Peckman, H.J.; Snyder, C.R. Seizures associated with fluoroquinolones. Ann. Pharmacother. 2001, 35, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, N.; Raghavendra, N.; Venkatarathnamma, P.N. Levofloxacin-induced acute psychosis. Indian J. Psychiatry 2008, 50, 57–58. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.S. Peripheral neuropathy associated with fluoroquinolones. Ann. Pharmacother. 2001, 35, 1540–1547. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Kepplinger, B. d-cycloserine lowers kynurenic acid formation–New mechanism of action. Eur. Neuropsychopharmacol. 2014, 24, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Akladios, F.N.; Nadvi, N.A.; Park, J.; Hanrahan, J.R.; Kapoor, V.; Gorrell, M.D.; Church, W.B. Design and synthesis of novel inhibitors of human kynurenine aminotransferase-I. Bioorg. Med. Chem. Lett. 2012, 22, 1579–1581. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Marugami, M.; Kondo, T. In vivo inhibition of kynurenine aminotransferase activity by isonicotinic acid hydrazide in rats. Biosci. Biotechnol. Biochem. 1996, 60, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Rizzo, R.C.; Costantino, G.; Marinozzi, M.; Amori, L.; Guidetti, P.; Wu, H.Q.; Schwarcz, R. Modulators of the kynurenine pathway of tryptophan metabolism: synthesis and preliminary biological evaluation of (S)-4-(ethylsulfonyl)benzoylalanine, a potent and selective kynurenine aminotransferase II (KAT II) inhibitor. ChemMedChem 2006, 1, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Slizik, L.U.; Berkes, D. A concise synthesis of (S)-ESBA, the first selective KAT II inhibitor and their analogs. 2012. Available online: http://www.usc.es/congresos/ecsoc/16/hall_b_BMNPC/b016/index.pdf (accessed on 1 May 2016).
- Pellicciari, R.; Venturoni, F.; Bellocchi, D.; Carotti, A.; Marinozzi, M.; Macchiarulo, A.; Amori, L.; Schwarcz, R. Sequence variants in kynurenine aminotransferase II (KAT II) orthologs determine different potencies of the inhibitor S-ESBA. Chemmedchem 2008, 3, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Schwarcz, R.; Guidetti, P. Preparation of 4-Sulfonyl-Substituted Benzoylalanine Derivatives Useful as Kynurenine-Aminotransferase Inhibitors. WIPO Patent WO2006013085A1, 9 February 2006. [Google Scholar]
- Schwarcz, R.; Varasi, M.; Della Torre, A.; Speciale, C.; Bianchetti, A. Preparation of Substituted Kynurenines as Kynurenine Aminotransferase (KAT) Inhibitors. WIPO Patent WO9504714A1, 16 February 1995. [Google Scholar]
- Varasi, M.; Della Torre, A.; Heidempergher, F.; Pevarello, P.; Speciale, C.; Guidetti, P.; Wells, D.R.; Schwarcz, R. Derivatives of kynurenine as inhibitors of rat brain kynurenine aminotransferase. Eur. J. Med. Chem. 1996, 31, 11–21. [Google Scholar] [CrossRef]
- Schwarcz, R.; Guidetti, P.; Pellicciari, R.; Opal, S. Inhibitors of Kynurenine Aminotransferase for Treatment of CNS Diseases. WIPO Patent WO2007064784A1, 7 June 2007. [Google Scholar]
- Wejksza, K.; Rzeski, W.; Parada-Turska, J.; Zdzisinska, B.; Rejdak, R.; Kocki, T.; Okuno, E.; Kandefer-Szerszen, M.; Zrenner, E.; Turski, W.A. Kynurenic acid production in cultured bovine aortic endothelial cells. Homocysteine is a potent inhibitor. N-S Arch. Pharmacol. 2004, 369, 300–304. [Google Scholar]
- Kocki, T.; Luchowski, P.; Luchowska, E.; Wielosz, M.; Turski, W.A.; Urbanska, E.M. l-cysteine sulphinate, endogenous sulphur-containing amino acid, inhibits rat brain kynurenic acid production via selective interference with kynurenine aminotransferase II. Neurosci. Lett. 2003, 346, 97–100. [Google Scholar] [CrossRef]
- Gramsbergen, J.B.; Hodgkins, P.S.; Rassoulpour, A.; Turski, W.A.; Guidetti, P.; Schwarcz, R. Brain-specific modulation of kynurenic acid synthesis in the rat. J. Neurochem. 1997, 69, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, E.M.; Kocki, T.; Saran, T.; Kleinrok, Z.; Turski, W.A. Impairment of brain kynurenic acid production by glutamate metabotropic receptor agonists. Neuroreport 1997, 8, 3501–3505. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Rassoulpour, A.; Wu, H.Q.; Hodgkins, P.S.; Kiss, C.; Nicoletti, F.; Schwarcz, R. Some metabotropic glutamate receptor ligands reduce kynurenate synthesis in rats by intracellular inhibition of kynurenine aminotransferase II. J. Neurochem. 2000, 75, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
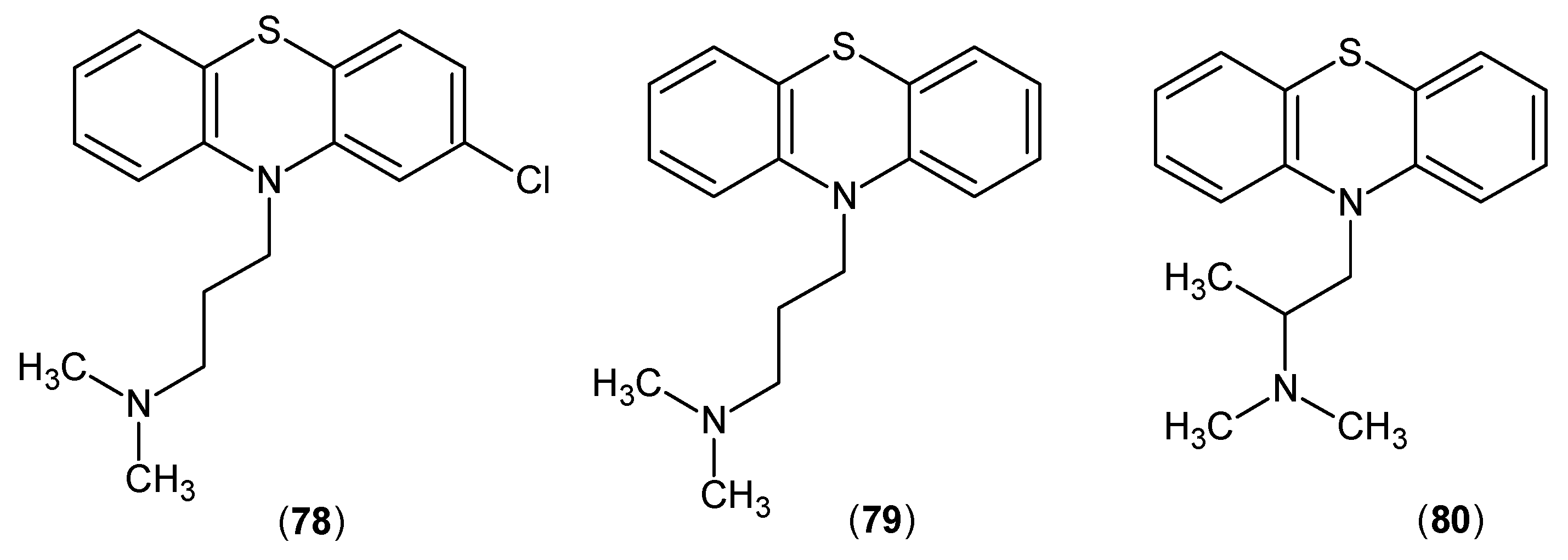
- Mostafa, M.H.; El-Sewedy, S.M.; El-Bassiouni, E.A.; Abdel-Tawab, G.A. In vivo and in vitro studies on the effects of some phenothiazines and sulpiride on kynurenine metabolism. Biochem Pharmacol 1982, 31, 2227–2230. [Google Scholar] [CrossRef]
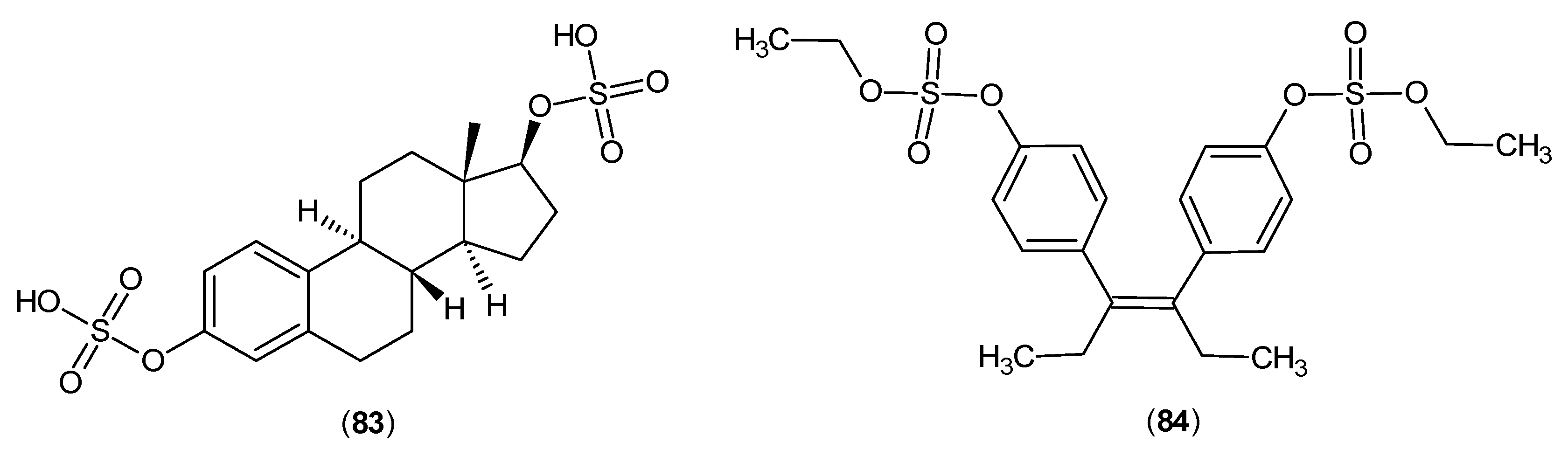
- El-Zoghby, S.M.; El-Sewedy, S.M.; Saad, A.A.; Mostafa, M.H.; Ebied, S.M.; Abdel-Tawab, G.A. In vitro trials to counteract the inhibitory effect of β-estradiol and ethynylestradiol on the B6-dependent kynurenine aminotransferase enzyme. Biochem. Pharmacol. 1976, 25, 2411–2413. [Google Scholar] [CrossRef]
- Chaverri, G.M. Kynurenic and xanthurenic acid production by kynurenine aminotransferase: Comparative study of these reactions and their inhibition by conjugated estrogens. Federation Am. Soc. Exp. Biol. 1970, 29, pA910. [Google Scholar]
- Mason, M.; Gullekson, E. Inhibition of pyridoxal phosphate-dependent enzymes by the sulfate esters of estradiol, estrone, and diethylstilbestrol. J. Am. Chem. Soc. 1959, 81, 1517. [Google Scholar] [CrossRef]
- Mason, M.; Gullekson, E. Estrogen-enzyme inhibitions: inhibition and protection of kynurenine transaminase by the sulfate esters of diethylstilbestrol, estradiol, and estrone. J. Biol. Chem. 1960, 235, 1312–1316. [Google Scholar] [PubMed]
- Rossi, F.; Han, Q.; Li, J.; Li, J.; Rizzi, M. Crystal structure of human kynurenine aminotransferase I. J. Biol. Chem. 2004, 279, 50214–50220. [Google Scholar] [CrossRef] [PubMed]
- Okuno, E.; Nakamura, M.; Schwarcz, R. Two kynurenine aminotransferases in human brain. Brain Res. 1991, 542, 307–312. [Google Scholar] [CrossRef]
- DeLano, W. The PyMOL Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Han, Q.; Robinson, H.; Cai, T.; Tagle, D.A.; Li, J. Biochemical and structural characterization of mouse mitochondrial aspartate aminotransferase, a newly identified kynurenine aminotransferase-IV. Biosci. Rep. 2011, 31, 323–332. [Google Scholar] [CrossRef] [PubMed]


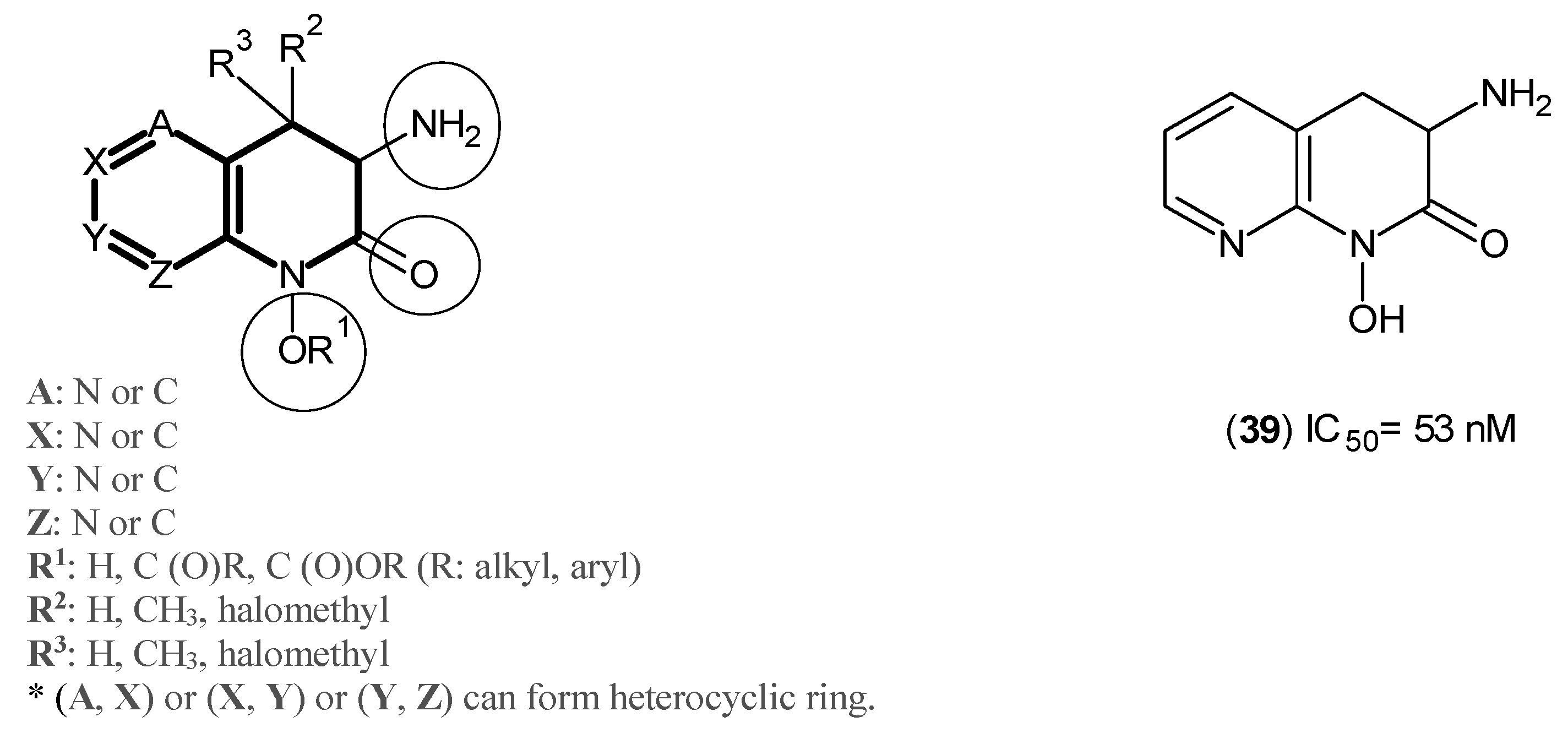
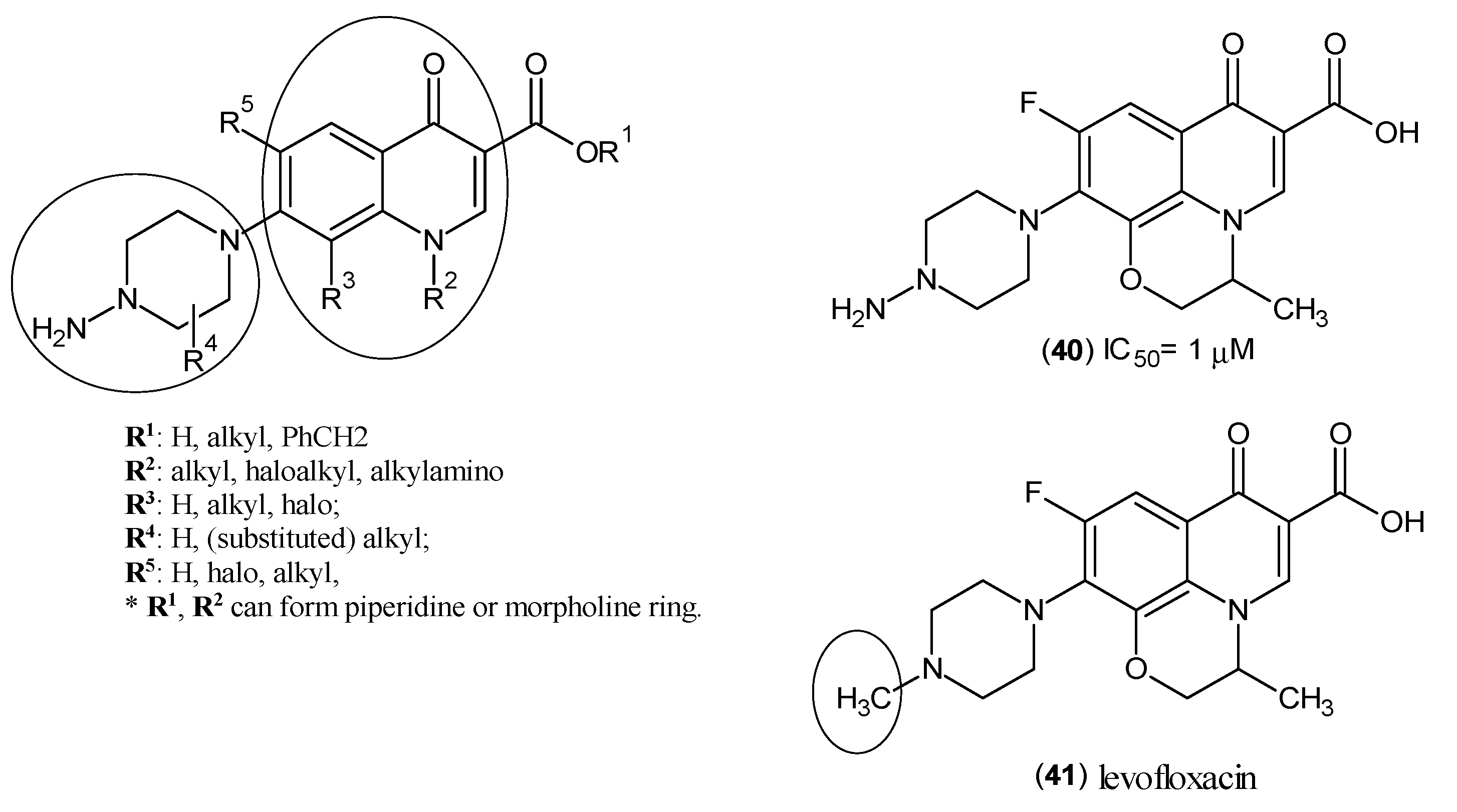
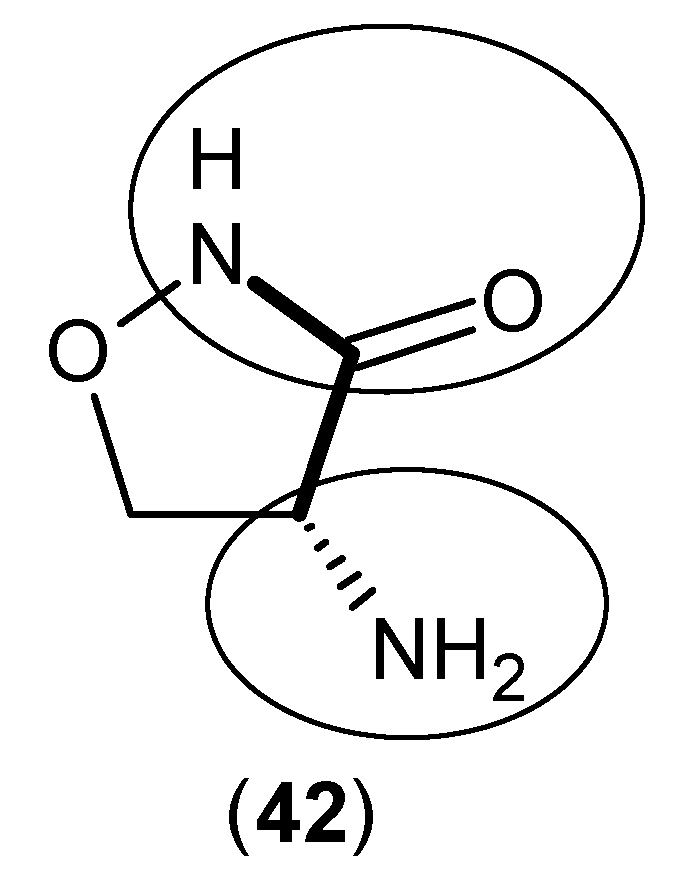
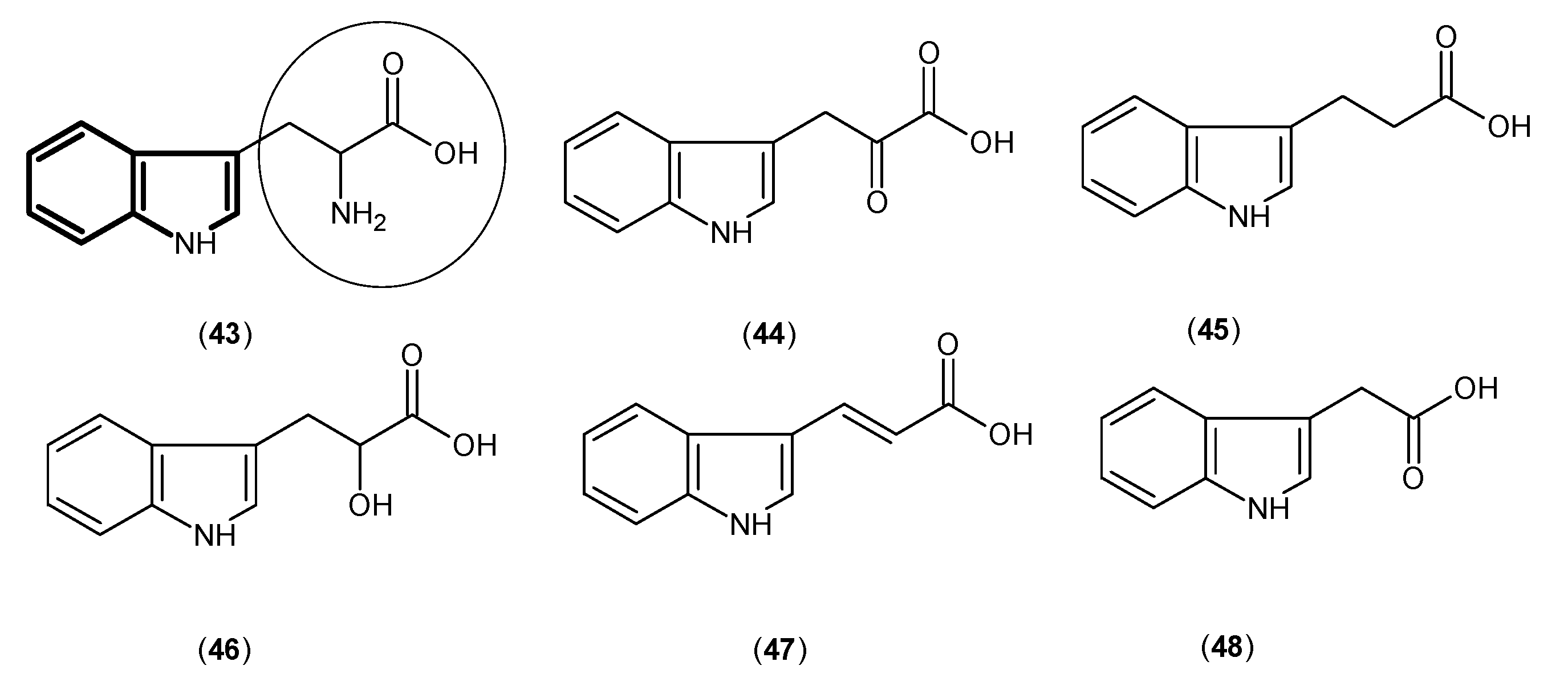



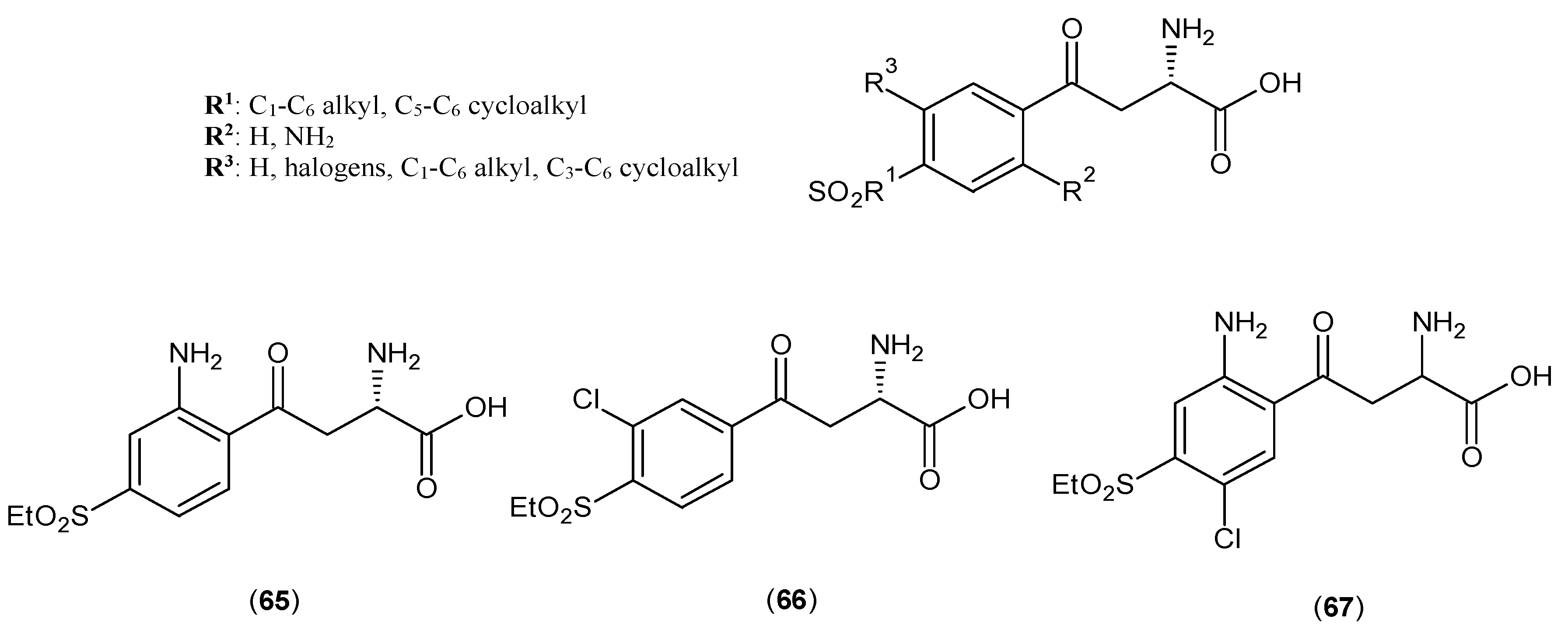
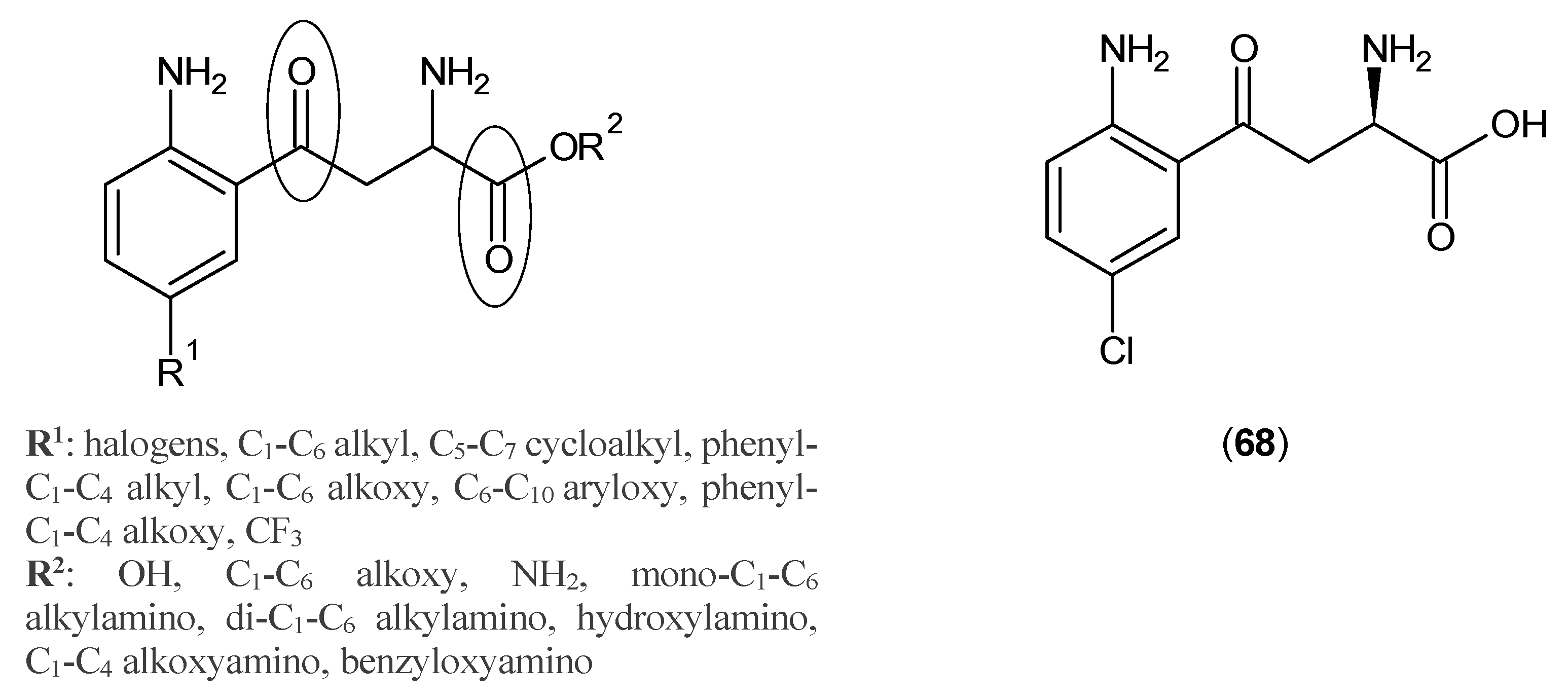
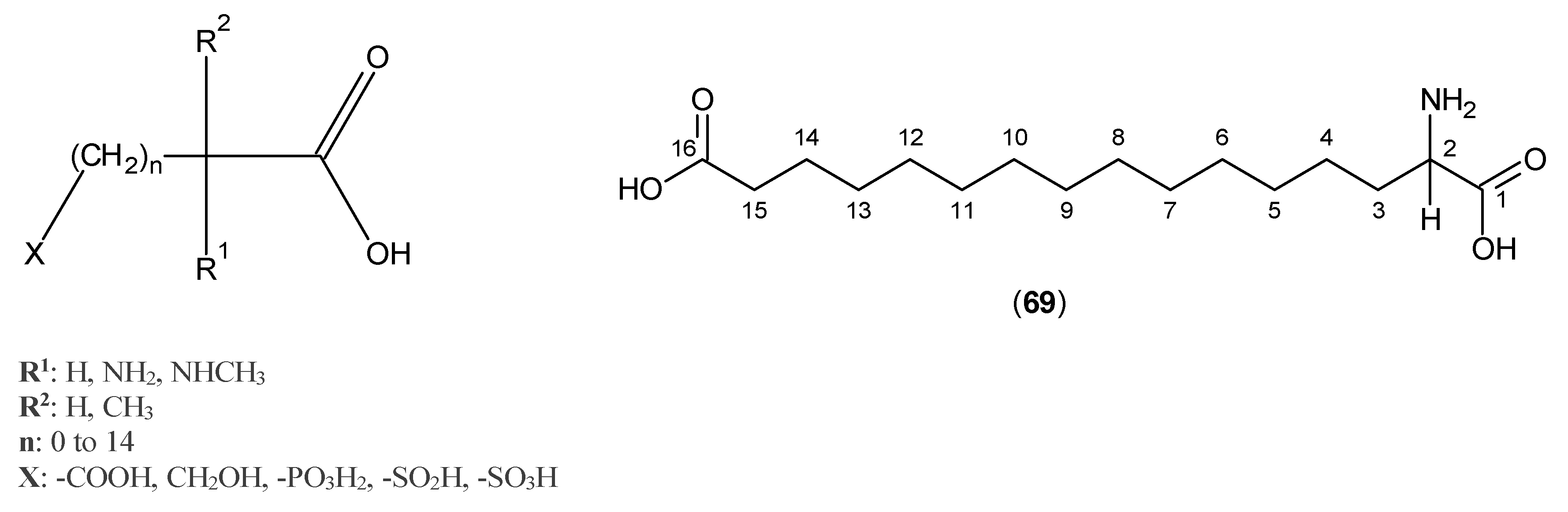
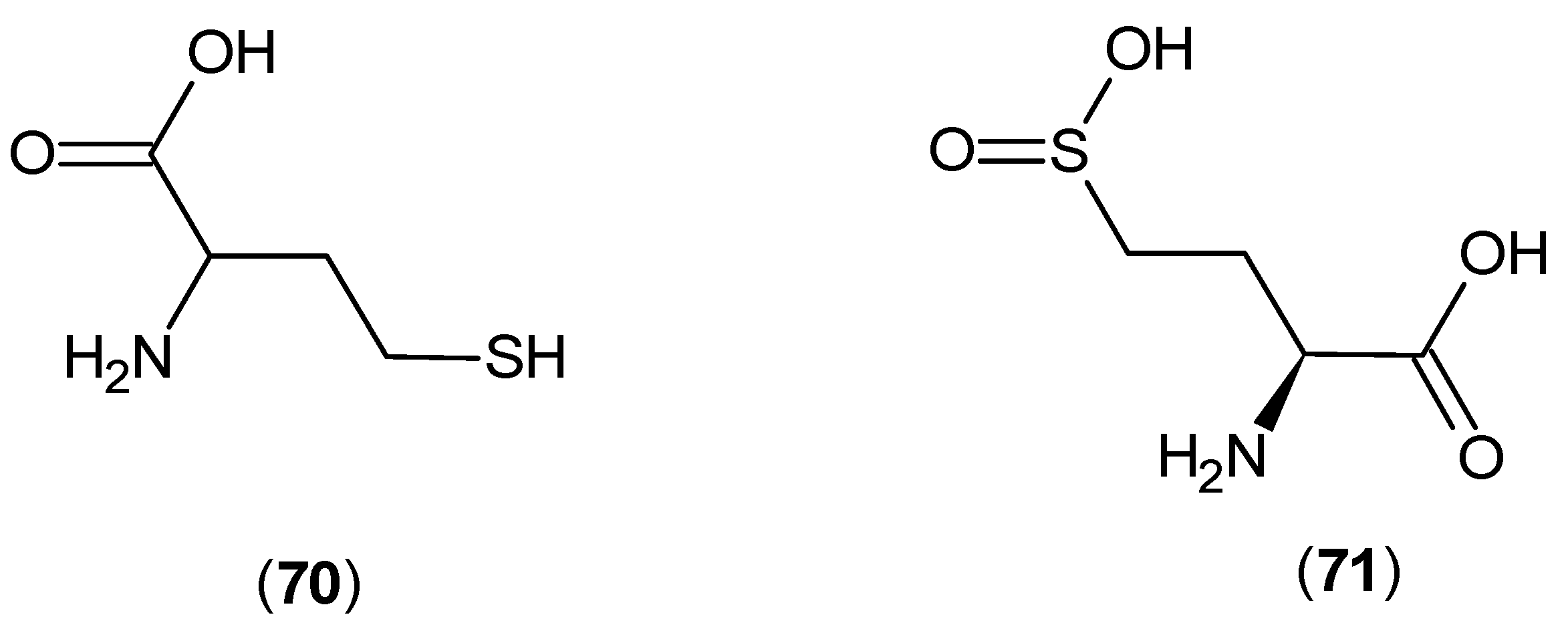

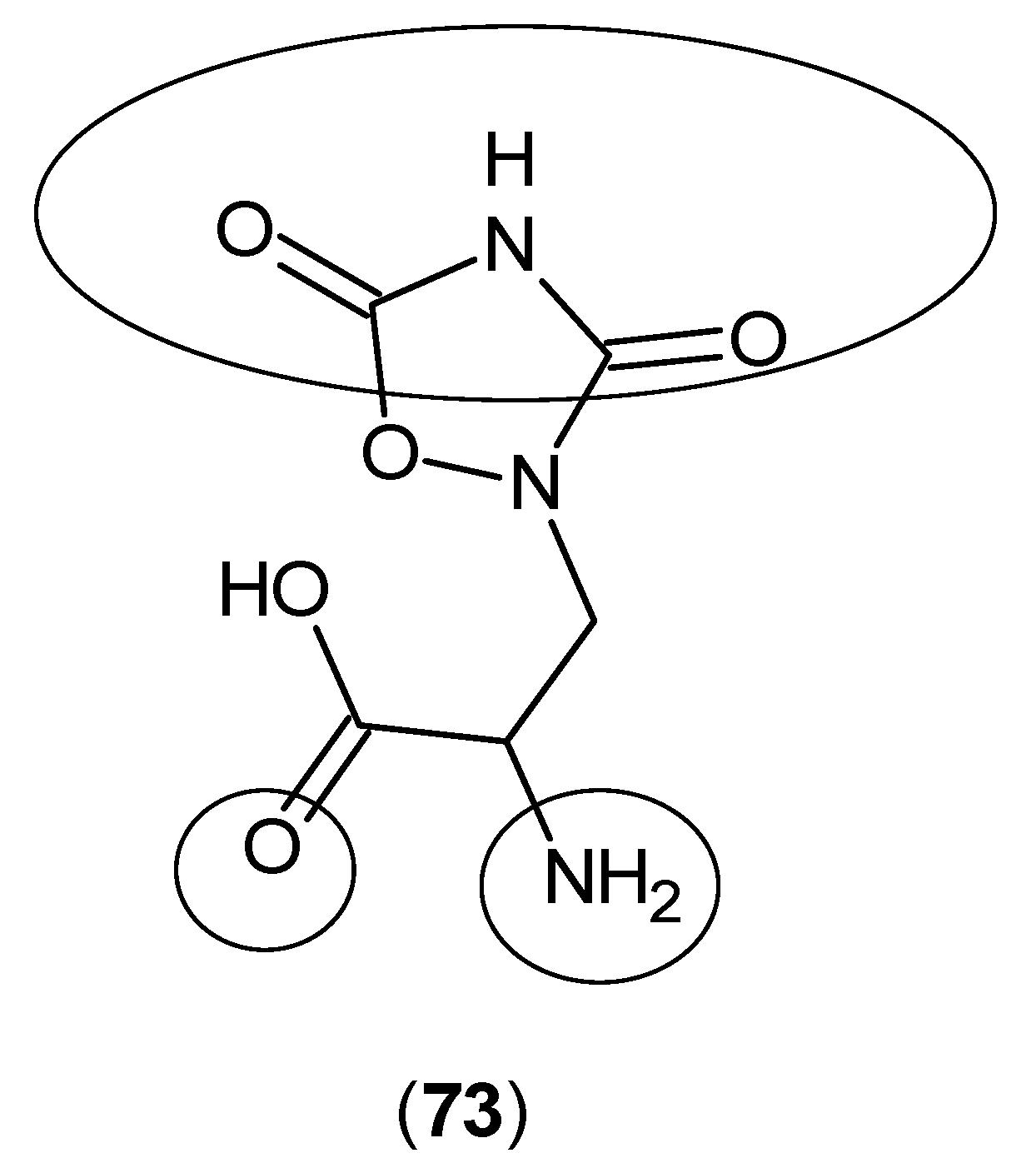


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nematollahi, A.; Sun, G.; Jayawickrama, G.S.; Church, W.B. Kynurenine Aminotransferase Isozyme Inhibitors: A Review. Int. J. Mol. Sci. 2016, 17, 946. https://doi.org/10.3390/ijms17060946
Nematollahi A, Sun G, Jayawickrama GS, Church WB. Kynurenine Aminotransferase Isozyme Inhibitors: A Review. International Journal of Molecular Sciences. 2016; 17(6):946. https://doi.org/10.3390/ijms17060946
Chicago/Turabian StyleNematollahi, Alireza, Guanchen Sun, Gayan S. Jayawickrama, and W. Bret Church. 2016. "Kynurenine Aminotransferase Isozyme Inhibitors: A Review" International Journal of Molecular Sciences 17, no. 6: 946. https://doi.org/10.3390/ijms17060946