Abstract
Breast cancer persists as the most common cause of cancer death in women worldwide. Ovarian cancer is also a significant source of morbidity and mortality, as the fifth leading cause of cancer death among women. This reflects the continued need for further understanding and innovation in cancer treatment. Though breast and ovarian cancer usually present as distinct clinical entities, the recent explosion of large-scale -omics research has uncovered many overlaps, particularly with respect to genetic and epigenetic alterations. We compared genetic, microenvironmental, stromal, and epigenetic changes common between breast and ovarian cancer cells, as well as the clinical relevance of these changes. Some of the most striking commonalities include genetic alterations of BRCA1 and 2, TP53, RB1, NF1, FAT3, MYC, PTEN, and PIK3CA; down regulation of miRNAs 9, 100, 125a, 125b, and 214; and epigenetic alterations such as H3K27me3, H3K9me2, H3K9me3, H4K20me3, and H3K4me. These parallels suggest shared features of pathogenesis. Furthermore, preliminary evidence suggests a shared epigenetic mechanism of oncogenesis. These similarities, warrant further investigation in order to ultimately inform development of more effective chemotherapeutics, as well as strategies to circumvent drug resistance.
1. Classification of Breast and Ovarian Cancer
Globally, breast cancer persists as the most common cause of cancer death in women, and the leading cause of death in women ages 40–49. Despite an over 95% sensitivity of current breast cancer screening, overall mortality has decreased by only 30%. Though less common, ovarian cancer is the fifth leading cause of cancer death in women in the US, owing both to its virulence, and our lack of effective screening. This reflects the continued, if not dire, need for further understanding and innovation in these respective fields of cancer treatment.
This need for more efficacious screening and therapeutics has fueled a rapidly evolving body of scientific work. This has led to an emerging understanding that these clinical entities encompass many heterogeneous cancers, resulting in a new classification schema. Breast cancer tumors were classically categorized by histological origin into infiltrating ductal (76%), invasive lobular (8%), ductal/lobular (7%), mucinous/colloid (2.4%), tubular (1.5%), medullary (1.2%), and papillary (1%). New definitions have simplified the sub groups as luminal A, luminal B, hormone receptor positive and triple negative, which more closely parallels clinical treatment algorithms.
This new schema also aligns with ovarian cancer classification, which includes surface, germ, stromal, and borderline tumors. Of these groups, epithelial carcinomas account for the vast majority (over 90%) and can be divided into serous I, serous II, endometroid, mucinous, Brenner, and clear cell [1]. Based on molecular markers, ovarian tumors can be further classified into Type I versus Type II tumors. Type I, or low-grade serous carcinoma, is associated with a ~55% 5 years survival. These tumors develop via the stepwise accumulation of carcinogenic modifications, including BRAF/KRAS. Type II tumors, on the other hand, are often high-grade serous carcinomas (~30% five years survival) at the time of presentation and are associated with various shared patterns of gene expression, as well as frequent p53 mutations. Interestingly, in both breast and ovarian cancer, the vast majority of tumors are of epithelial origin; less commonly, tumors arise from functional and stromal cells. This is further supported by the emerging body of evidence that a significant subset of ovarian cancers in fact originate in the fallopian tubes, adding to the epithelial predominance [1].
2. Comparison of the Genetic and microRNA Environments of Breast and Ovarian Cancer
Though breast and ovarian cancer are more often distinct clinical entities, the recent explosion of genomics, proteomics, metabolomics, and other large-scale-omics research, has uncovered many overlaps between the two, including what we and others posit to be relevant shared genetic alterations [2]. These similarities are particularly striking between epithelial-origin triple negative basal cell breast cancer (TNBC) and high-grade serous ovarian cancer (HGS-OvCa). A summary of common mutations in breast and ovarian cancers is listed in Table 1.

Table 1.
Comparison of significant genetic alterations in breast and ovarian cancers.
As one might expect, the seemingly disparate genetic alterations are directly or indirectly related to cell cycle control, growth, development, differentiation, DNA damage repair, and apoptosis (Figure 1). Thus, it follows that many of these key genetic alterations have been found in multiple types of cancer, in addition to breast and ovarian [18].
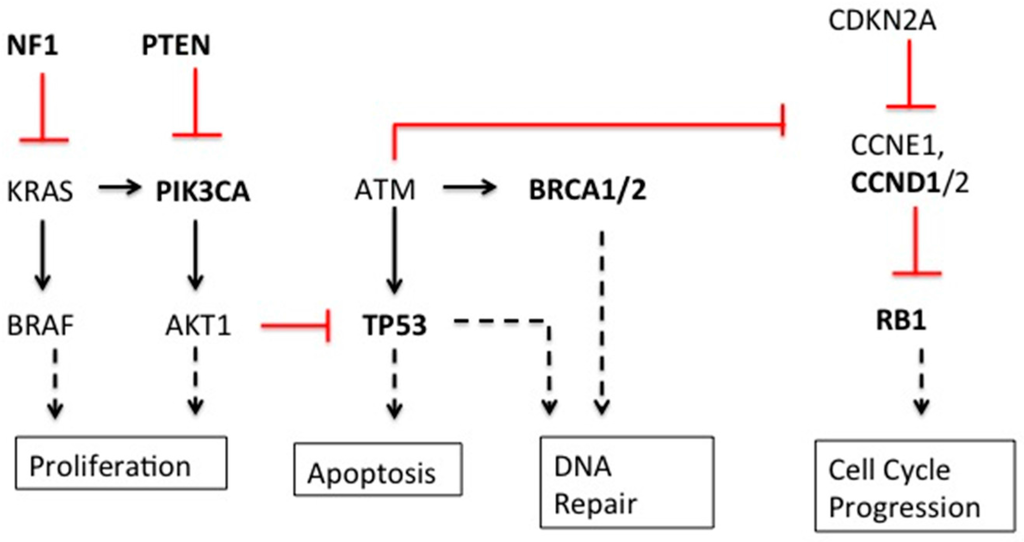
Figure 1.
Mutated genes in breast and ovarian cancer. Bolded genes are those significantly mutated in breast and ovarian cancer. Red lines show inhibition of expression; solid black lines show induction of expression; dashed black lines show effector events.
The Human Cancer Genome Atlas, an extensive study investigating gene copy numbers, exon sequences, DNA methylation, and the expression of miRNA, and proteins in breast cancer tumors, identified numerous common genetic alterations among seemingly heterogeneous breast cancers. For example, TP53, PIK3CA, and GATA3 mutations occurred at an incidence greater than 10% across all breast cancers. When separated into molecular subtypes, more commonalities were identified. In basal-like tumors and TBNC (ER-, PR-, HER2-), RB1, BRCA1, and TP53 commonly contained loss-of-function mutations [3]. Interestingly, these mutations are also prevalent in HGS-OvCa tumors [5]. Although mutations in these genes are often found in cancers, the high frequency at which mutations in RB1, BRCA1, and TP53 occur in TNBC and HGS-OvCa may indicate molecular similarities in the development or progression of these two different, aggressive cancers [3].
By far, the most commonly mutated gene in both TNBC and HGS-OvCa is TP53. The Cancer Genome Atlas found that TP53 was mutated in 80% of TNBC tumors and 96% of HGS-OvCa tumors analyzed [3]. Other groups have reported a similarly high prevalence of TP53 mutations in these and other aggressive cancer subtypes [2,3,19] compared to the lower frequency of somatic TP53 mutations seen in many other cancers. For example TP53 mutations have been identified in 27% of brain tumors and 5.8% of cervical cancers [20]. An extensively studied gene, TP53, encodes the transcription factor p53, a protein involved in many signaling pathways that halt cell cycle progression, activate DNA repair mechanisms, and induce apoptosis when activated in response to cellular stress [19,21,22]. Thus, as an anti-proliferative or even pro-apoptotic protein, p53 acts as an important tumor suppressor gene in response to DNA damage. In the context of cancer, it is apparent that tumors with mutations in TP53 are more susceptible to retaining and passing down DNA damage, allowing for the selection of more aggressive tumors. The significance of the shared, high TP53 mutation rate in TNBC and HGS-OvCa is striking and suggestive of shared fundamental features of pathogenesis.
Another commonly mutated gene in breast and ovarian cancers, functional BRCA1 protects the cell from double-stranded DNA damage through homologous recombination. BRCA1/2 mutations are present in 5%–15% of all breast and ovarian cancers [3,4,5] and have been found in other hereditary diseases [23,24,25,26], in the absence of homolous recombination, cells are more vulnerable to genomic instability. Accordingly, cancers containing mutations in BRCA1 have numerous copy number aberrations (CNAs) and are more likely to be TNBC or HGS-OvCa. Interestingly, many BRCA1-mutated TNBC show characteristic patterns in CNAs leading to the classification of BRCA1-mutated cancers as BRCA1-like or non-BRCA1-like TNBC [27,28]. These and other studies comparing copy number landscapes of breast and ovarian cancers have reported several common features between TNBC tumors and HGS-OvCa tumors, including common gains within 1q, 3q, 8q, and 12p (in particular HIF1A and MYC) and loss within 4q, 5q, 8p, and 10q (in particular FOXA1, ER, and PTEN) [28].
3. Comparison of the Respective MicroRNA Environments
In addition to coding-DNA mutations, breast and ovarian cancers contain an array of miRNA expression alterations, which contribute to the development and progression of cancer [29]. In healthy cells, miRNA regulate mRNA stability, ensuring proper timing in the formation of functional proteins. Thus, when misregulated, miRNAs can lead to improper protein translation. Table 2 lists misregulated miRNAs implicated in breast and ovarian cancers, respective gene targets and the functional implications in cancer. For example, miR-100, miR-214, miR-206, and miR-233 are involved in diverse pro-growth and anti-apoptotic pathways. miR-100 targets the pro-growth proteins FRAP1/mTOR and FGFR3 for degradation and when downregulated, these genes are re-expressed and function as oncogenes (Figure 2A, [30,31,32,33,34,35,36,37,38,39,40,41,42]). miR-214 deregulation induces cell survival and cisplatin resistance. In breast cancer, downregulation of miR-214 leads to increased EZH2 expression which causes increased cell proliferation [32]. EZH2 is an important epigenetic regulator that positively regulates insulation zone formation for selective gene silencing by enhancing H3K27me3. In ovarian cancer, miR-214 targets PTEN, leading to activation of the Akt cell survival pathway. Akt inhibitors have been found to nullify miR-214 induced cell survival [32]. Interestingly, EZH2 is a part of the polycomb group of proteins that helps to regulate gene expression during normal development. On the other hand, miR-206 is upregulated in breast cancer and targets the ERα gene [41].
Table 2.
miRNA implicated in breast and ovarian cancers.
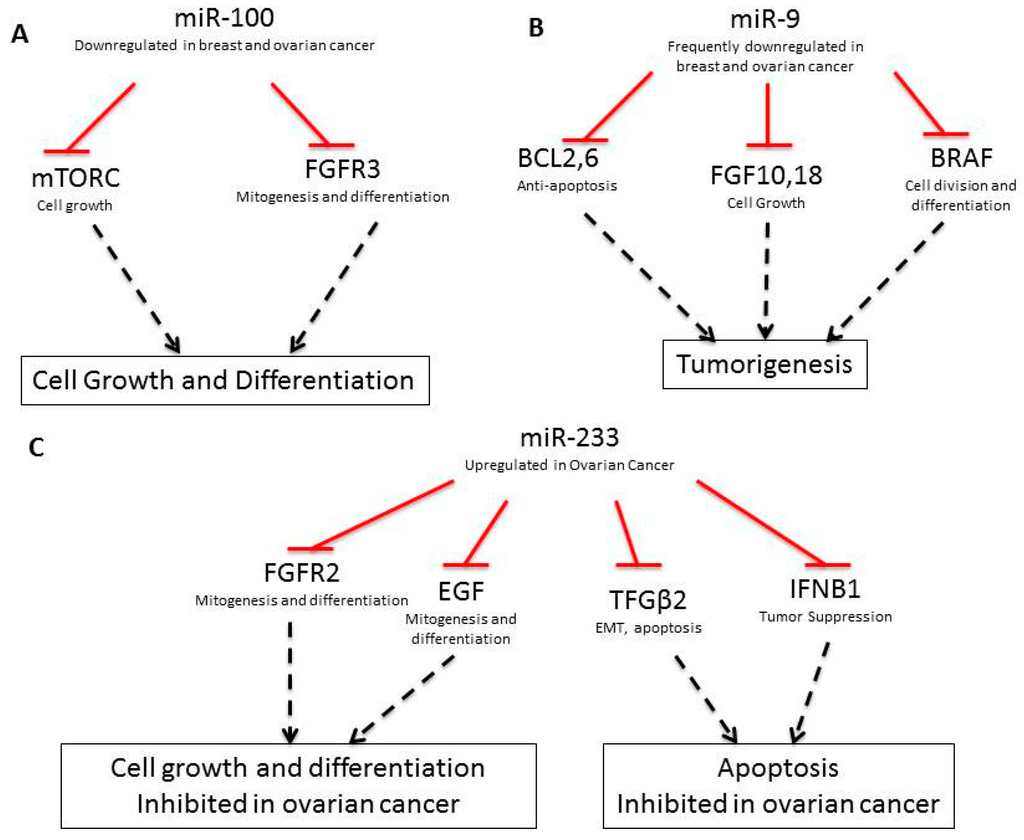
Figure 2.
Pathways regulated by miR-100 (A), miR-9 (B), and miR-233 (C). Red lines show inhibition; dashed black lines show effector events.
As shown in Table 2, several miRNAs are also involved in the increased invasiveness of breast and ovarian cancers and the development of higher grade tumors. For example, the downregulation of miR-9-3, is implicated in breast cancer metastasis, vascular invasion, angiogenesis, and drug resistance development [31,40]. miR-9 expression downregulates BCL2, BCL6, FGFR10, FGFR18, and BRAF, which are involved in anti-apoptosis, cell-growth, and differentiation. Downregulation of miR-9 upregulates these pathway (Figure 2B, [43]).
Additionally, miR-233 downregulates FGFR2 and EGF, which are involved in cell growth and differentiation, as well as TGF-β2 and IFNB1, which are involved in apoptosis. miR-233 is upregulated in ovarian cancer, downregualting these pathways (Figure 2C, [44]). Other miRNAs, including miR-34c, miR-125a, miR-125b, miR-200a, and miR-200c, are downregulated during the epithelial-to-mesenchymal (EMT) transition in ovarian cancer cells [45,46,47,48]. Downregulation of miR-34c, miR-125a, and miR-125b also induces EMT in breast cancer-initiating cells [36]. In particular, miR-34c gene down-regulation via DNA methylation has been shown to promote self-renewal and EMT in breast tumor-initiating cells [36]. Downregulation of miR-125a suppresses ARID3B, a gene which is overexpressed in human ovarian cancer [49]. Furthermore, sequence alternations in miRNAs interact pathologically with BRCA1 and BRCA2. For example, a polymorphism found within the miR-146a sequence has been found to alter the stem region of this miRNA from G:U to C:U. This variant allele produces more mature miR-146a than the wild-type, and the mutant variant has a significantly stronger binding capability to its target BRCA1 and BRCA2 mRNA sequences, which are, themselves, arguably the most studied shared mutations of breast and ovarian cancer [50].
4. Epigenetic Comparison of Breast and Ovarian Cancer
In addition to genetic alterations and miRNA, breast and ovarian cancers frequently share key epigenetic patterns. Research has identified consistent methylation patterns that often precede breast cancer progression. For example, moderate to low levels of lysine acetylation (H3K9ac, H3K18ac, and H4K12ac), lysine methylation (H3K4me2 and H4K20me3), and arginine methylation (H4R3me3) were observed in HER2-positive tumors [51]. Similar data with respect to ovarian cancer is still emerging. The level of histone methylation depends on the activation or deactivation of specific histone methylases or demethylases, and alterations of the methylation status of histones has been clearly demonstrated in breast cancer [52,53]. In the case of ovarian cancers, histone splicing is observed. For example, alternative splicing of H2A type histone variants have been observed in ovarian cancers. Additionally, investigations on nuclear dynamics changes are currently underway in ovarian cancer [54] and breast cancer [55]. Importantly, common histone methylation patterns in breast and ovarian cancer include H3K27me3, H3K9me2, H3K9me3, H4K20me3, and H3K4me. In fact, Elisheikh et al. [51] have found a consistent correlation between H4K16me3 and 78.9% of all breast cancers. Recent studies showed that mutation of DNA methylases can increase methylation of specific lysine residues in histone 3, causing changes which favor carcinogenesis [56]. For example, the DNA methyltransferase EZH2, which causes H3K27me3, is mutated in breast cancer [56]. Interestingly, H3K27me3 recruits the CCCTC motif binding protein CTCF, which creates an insulation zone and inhibits enhancer and promoter interactions to silence specific genes during development [57,58,59]. These interactions are disrupted by DNA methylation around the interacting sites. For example, in breast cancer, histone modifications at enhancers were observed to regulate genes even up to 750 kilobase. In addition, about 50% of active enhancers were found in nucleosome depleted regions. Expression data analysis identified 600 active enhancers. The genes regulated by these enhancers have functions which include proteolysis, epidermis development, cell adhesion, mitosis, cell cycle, and DNA replications [55].
In addition to histone modifcations, global modification of DNA sequences by methylation at CpG residues plays an important role in oncogenesis of both breast and ovarian cancer. As described above, methylation of the enhancer sites reverse CTCF regulated DNA silencing [60], but methylation of specific CpG islands in the promoter regions of genes silence them [61,62]. Shared targets of methylation include the promotor regions of p21, p16, MGMT, BRAC1, MLH1, HOXD11, CDH1 (E-cadherin), TGF-R, ARHI, and RASSF1A in breast and ovarian cancer [63].
In the case of breast cancer, 30% of tumors are associated with an overexpression of HER2 due to amplification of gene copy number [64]. Interestingly, overexpression of HER2-mediated signaling has been shown to alter the genome-wide methylation pattern [65,66]. Specifically, the genes most commonly methylated in these tumors were relevant to development and transcription (AKT3, HK1, PFKP, AKR1B1, INA, FOXC2, NEUROD1, CDKL2, IRF4) or were Homeobox genes (DBX1, NX-6, SIX6) [67]. Other well-known drivers of breast cancer metastasis include upstream methylation of E-cadherin, RARRF1A, RAR-2, APC, TWIST, and GSTP1 [68]. Another important example, cell adhesion receptor integrins including α5β6, may also regulate methylation in breast cancer cells [69,70]. Interestingly, it has been shown that HER2 and integrin receptors associate to enhance downstream signaling for breast cancer progression [71]. It is reasonable to believe that these downstream signaling processes regulate DNA methylation. In support of this notion, a recent study showed that ERK kinase regulated DNA Methyltransferase 1 (DNMT1) and methylation of specific genes in prostate cancer cells [72]. In addition to ERK, Akt DNA methylation also regulates H3K27me3 mediated gene silencing [73]. This suggests that intracellular signaling plays a significant role in the methylation process regulating gene silencing. Usually, DNMTI is highly expressed in cancer cells compared to normal cells where its expression varies at different stages of the cell cycle [74]. A recent system biology study showed that higher expression of DNMTI does not necessarily methylate and silence all types of genes in cancer cells. DNMTI is allosterically activated at the locations of genes it silences [75]. Taken together, this suggests a complicated but well-orchestrated mechanism by which methylation and thereby gene silencing are regulated. Changes in the balance can, thus, perturb epigenetic regulation and gene expression.
Figure 3 depicts a simplistic model of possible epigenetic regulation of tumor suppressor genes and growth promoting genes in breast and ovarian cancer progression. Many of the epigenetically-regulated genes are common in both types of cancers and some are different but the regulation could be similar. Hypermethylation of promoter regions blocks POLII binding inhibiting transcription of tumor suppressor genes (Figure 3A). As this process is reversible, when these regions are demethylated by treatment with epigenetic drugs, POLII binds and tumor suppressor genes are re-expressed (Figure 3B), which is an important aspect of current improvement as combination therapy. In contrast, growth promoting genes are kept under controlled expression by CTCF binding to the H3K27me2/me3 regions creating an insulation zone and inhibiting enhancer-promoter interaction necessary for POLII binding and transcription initiation (Figure 3C). In both breast and ovarian cancer this control could be lost because of hypermethylation around the region where CTCF binds (Figure 3D), which causes an above normal expression of growth promoting genes. Demethylation as a result of epigenetic drug treatment, allows CTCF to bind in these regions controlling above normal growth promoting gene expression. Since the formation of breast cancer or ovarian cancer progenitor cells is a long term process, gradual epigenetic changes which cause alteration of gene expression slowly convert a few pre-disposed cells to breast or ovarian cancer progenitor cells. Other genetic alterations help advance and speed up this progression. Overall, this model shows that a change of balance in CpG DNA methylation in upstream promoter regions and in enhancers along with differential histone methylations regulates the initial steps in tumorigenesis. Although not currently included in this model, histone modifications, such as H3K4, are known to facilitated gene expression. Additionally, lnc RNA may also have a role in this process [76]. However, future studies will reveal how much these factors contribute to the described model.
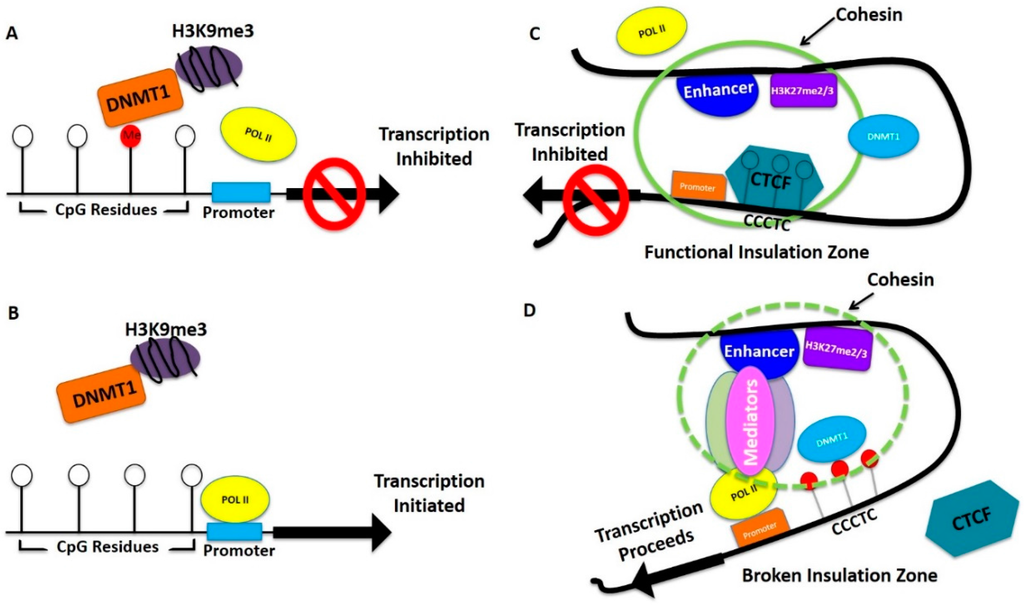
Figure 3.
Model of DNA methylation and histone methylation regulating gene expression for breast and ovarian cancer initiation. (A,B) Hypermethylation is indicated by red balls (CpG residue). White balls represent unmethylated CpG residues. DNMT1 represents DNA methyl transferase 1, POLII represents RNA polymares II; (B) In the absence of hypermethylation, POLII is able to bind to the promoter region and initiate transcription; (C,D) White balls represent unmethylated, and red balls represent hypermethylated CpG residues. CCCTC is CTCF binding motif. The green circle represents effective insulation zone. Break of insulated zone is shown by dotted green circle.
5. Role of Stroma in Breast and Ovarian Cancer
Epigenetic regulation originates both intracellularly and extracellularly. Stromal cells are central to this process, providing extracellular signals, and inducing intracellular changes via cell-cell interactions. As described above, signaling pathways (e.g., ERK and AKT) regulate methylation. Cytokines, growth factors, and hormones from the extracellular milieu, as well as direct attachment of stromal cells to breast or ovarian epithelial cells, regulate intracellular signaling. The relationship between stromal mutations and carcinogenesis is well known [14,68,77]. Furthermore, ovarian cancers have been associated with higher numbers of fibroblasts, lymphocytes and other inflammatory cells. These cell types may receive deranged regulatory signals from surrounding pathological cells. For example, it has been shown that ovarian surface epithelial cells that have been immortalized by Ras may reprogram the stromal microenvironment through the senescence of fibroblasts, and through this mechanism, support tumorigenesis [77]. Furthermore, expression of the chemokine CXCR4 has been found in 59% of ovarian cancers, and CXCL12 in 91% of ovarian cancers; both have been associated with decreased disease free survival [78]. Finally, as with tumor genomes, studies have shown that methylation levels are elevated in oncogenic breast stromal environments, often even before there are detectable tumors [79]. Taken together, these examples support the notion that stromal changes produce alterations in signaling that lead to increased methylation and histone modifications, which in turn drives the formation of cancer progenitor cells (Figure 3). Although breast and ovarian stromal environments are morphologically and functionally distinct, there are many shared common epigenetic signaling pathways. Furthermore, as previously discussed, continued study of the fallopian (epithelial) origin of various ovarian cancers my further our understanding of the pathogenicity of the respective stromal environments.
6. Current and Future Treatment of Breast and Ovarian Cancer
In the case of breast cancer, clinical management is primarily driven by the hormonal and receptor status of the tumor, as well as stage and grade. At this time, the standard of care necessitates genetic testing of all breast cancer tumors. In both hormone receptor positive and negative tumors, standard treatment begins with surgical excision, then local radiation, and finally chemotherapy guided by the specific genetic diagnosis. Common agents include Tamoxifen (estrogen receptor antagonist), Herceptin (HER2 inhibitor), and proteasome inhibitors. Thus, in breast cancer, depending on the origin or the classification, target-specific drugs are part of the standard of care. In patients that have an underlying predisposition to cancer, prophylactic surgery may also be considered.
The clinical manifestations of ovarian cancer are distinct among the gynecological malignancies in that there is no standard screening protocol, and at best, subtle clinical signs; diagnosis can only be confirmed by surgery. As such, at the time of presentation, the vast majority of neoplasms have metastasized beyond the pelvis (grade III). Management includes surgical debulking, followed by systemic chemotherapy. Since most cancers have metastasized before presentation, local radiation is not the norm, except in select clinical scenarios including noninvasive tumor subtypes such as benign teratomas. Standard regimens often include an intercalating agent, an alkalating agent (e.g., cis-platin, carboplatin), an anti-growth agent (e.g., vincristine) and in some cases an anti-tetrahydrofolate reductase (e.g., methotrexate). In contrast to breast cancer, chemotherapies used in the management of ovarian cancer are often not target specific.
7. Drug Resistance
One of the major problems associated with breast and ovarian cancer chemotherapeutics is the development of drug resistance. Clinically, this manifests as frequent relapse, and even more importantly, relapse of tumors of higher virulence and intractability. For example, although most women with advanced stage ovarian cancer initially respond to cytoreductive surgery and platinum based chemotherapy, over 70% of women relapse. Moreover, platinum-resistant epithelial ovarian cancer is considered uniformly fatal [79]. There are numerous pathways by which tumors may gain drug resistance, including efflux by p-glycoproteins (MDR gene overexpression), rapid drug metabolism, and alteration of the target site. In addition to traditional selective mechanisms, recent studies have indicated that methylation plays a significant role in this process [80,81,82]. For example, ER-i has been associated with tamoxifen resistance, and is methylated in 50% of breast cancers [83,84]. Methylation of the MCJ gene has been associated with poor response to therapy, and poor overall survival [85]. Demethylation of Fanconi anemia complementation group F (FANCF) with decitabine has been shown to decrease sensitivity to cisplatin in various cell models [15]. Methylation of hMLHI (a DNA mismatch repair protein) and subsequent loss of expression is associated with resistance to cis-platin [85]. Importantly, demethylation and, thus, reversal of this and other pathological modifications, has been associated with resensitization to therapy [86]. These various examples are supported by the more general observation that DNMT1 inhibitors (AZA derivatives) and histone deacetylase inhibitors (HDACi) have been shown to resensitize resistant ovarian cancer cells to cisplatin by demethylation and re-epression of the RGS10 [87,88]. Though epigenetic changes are observed in breast and ovarian cancers, no systematic study has been performed to compare the global methylation status before versus after remission of both types of cancers.
Drawing inferences from similar studies in leukemia patients in which methylation levels remained high throughout treatment, it is likely that a durable chemotherapeutic response will require treatment that includes agents that modify global methylation and, thus, attack cancer progenitor cells [61,81,84,89,90]. Furthermore, these agents must have the capacity to modulate chromatin opening, as well as inhibit CpG methylation, in order to render drug resistant cells vulnerable to treatment, while at the same time inhibiting the formation of new cancer progenitor cells. In support of this notion, it has been shown that HDAC inhibitors (Class I and Class III) possesses the ability to successfully perform both of these functions [90]. When these agents are combined with traditional chemotherapeutics, even in suboptimal doses, they produce additive, or even synergetic death of breast and ovarian cell lines [91,92].
Several clinical trials of such combination therapies are underway. It has been demonstrated by these authors and others that the efficiency of demethylation increases when HDACi are used in combination with DNMT1i [61,88,89,90,91,92,93]. Others have shown that such combinations are highly effective against breast cancer in mouse models [94]. Furthermore reversal of platinum drug resistant ovarian cancer after treatment with HDACi [88], and similarly reduction in the relapse of lung cancer patients with use of epigenetic chemotherapeutics, have been reported [95]. As chemotherapy of ovarian cancer is mainly target non-specific, and because epigenetic modifications possibly play similar types of roles in ovarian and breast cancers, current investigators are extremely encouraged to develop epigenetic therapies in combination with traditional drugs for breast and ovarian cancers. [50,61,62,91,92,94,96,97,98,99].
8. Inferences from a Comparison of Breast and Ovarian Cancer: An Epigenetic Link
Research continues to reveal that breast and ovarian cancer have a surprising number of shared genetic, stromal, and epigenetic features, despite profoundly different clinical presentations. This has led us to consider shared features of pathogenesis. Amidst the seemingly profound heterogeneity of breast cancers, it has been suggested that all solid tumors may originate from 2 to 3 types of breast cancer stem cells [2,3,100]. This is at odds with the classical notion that breast cancer development is driven from myriad distinct genetic alterations. Rather, it is suggestive of a shared pathogenic mechanism, which is then followed by the generation of diverse passenger mutations. There is a significant body of indirect evidence that the shared mechanism for these discrete cancer events is epigenetically driven. For example, methylation increases in breast and ovarian stromal cells prior to carcinogenesis. This may suggest that oncogenesis is driven by expression of stromal signaling molecules, which in turn induces epigenetic changes in target breast or ovarian cells, as described in previous sections [50,101]. Furthermore, it has been reported that cancer progenitor cells are only irradiated when treated with epigenetic modulating drugs [2]. Therefore, further research is likely to reveal that the most significant commonalities between breast and ovarian carcinogenesis are shared anteceding and perhaps causational patterns of epigenetics.
Acknowledgments
Mckenna Longacre, Nicole A. Snyder, Genevieve Housman, Meghan Leary, Karolina Lapinska, Sarah Heerboth, and Amber Willbanks were supported by Undergraduate Research Opportunity Program (UROP), Boston University (BU).
Author Contributions
Sibaji Sarkar conceived the concept for this paper. Sibaji Sarkar, Mckenna Longacre and Genevieve Housman developed the outline. All authors (Mckenna Longacre, Nicole A. Snyder, Genevieve Housman, Meghan Leary, Karolina Lapinska, Sarah Heerboth, Amber Willbanks, and Sibaji Sarkar) contributed equally to the writing of this paper.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AKR1B1 | Aldo-keto reductase family 1, member B1 |
| APC | Adenomatous polyposis coli |
| ARHI | Aplysia ras homology member I (GTP binding protein Di-RAS3) |
| ARID3B | AT-rich interactive domain-containing protein 3B |
| CBFB | Core-binding factor subunit beta |
| CDH1 | Cadherin-1 |
| CDK | Cyclin kependent kinase |
| CDKL2 | Cyclin-dependent kinase-like 2 |
| CREBBP | CREB-binding protein |
| DBX1 | developing brain homeobox protein 1 |
| DNMT | DNA methyltransferase |
| DNMTi | DNA methyltransferase inhibitor |
| EGF | Epidermal growth factor |
| EMT | Epithelial mesenchymal transition |
| ER | Estrogen receptor |
| ER- | Estrogen receptor negative |
| EZH2 | Enhancer of zeste homolog 2 |
| FANCF | Fanconi anemia complementation group F |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FRAP1 | FK506-binding protein 12-rapamycin-associated protein 1 (Mammalian target of rapamycin, MTOR) |
| GABRA6 | Gamma-aminobutyric acid receptor subunit alpha-6 |
| GSTP1 | Glutathione S-transferase P |
| HDACi | Histone deacetylase inhibitor |
| HGS-OvCa | high-grade serous ovarian cancer |
| HIF | Hypoxia induced factor |
| HK1 | Hexokinase-1 |
| IRF2BP2 | Interferon regulatory factor 2-binding protein 2 |
| IRF4 | interferon regulatory factor 4 |
| KMT2C | Lysine N-methyltransferase 2C (Mixed-lineage leukemia protein 3, MLL3) |
| MAP3K1 | (Mitogen-Activated Protein Kinase Kinase Kinase 1 |
| MDR | Multidrug resistant |
| MECOM | MDS1 and EVI1 complex locus protein EVI1 |
| MGMT | O-6-Methylguanine-DNA methyltransferase |
| miR | microRNA |
| miRNA | microRNA |
| MLH1 | MutL homolog 1 |
| MMP9 | Matrix metallopeptidase 9 |
| NEUROD1 | Neuronal differentiation 1 |
| NF1 | Neurofibromin 1 |
| PAX8 | Paired box gene 8 |
| PFKP | Phosphofructokinase, platelet |
| PIK3 | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PTEN | Phosphatase and tensin homolog |
| PTPRD | Protein tyrosine phosphatase, receptor type D |
| PVRL2 | Poliovirus receptor-related 2 |
| RASSF1A | Ras association domain-containing protein 1 |
| RB1 | Retinoblastoma 1 |
| RUNX1 | Runt-related transcription factor 1 |
| SF3B1 | Splicing factor 3B subunit 1 |
| TERT | Telomerase reverse transcriptase |
| TGF | Transforming growth factor |
| TNBC | triple negative basal cell breast cancer |
| TxF | Transcription factor |
| USF2 | Upstream stimulatory factor 2 |
| ZEB | Zinc finger E-box-binding homeobox 1 |
| ZMYND8 | Protein kinase C-binding protein 1 |
References
- Langerød, A.; Zhao, H.; Borgan, Ø.; Nesland, J.M.; Bukholm, I.R.; Ikdahl, T.; Kåresen, R.; Børresen-Dale, A.-L.; Jeffrey, S.S. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Burgess, M.; Puhalla, S. BRCA 1/2-Mutation related and sporadic breast and ovarian cancers: More alike than different. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Sadeqzadeh, E.; de Bock, D.E.; Thorne, R.F. Sleeping giants: Emerging roles for cadherins in health and disease. Med. Res. Rev. 2014, 34, 190–221. [Google Scholar] [CrossRef] [PubMed]
- Oeschger, F.M.; Wang, W.; Lee, S.; Garcia-Moreno, F.; Goffinet, A.M.; Arbones, M.I.; Rakic, S.; Molnar, Z. Gene expression analysis of the embryonic subplate. Cereb. Cortex 2012, 6, 1343–1359. [Google Scholar] [CrossRef] [PubMed]
- Ekumi, K.M.; Paculova, H.; Lenasi, T.; Popichalova, V.; Bosken, C.A.; Rybarikova, J.; Bryja, V.; Geyer, M.; Blazek, D.; Barboric, M. Ovarian carcinoma CKD12 mutations misregulate expression of DNA repair genes via Cdk12/CycK complex. Nucleic Acids Res. 2015, 5, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Koff, A.; Giordano, A.; Desai, D.; Yamashita, K.; Harper, J.W.; Elledge, S.; Nishimoto, T.; Morgan, D.O.; Franza, B.R.; Roberts, J.M. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992, 257, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.; Wilson, M.; Gonzalez, R.; Zhang, Y.; Perkins, A.S. The role of EVI1 in myeloid malignancies. Blood Cells Mol. Dis. 2014, 53, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Chiu, L.-Y.; Cox, B.; Aymard, F.; Clouaire, T.; Leung, J.W.; Cammarata, M.; Perez, M.; Agarwal, P.; Brodbelt, J.S.; et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015, 29, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Ling, F.; Kang, B.; Sun, X. Id proteins: Small molecules, mighty regulators. Curr. Top. Dev. Biol. 2014, 110, 189–216. [Google Scholar] [PubMed]
- Mittag, J.; Winterhager, E.; Bauer, K.; Grummer, R. Congenital hypothyroid femail pax8-deficient mice are infertile despite thyroid hormone replacement therapy. Endocrinology 2007, 148, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Dydensborg, A.B.; Rose, A.A.N.; Wilson, B.J.; Grote, D.; Paquet, M.; Giguère, V.; Siegel, P.M.; Bouchard, M. GATA3 inhibits breast cancer growth and pulmonary breast cancer metastasis. Oncogene 2009, 28, 2634–2642. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.J.; Dingwall, A.K. The cancer COMPASS: Navigating the functions of MLL complexes in cancer. Cancer Genet. 2015, 208, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.; Grothey, E.; Couch, F.J. The role of Tbx2 and Tbx3 in mammary development and tumorigenesis. J. Mammary Gland Biol. Neoplasia 2004, 2, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Melko, M.; Nguyen, L.S.; Shaw, M.; Jolly, L.; Bardoni, B.; Gecz, J. Loss of FMR2 further emphasizes the link between deregulation of immediate early response genes FOS and JUN and intellectual disability. Hum. Mol. Genet. 2013, 22, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Prat, J. New insights into ovarian cancer pathology. Ann. Oncol. 2012, 23, x111–x117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Helland, A.; Holm, R.; Skomedal, H.; Abeler, V.M.; Danielsen, H.E.; Tropé, C.G.; Børresen-Dale, A.-L.; Kristensen, G.B. TP53 mutations in early-stage ovarian carcinoma, relation to long-term survival. Br. J. Cancer 2004, 90, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kringen, P.; Kristensen, G.B.; Holm, R.; Baekelandt, M.M.O.; Olivier, M.; Skomedal, H.; Hainaut, P.; Tropé, C.G.; Abeler, V.M.; et al. Effect of the codon 72 polymorphism (c.215G>C, p.Arg72Pro) in combination with somatic sequence variants in the TP53 gene on survival in patients with advanced ovarian carcinoma. Hum. Mutat. 2004, 24, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.; Beiner, M.; Lubinski, J.; Lynch, H.T.; Moller, P.; Rosen, B.; Murphy, J.; Ghadirian, P.; Friedman, E.; Foulkes, W.D.; et al. Hereditary Ovarian Cancer Clinical Study Group. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 mutation. JAMA 2006, 296, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, C.R.; Levine, D.A.; Tang, L.H.; Allen, P.J.; Jarnagin, W.; Brennan, M.F.; Offit, K.; Robson, M.E. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2009, 27, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Tian, L.; Kähkönen, M.; Schwartzentruber, J.; Kircher, M.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium; Majewski, J.; Dyment, D.A.; Innes, A.M.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Severson, T.M.; Peeters, J.; Majewski, I.; Michaut, M.; Bosma, A.; Schouten, P.C.; Chin, S.F.; Pereira, B.; Goldgraben, M.A.; Bismeijer, T.; et al. BRCA1-line signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Mol. Oncol. 2015, 9, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Laddach, N.; Savola, S.P.; Vollebergh, M.A.; Oonk, A.M.; Imholz, A.L.; Wessels, L.F.; Wesseling, J.; Nederlof, P.M.; Rodenhuis, S. Quantitative copy number analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res. 2011, 13, R107. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol. Endocrinol. 2010, 24, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.-G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Derfoul, A.; Juan, A.H.; Difilippantonio, M.J.; Palanisamy, N.; Ried, T.; Sartorelli, V. Decreased microRNA-214 levels in breast cancer cells coincides with increased cell proliferation, invasion and accumulation of the Polycomb Ezh2 methyltransferase. Carcinogenesis 2011, 32, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- O’Day, E.; Lal, A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. microRNA-antagonism regulates breast cancer stemness and metastasis via TET family dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Jiao, Y.; Zhu, Y.; Wang, Y.; Zhu, J.; Cui, X.; Liu, Y.; He, Y.; Park, E.-Y.; Zhang, H.; et al. MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J. Biol. Chem. 2012, 287, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kong, W.; He, L.; Zhao, J.-J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V.; et al. MicroRNA expression profiling in human ovarian cancer: MiR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L.; et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther. 2008, 7, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Laios, A.; O’Toole, S.; Flavin, R.; Martin, C.; Kelly, L.; Ring, M.; Finn, S.P.; Barrett, C.; Loda, M.; Gleeson, N.; et al. Potential role of miR-9 and miR-223 in recurrent ovarian cancer. Mol. Cancer 2008, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Furneaux, H.; White, B.A. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol. 2007, 21, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Huang, R.; Wang, Z. Potential role of miR-100 in cancer diagnosis, prognosis, and therapy. Tumor Biol. 2015, 36, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wei, W.; Sun, Y.; Gao, J.; Wang, Q.; Zheng, J. MicroRNA-9 promotes tumorigenesis and mediates sensitivity to cisplatin in primary epithelial ovarian cancer. Tumor Biol. 2015, 36, 6867–6873. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Gao, Y.; Zhou, J.; Wang, J.; Zheng, F.; Guo, F.; Chang, A.; Li, X.; Wang, B. miR-233 regulates adipogenic and osteogenic differentiation of mesenchymal stem cells through a C/EBPs/miR-233/FGFR2 regualtory feedback loop. Stem Cells 2015, 33, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, M.; Mir, R.; Das, J.; Ahmad, I.; Javid, J.; Yadav, P.; Masroor, M.; Ahmad, S.; Ray, P.C.; Saxena, A. Expression of serum miR-200a, miR-200b, and miR-200c as candidate biomarkers in epithelial ovarian cancer and their association with clinicopathological features. Clin. Transl. Oncol. 2015, 17, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Mukohyama, J.; Nakamura, S.; Minami, H. MicroRNA regulation of human breast cancer stem cells. J. Clin. Med. 2016, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Guo, Y.; Chen, Z.; Wang, Y.; Yang, C.; Dudas, A.; Du, Z.; Liu, W.; Zou, Y.; Szabo, E.; et al. miR-203 functions as a tumor suppressor by inhibiting epithelial to mesenchymal transition in ovarian cancer. J. Cancer Sci. Ther. 2015, 7, 34–43. [Google Scholar] [PubMed]
- Zhang, S.; Lu, Z.; Unruh, A.K.; Ivan, C.; Baggerly, K.A.; Calin, G.A.; Li, Z.; Bast, R.C.; Le, X. Clinically relevant microRNAs in ovarian cancer. Mol. Cancer Res. 2015, 13, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.D.C.; Dahl, R.; Kruichak, J.N.; Hudson, L.G. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia 2009, 11, 1208–1215. [Google Scholar] [CrossRef]
- Byler, S.; Sarkar, S. Do epigenetic drug treatments hold the key to killing cancer progenitor cells? Epigenomics 2014, 6, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.Q. Genetic alterations of histone lysine methyltransferases and their significance in breast cancer. Oncotarget 2014, 6, 2466–2482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.Y.; Lei, P.J.; Zhang, X.; Zheng, J.Y.; Wang, H.Y.; Zhao, J.; Li, Y.M.; Ye, M.; Li, L.; Wei, G.; et al. Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clin. Epigenet. 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.J.; Shah, J.S.; Cole, A.J. Histones and Their Modifications in Ovarian Cancer—Drivers of Disease and Therapeutic Targets. Front. Oncol. 2014, 4, 144. [Google Scholar] [PubMed]
- Rhie, S.K.; Hazelett, D.J.; Coetzee, S.G.; Yan, C.; Noushmehr, H.; Coetzee, G.A. Nucleosome positioning and histone modifications define relationships between regulatory elements and nearby gene expression in breast epithelial cells. BMC Genom. 2014, 15, 331. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Cross, N.C.P. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Kron, K.J.; Bailey, S.D.; Lupien, M. Enhancer altertions in cancer: A source for a cell identity crisis. Genome Med. 2014, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Niederriter, A.R.; Varshney, A.; Parker, S.C.; Martin, D.M. Super enhancers in cancers, complex diseases, and developmental disorders. Genes (Basel) 2015, 4, 1183–1200. [Google Scholar] [CrossRef] [PubMed]
- Dowen, J.M.; Fan, Z.P.; Hnisz, D.; Ren, G.; Abraham, B.J.; Zhang, L.N.; Weintraub, A.S.; Schuijers, J.; Lee, J.I.; Zhao, K.; et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 2014, 159, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maurano, M.T.; Qu, H.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sandstrom, R.; et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Goldgar, S.; Byler, S.; Rosenthal, S.; Heerboth, S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: Sensitization of cancer cells by combination therapy. Epigenomics 2013, 5, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Koukoura, O.; Spandidos, D.A.; Daponte, A.; Sifakis, S. DNA methylation profiles in ovarian cancer: Implication in diagnosis and therapy (Review). Mol. Med. Rep. 2014, 10, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lavaud, P.; Andre, F. Strategies to overcome trastuzumab resistance in HER2-overexpressing breast cancers: Focus on new data from clinical trials. BMC Med. 2014, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Deftereos, G. DNA hypermethylation, Her-2/neu overexpression and p53 mutations in ovarian carcinoma. Gynecol. Oncol. 2008, 111, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, H.; Millinger, S.; Goebel, G.; Müller-Holzner, E.; Marth, C.; Laird, P.W.; Widschwendter, M. Breast cancer DNA methylation profiles in cancer cells and tumor stroma: Association with HER-2/neu status in primary breast cancer. Cancer Res. 2006, 66, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, B.M.; Wingren, S.; Motlagh, P.B.; Nilsson, T.K. Whole genome DNA methylation signature of HER2-positive breast cancer. Epigenetics 2014, 9, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Rodenhiser, D.I.; Andrews, J.; Kennette, W.; Sadikovic, B.; Mendlowitz, A.; Tuck, A.B.; Chambers, A.F. Epigenetic mapping and functional analysis in a breast cancer metastasis model using whole-genome promoter tiling microarrays. Breast Cancer Res. 2008, 10. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar] [PubMed]
- Do, S.-I.; Ko, E.; Kang, S.Y.; Lee, J.E.; Nam, S.J.; Cho, E.Y.; Kim, D.-H. Aberrant DNA methylation of integrin α4 in human breast cancer. Tumour Biol. 2014, 35, 7079–7084. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Kersch, C.; Beeson, K.A.; Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. Interactions between αv-integrin and HER2 and their role in the invasive phenotype of breast cancer cells in vitro and in rat brain. PLoS ONE 2015, 10, e0131842. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011, 31, 2723–2732. [Google Scholar] [PubMed]
- Zuo, T.; Liu, T.-M.; Lan, X.; Weng, Y.-I.; Shen, R.; Gu, F.; Huang, Y.-W.; Liyanarachchi, S.; Deatherage, D.E.; Hsu, P.-Y.; et al. Epigenetic silencing mediated through activated PI3K/AKT signaling in breast cancer. Cancer Res. 2011, 71, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Keyomarsi, K.; Gonzales, F.A.; Velicescu, M.; Jones, P.A. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000, 28, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Samorodnitsky, E.; Ghosh, E.; Mazumder, S.; Sarkar, S. Methylation by DNMT1 is more Efficient in Chronic Lymphocytic Lymphoma Cells than in Normal Cells. J. Proteom. Bioinform. 2014, 1. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, S.; Song, E.; Liu, S. The roles of ncRNAs and histone-modifiers in regulating breast cancer stem cells. Protein Cell 2016, 7, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16472–16477. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gauthier, A.; Postma, M.J.; Ribassin-Majed, L.; Largeron, N.; Bresse, X. A critical review of cost-effectiveness analyses of vaccinating males against human papillomavirus. Hum. Vaccines Immunother. 2013, 9, 2285–2295. [Google Scholar] [CrossRef]
- Matei, D.; Sill, M.W.; Lankes, H.A.; DeGeest, K.; Bristow, R.E.; Mutch, D.; Yamada, S.D.; Cohn, D.; Calvert, V.; Farley, J.; et al. Activity of sorafenib in recurrent ovarian cancer and primary peritoneal carcinomatosis: A gynecologic oncology group trial. J. Clin. Oncol. 2011, 29, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Longacre, M.; Tatur, N.; Heerboth, S.; Lapinska, K. Encyclopedia of Analytical Chemistry; Histone Decatylases (HDACs): Function, Mechanism & Inhibition; Wiley Online Library: London, UK, 2014; pp. 1–9. [Google Scholar]
- Chang, H.G.; Kim, S.J.; Chung, K.-W.; Noh, D.-Y.; Kwon, Y.; Lee, E.S.; Kang, H.-S. Tamoxifen-resistant breast cancers show less frequent methylation of the estrogen receptor beta but not the estrogen receptor alpha gene. J. Mol. Med. 2005, 83, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Glasspool, R.M.; Teodoridis, J.M.; Brown, R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br. J. Cancer 2006, 94, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, G.; Vass, J.K.; Oien, K.A.; Siddiqui, N.; Curto-Garcia, J.; Brown, R. Demethylation of the MCJ gene in stage III/IV epithelial ovarian cancer and response to chemotherapy. Gynecol. Oncol. 2005, 97, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Gifford, G.; Paul, J.; Vasey, P.A.; Kaye, S.B.; Brown, R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin. Cancer Res. 2004, 10, 4420–4426. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Unterberg, M.; Koschmieder, S.; zur Stadt, U.; Brunnberg, U.; Verbeek, W.; Büchner, T.; Berdel, W.E.; Serve, H.; Müller-Tidow, C. DNA methylation of tumor suppressor genes in clinical remission predicts the relapse risk in acute myeloid leukemia. Cancer Res. 2007, 67, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.; Huang, T.H.-M.; Brown, R.; Nephew, K.P. The epigenetics of ovarian cancer drug resistance and resensitization. Am. J. Obstet. Gynecol. 2004, 191, 1552–1572. [Google Scholar] [CrossRef] [PubMed]
- Mataga, M.A.; Rosenthal, S.; Heerboth, S.; Devalapalli, A.; Kokolus, S.; Evans, L.R.; Longacre, M.; Housman, G.; Sarkar, S. Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res. 2012, 32, 2523–2529. [Google Scholar] [PubMed]
- Lapinska, K.; Housman, G.; Heerboth, S.; Longacre, M.; Sarkar, S. Anticancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res. 2012, 32, 2523–2529. [Google Scholar]
- Fujji, S.; Luo, R.Z.; Yuan, J.; Kadota, M.; Oshimura, M.; Dent, S.R.; Kondo, Y.; Issa, J.J.; Bast, R.C.; Yu, Y. Reactivation of the silenced and imprinted alleles of ARHI is associated with increased histone H3 acetylation and decreased histone H3 lysine 9 methylation. Hum. Mol. Genet. 2003, 12, 1791–1800. [Google Scholar] [CrossRef]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P.; Clarke, C.J.P.; Sharkey, J.; Anthony, D.A.; Banks, K.-M.; Haynes, N.M.; Gangatirkar, P.; Stanley, K.; et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl. Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar] [CrossRef] [PubMed]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non–small cell lung cancer. Cancer Discov. 2011. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Faller, D.T. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleotides 2011, 21, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Alfraidi, A.; Sacco, K.; Alshaker, H.; Muhammad, A.; Monzon, L. Histone deacetylases as new therapy targets for platinum-resistant epithelial ovarian cancer. J. Cancer Res. Clin. Oncol. 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gyparaki, M.T.; Papavassiliou, A.G. Epigenetic pathways offer targets for ovarian cancer treatment. Clin. Ovarian Other Gynecol. Cancer 2014, 4, 71. [Google Scholar] [CrossRef]
- Dong, Y.; Batra, J.; Anand, K.S.; Bapat, S.; Clements, A.J. Transforming the future of treatment for ovarian cancer. Clin. Exp. Pharmacol. 2014, 4, 3. [Google Scholar]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Byler, S.; Goldgar, S.; Heerboth, S.; Leary, M.; Housman, G.; Moulton, K.; Sarkar, S. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014, 34, 1071–1077. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).