
Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Kidney Physiology Relevant to Acute Kidney Injury (AKI)
3. Animal Models of AKI
4. Pathophysiological Process of Acute Kidney Injury
5. Mitochondria in Kidney Health and Disease
5.1. Mitochondrial Reactive Oxygen Species (ROS) Production
5.2. Regulation of Mitochondria Dynamics
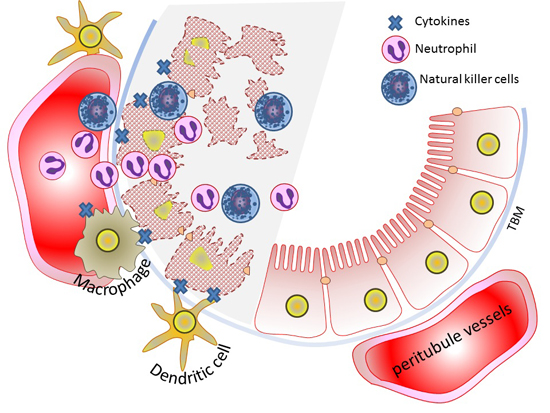
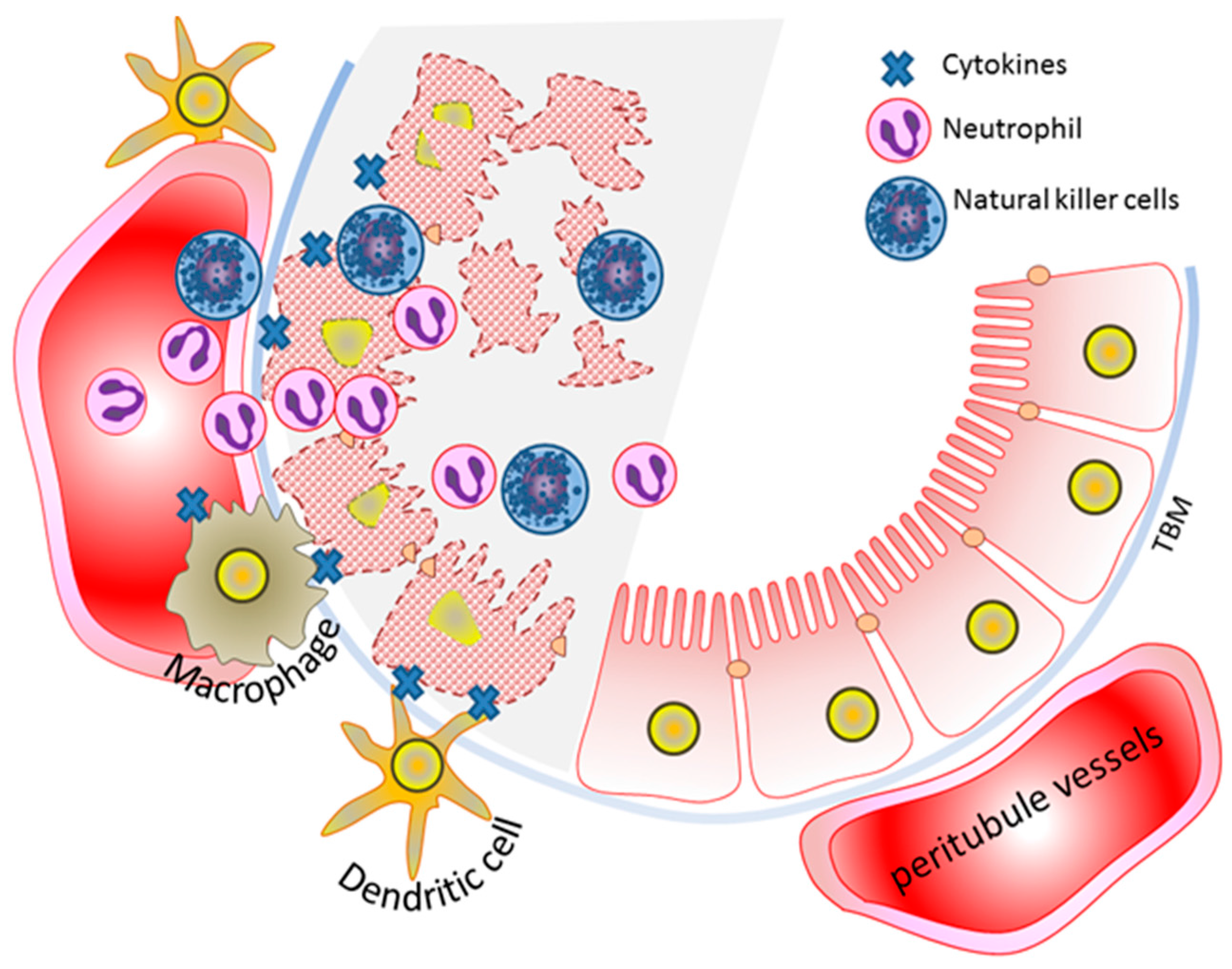
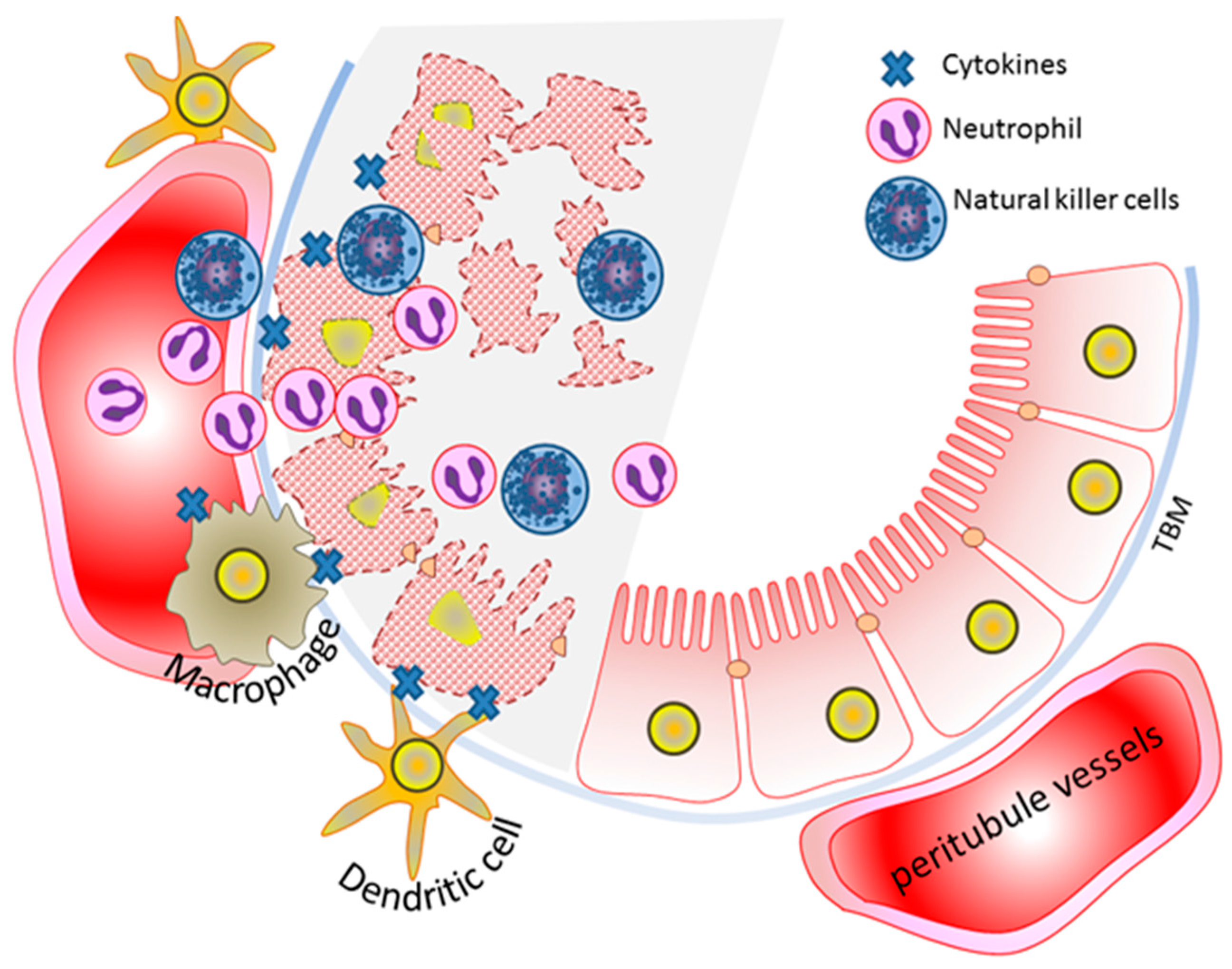
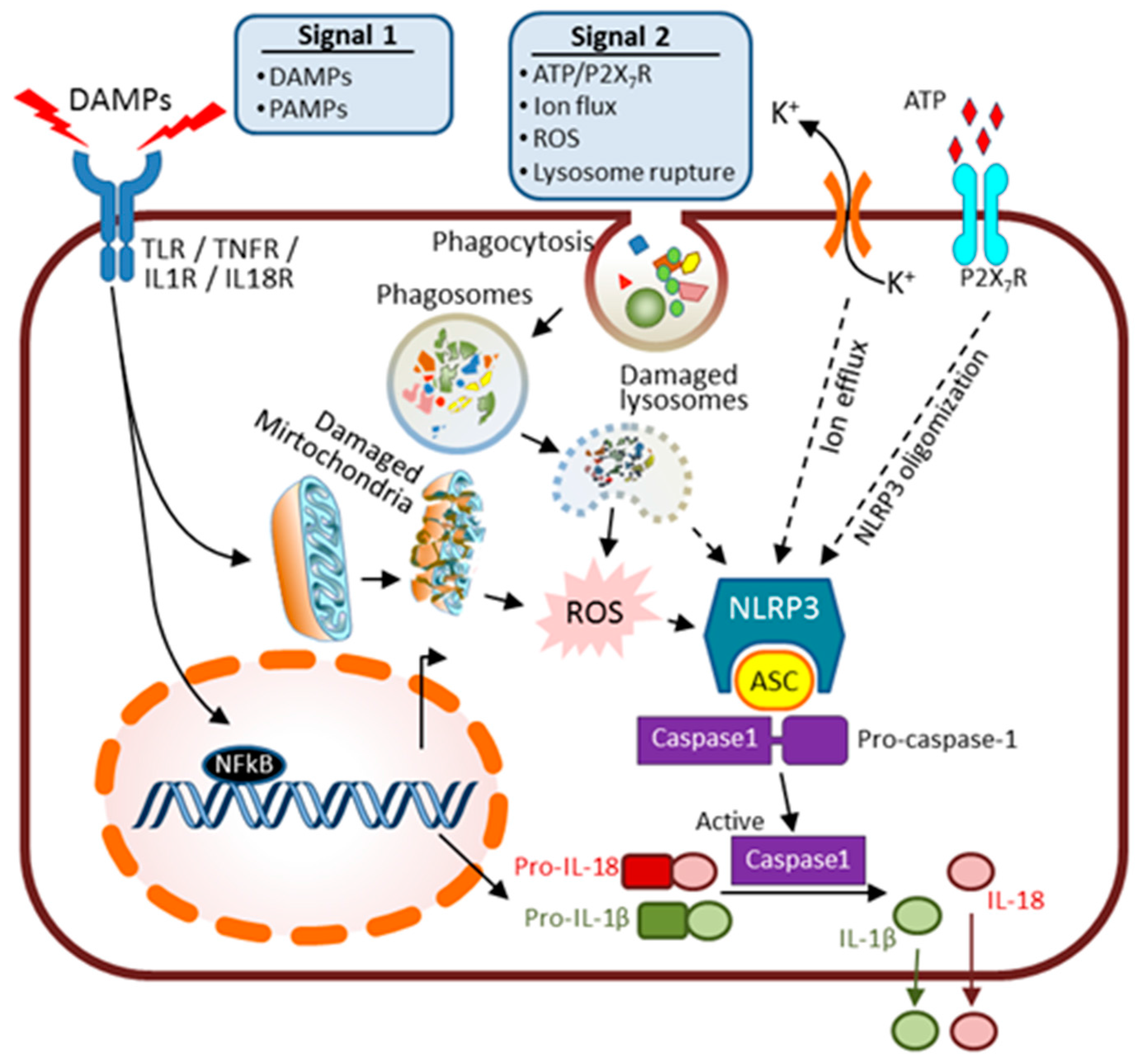
6. Innate Immunity and AKI
7. Kidney Repair and Regeneration after AKI
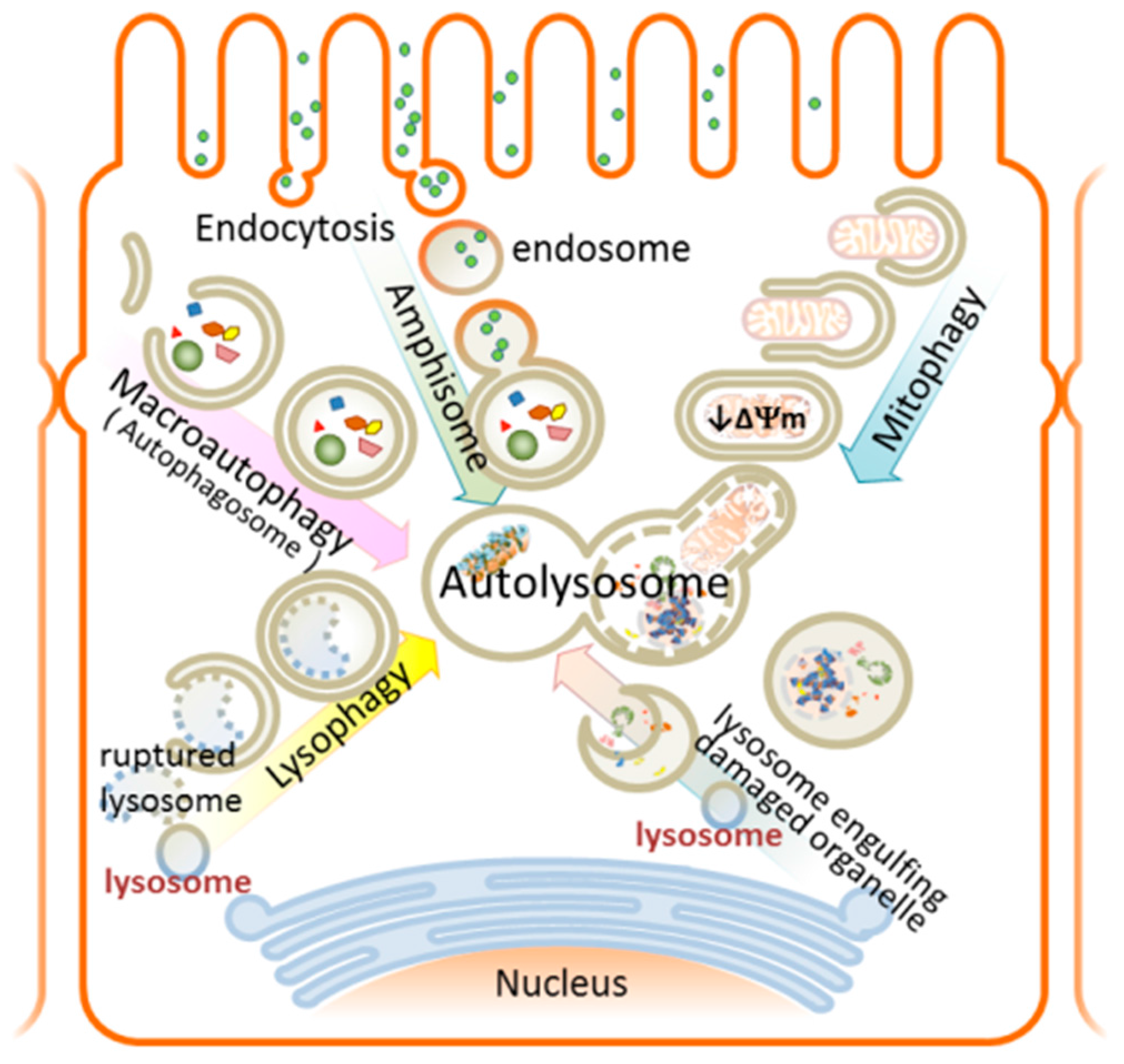
8. Autophagy
9. Autophagy and AKI—Overview
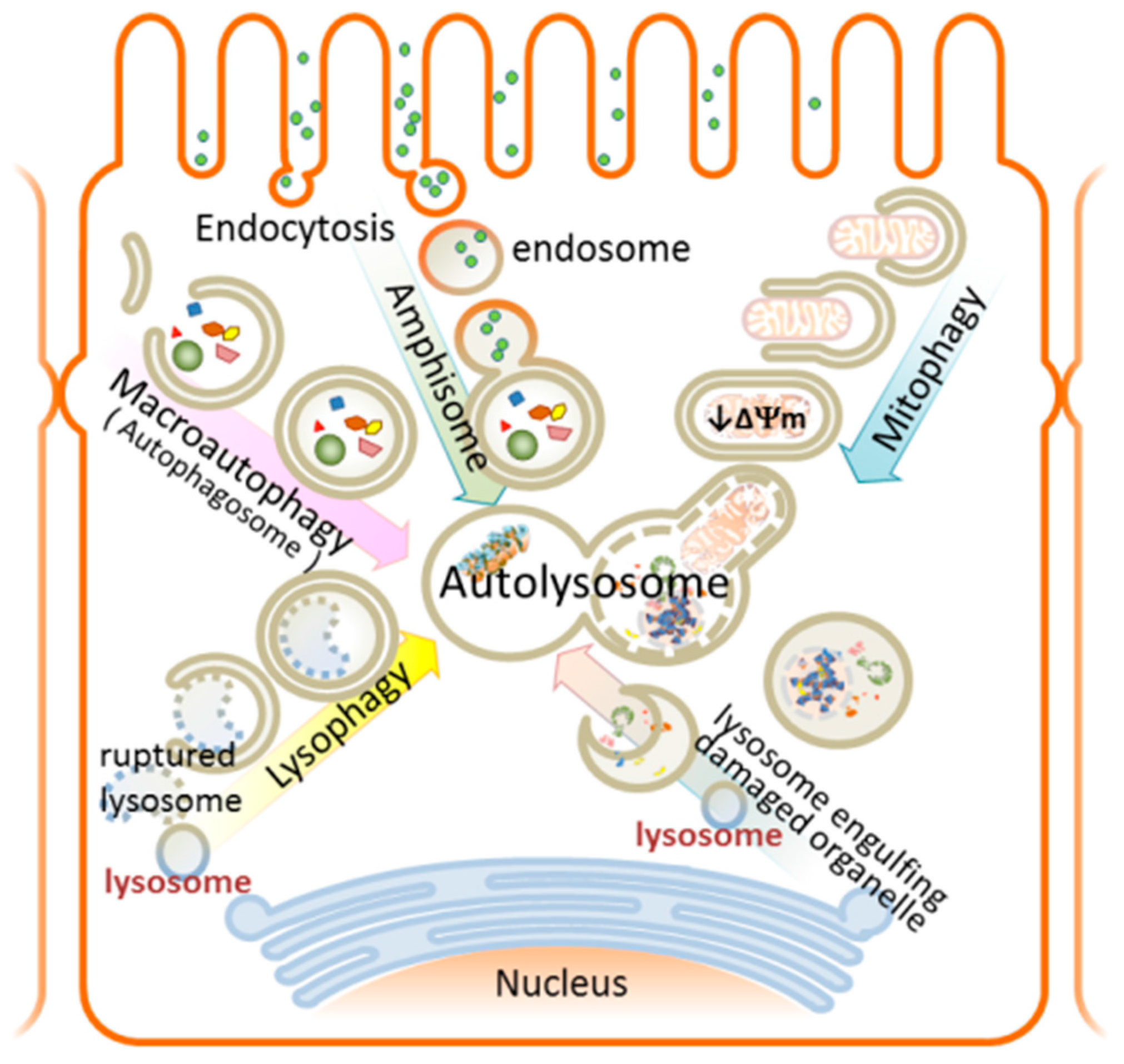
9.1. Selective Autophagy in AKI
9.1.1. Mitophagy—Mechanism and Involvement in AKI
9.1.2. Lysophagy—Mechanism and Involvement in AKI
10. Innovative Preclinical AKI Therapy—Targeting Cell Death and Tissue Regeneration
11. Concluding Remarks and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AKI | Acute kidney injury |
| CKD | Chronic kidney disease |
| IRI | Ischemic reperfusion injury |
| PTE | Proximal tubule epithelium |
| ROS | Reactive oxygen species |
| CLP | Cecal ligation and puncture model of sepsis |
| DAMPs | Damage-associated molecular patterns |
| PAMPs | Pathogen-associated molecular patterns |
| TLR | Toll-like receptors |
| OMM | Outer mitochondrial membrane |
| IMM | Inner mitochondrial membrane |
References
- Lameire, N.H.; Bagga, A.; Cruz, D.; de Maeseneer, J.; Endre, Z.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Cerda, J.; Burdmann, E.A.; Tonelli, M.; Garcia-Garcia, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): A human rights case for nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Rewa, O.; Bagshaw, S.M. Acute kidney injury-epidemiology, outcomes and economics. Nat. Rev. Nephrol. 2014, 10, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xing, G.; Wang, L.; Wu, Y.; Li, S.; Xu, G.; He, Q.; Chen, J.; Chen, M.; Liu, X.; et al. Acute kidney injury in China: A cross-sectional survey. Lancet 2015, 386, 1465–1471. [Google Scholar] [CrossRef]
- Silver, S.A.; Cardinal, H.; Colwell, K.; Burger, D.; Dickhout, J.G. Acute kidney injury: Preclinical innovations, challenges, and opportunities for translation. Can. J. Kidney Health Dis. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Kdigo, A.K.I. Kidney Disease Improving Global Outcomes. AKI Work Group: Clinical practice guideline for acute kidney injury. Kidney Int. Suppl. 2012, 2, 1–138. [Google Scholar]
- Sancho-Martinez, S.M.; Lopez-Novoa, J.M.; Lopez-Hernandez, F.J. Pathophysiological role of different tubular epithelial cell death modes in acute kidney injury. Clin. Kidney J. 2015, 8, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.P.; Zeidel, M.L. Homeostasis, the milieu interieur, and the wisdom of the nephron. Clin. J. Am. Soc. Nephrol. 2014, 9, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Unwin, R.J.; Parker, N.; Duchen, M.R. Multiphoton imaging reveals differences in mitochondrial function between nephron segments. J. Am. Soc. Nephrol. 2009, 20, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosenberger, C.; Rosen, S. Acute kidney injury: Lessons from experimental models. Contrib. Nephrol. 2011, 169, 286–296. [Google Scholar] [PubMed]
- Ueno, K.; Sato, H. Sex-related differences in pharmacokinetics and pharmacodynamics of anti-hypertensive drugs. Hypertens. Res. 2012, 35, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Nicolson, T.J.; Hammons, G.; Word, B.; Green-Knox, B.; Lyn-Cook, B. Expression of drug transporters in human kidney: Impact of sex, age, and ethnicity. Biol. Sex Differ. 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Breljak, D.; Brzica, H.; Sweet, D.H.; Anzai, N.; Sabolic, I. Sex-dependent expression of Oat3 (Slc22a8) and Oat1 (Slc22a6) proteins in murine kidneys. Am. J. Physiol. Ren. Physiol. 2013, 304, F1114–F1126. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, S.M.; Langenberg, C.; Wan, L.; May, C.N.; Bellomo, R. A systematic review of urinary findings in experimental septic acute renal failure. Crit. Care Med. 2007, 35, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Junemann, A.; Muthuraman, A.; Jaggi, A.S.; Singh, N.; Grover, K.; Dhawan, R. Animal models of acute renal failure. Pharmacol. Rep. 2012, 64, 31–44. [Google Scholar] [CrossRef]
- Ortiz, A.; Sanchez-Nino, M.D.; Izquierdo, M.C.; Martin-Cleary, C.; Garcia-Bermejo, L.; Moreno, J.A.; Ruiz-Ortega, M.; Draibe, J.; Cruzado, J.M.; Garcia-Gonzalez, M.A.; et al. Translational value of animal models of kidney failure. Eur. J. Pharmacol. 2015, 759, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Sander, V.; Davidson, A.J. Kidney injury and regeneration in zebrafish. Semin. Nephrol. 2014, 34, 437–444. [Google Scholar] [CrossRef] [PubMed]
- McKee, R.A.; Wingert, R.A. Zebrafish renal pathology: Emerging models of acute kidney injury. Curr. Pathobiol. Rep. 2015, 3, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Trevino, A.E.; Zhang, F. Genome editing using Cas9 nickases. Methods Enzymol. 2014, 546, 161–174. [Google Scholar] [PubMed]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [PubMed]
- Togel, F.; Westenfelder, C. Recent advances in the understanding of acute kidney injury. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bonventre, J.V.; Parrish, A.R. The aging kidney: Increased susceptibility to nephrotoxicity. Int. J. Mol. Sci. 2014, 15, 15358–15376. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef] [PubMed]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Schaefer, L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Stallons, L.J.; Funk, J.A.; Schnellmann, R.G. Mitochondrial homeostasis in acute organ failure. Curr. Pathobiol. Rep. 2013, 1, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [PubMed]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Ren. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef] [PubMed]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Lenaers, G.; Hajnoczky, G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J. Cell Biol. 2014, 205, 179–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Jin, P.; Yu, L.; Wang, Y.; Han, L.; Shi, T.; Li, X. Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS ONE 2014, 9, e92810. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126 Pt 3, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; de Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. The regulation of mitochondrial morphology: Intricate mechanisms and dynamic machinery. Cell Signal. 2011, 23, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M. Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Ren. Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef] [PubMed]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Molitoris, B.A. Dynamic multiphoton microscopy: Focusing light on acute kidney injury. Physiology (Bethesda) 2014, 29, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Rhodes, G.J.; Sandoval, R.M.; Corridon, P.R.; Molitoris, B.A. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013, 83, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Crawford, C.; Unwin, R.J.; Duchen, M.R.; Peppiatt-Wildman, C.M. Multiphoton imaging of the functioning kidney. J. Am. Soc. Nephrol. 2011, 22, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Valles, P.G.; Lorenzo, A.G.; Bocanegra, V.; Valles, R. Acute kidney injury: What part do Toll-like receptors play? Int. J. Nephrol. Renovasc. Dis. 2014, 7, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.; Kirschning, C.J.; Akira, S.; van der Poll, T.; Weening, J.J.; Florquin, S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Investig. 2005, 115, 2894–2903. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, H.; Pollock, C.; Komala, M.G.; Chadban, S.; Wu, H.; Panchapakesan, U. The role of Toll-like receptor proteins (TLR) 2 and 4 in mediating inflammation in proximal tubules. Am. J. Physiol. Ren. Physiol. 2013, 305, F143–F154. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. NLRP3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Deplano, S.; Cook, H.T.; Russell, R.; Franchi, L.; Schneiter, S.; Bhangal, G.; Unwin, R.J.; Pusey, C.D.; Tam, F.W.; Behmoaras, J. P2X7 receptor-mediated NLRP3-inflammasome activation is a genetic determinant of macrophage-dependent crescentic glomerulonephritis. J. Leukoc. Biol. 2013, 93, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Pirenne, J.; Jochmans, I. Autophagy in renal ischemia-reperfusion injury: Friend or foe? Am. J. Transplant. 2014, 14, 1464–1465. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Latz, E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Matsuzawa, A.; Yoshimura, A.; Ichijo, H. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J. Biol. Chem. 2014, 289, 32926–32936. [Google Scholar] [CrossRef] [PubMed]
- Hauenstein, A.V.; Zhang, L.; Wu, H. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr. Opin. Struct. Biol. 2015, 31, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Hu, C.; Ding, G.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. Albumin impairs renal tubular tight junctions via targeting the NLRP3 inflammasome. Am. J. Physiol. Ren. Physiol. 2015, 308, F1012–F1019. [Google Scholar] [CrossRef] [PubMed]
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef] [PubMed]
- Darisipudi, M.N.; Knauf, F. An update on the role of the inflammasomes in the pathogenesis of kidney diseases. Pediatr. Nephrol. 2015, 31, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Leemans, J.C.; Kors, L.; Anders, H.J.; Florquin, S. Pattern recognition receptors and the inflammasome in kidney disease. Nat. Rev. Nephrol. 2014, 10, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, J.H.; Haegele, H.; Muller, S.; Anders, H.J. Danger control programs cause tissue injury and remodeling. Int. J. Mol. Sci. 2013, 14, 11319–11346. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Grobmayr, R.; Ryu, M.; Lorenz, G.; Hartter, I.; Mulay, S.R.; Susanti, H.E.; Kobayashi, K.S.; Flavell, R.A.; Anders, H.J. Macrophage phenotype controls long-term AKI outcomes—Kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 2014, 25, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Honma, S. Regeneration of injured renal tubules. J. Pharmacol. Sci. 2014, 124, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, T.; Humphreys, B.D. Controversies on the origin of proliferating epithelial cells after kidney injury. Pediatr. Nephrol. 2014, 29, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Tetta, C. Paracrine/endocrine mechanism of stem cells on kidney repair: Role of microvesicle-mediated transfer of genetic information. Curr. Opin. Nephrol. Hypertens. 2010, 19, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sabin, K.; Kikyo, N. Microvesicles as mediators of tissue regeneration. Transl. Res. 2014, 163, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Humphreys, B.D.; Bonventre, J.V. Pathophysiology of acute kidney injury to chronic kidney disease: Maladaptive repair. Contrib. Nephrol. 2011, 174, 149–155. [Google Scholar] [PubMed]
- Barnes, J.L.; Glass, W.F., 2nd. Renal interstitial fibrosis: A critical evaluation of the origin of myofibroblasts. Contrib. Nephrol. 2011, 169, 73–93. [Google Scholar] [PubMed]
- Chuang, P.Y.; Menon, M.C.; He, J.C. Molecular targets for treatment of kidney fibrosis. J. Mol. Med. (Berl.) 2013, 91, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [PubMed]
- Wu, X.; Won, H.; Rubinsztein, D.C. Autophagy and mammalian development. Biochem. Soc. Trans. 2013, 41, 1489–1494. [Google Scholar] [PubMed]
- Paulus, G.L.; Xavier, R.J. Autophagy and checkpoints for intracellular pathogen defense. Curr. Opin. Gastroenterol. 2015, 31, 14–23. [Google Scholar] [PubMed]
- Steele, S.; Brunton, J.; Kawula, T. The role of autophagy in intracellular pathogen nutrient acquisition. Front. Cell. Infect. Microbiol. 2015, 5, 51. [Google Scholar] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Autophagy in Cell Life and Cell Death. Curr. Top. Dev. Biol. 2015, 114, 67–91. [Google Scholar] [PubMed]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Chittaranjan, S.; Bortnik, S.; Gorski, S.M. Monitoring Autophagic Flux by Using Lysosomal Inhibitors and Western Blotting of Endogenous MAP1LC3B. Cold Spring Harb. Protoc. 2015, 2015, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Martinez, L.; Boya, P. Autophagic flux determination in vivo and ex vivo. Methods 2015, 75, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Andres, A.M.; Sin, J.; Taylor, D.P. Untangling autophagy measurements: All fluxed up. Circ. Res. 2015, 116, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Z.V.; Hill, J.A.; Lin, F. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J. Am. Soc. Nephrol. 2014, 25, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.; Isaka, Y.; Takabatake, Y.; Tanaka, H.; Koike, M.; Shibata, M.; Uchiyama, Y.; Takahara, S.; Imai, E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2008, 368, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Liu, K.; Luo, J.; Dong, Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am. J. Pathol. 2010, 176, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N.; et al. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Z.; Jiang, L.; Mao, J.; Zeng, Z.; Fang, L.; He, W.; Yuan, W.; Yang, J.; Dai, C. Rictor/mTORC2 protects against cisplatin-induced tubular cell death and acute kidney injury. Kidney Int. 2014, 86, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Grahammer, F.; Haenisch, N.; Steinhardt, F.; Sander, L.; Roerden, M.; Arnold, F.; Cordts, T.; Wanner, N.; Reichardt, W.; Kerjaschki, D.; et al. mTORC1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc. Natl. Acad. Sci. USA 2014, 111, E2817–E2826. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.W.; Tsai, K.L.; Wang, L.F.; Chen, Y.H.; Chiang, P.C.; Chuang, S.M.; Hsu, C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 2012, 37, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Howell, G.M.; Guo, L.; Collage, R.D.; Loughran, P.A.; Zuckerbraun, B.S.; Rosengart, M.R. CaMKIV-dependent preservation of mTOR expression is required for autophagy during lipopolysaccharide-induced inflammation and acute kidney injury. J. Immunol. 2014, 193, 2405–2415. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.M.; Gomez, H.; Collage, R.D.; Loughran, P.; Zhang, X.; Escobar, D.A.; Billiar, T.R.; Zuckerbraun, B.S.; Rosengart, M.R. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS ONE 2013, 8, e69520. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Ceulemans, L.J.; Agostinis, P.; Monbaliu, D.; Naesens, M.; Pirenne, J.; Jochmans, I. Autophagy and the kidney: Implications for ischemia-reperfusion injury and therapy. Am. J. Kidney Dis. 2015, 66, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Choi, M.E. Autophagy in kidney health and disease. Antioxid. Redox Signal. 2014, 20, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial function and disturbances in the septic kidney. Semin. Nephrol. 2015, 35, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar]
- Ney, P.A. Normal and disordered reticulocyte maturation. Curr. Opin. Hematol. 2011, 18, 152–157. [Google Scholar]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar]
- Wei, Q.; Dong, G.; Chen, J.K.; Ramesh, G.; Dong, Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013, 84, 138–148. [Google Scholar]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar]
- Christensen, E.I.; Verroust, P.J.; Nielsen, R. Receptor-mediated endocytosis in renal proximal tubule. Pflugers Arch. 2009, 458, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Gleich, K.; Fraser, S.A.; Katerelos, M.; Mount, P.F.; Power, D.A. Limited capacity of proximal tubular proteolysis in mice with proteinuria. Am. J. Physiol. Ren. Physiol. 2013, 304, F1009–F1019. [Google Scholar] [CrossRef] [PubMed]
- Pourahmad, J.; Hosseini, M.J.; Eskandari, M.R.; Shekarabi, S.M.; Daraei, B. Mitochondrial/lysosomal toxic cross-talk plays a key role in cisplatin nephrotoxicity. Xenobiotica 2010, 40, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Wang, Y.; Minto, A.W.; Quigg, R.J.; Cunningham, P.N. Acute renal failure in endotoxemia is dependent on caspase activation. J. Am. Soc. Nephrol. 2004, 15, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Linkermann, A.; Anders, H.J. Necroinflammation in kidney disease. J. Am. Soc. Nephrol. 2015, 27, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Chen, Y.; Zhang, L.; Jiang, F.; Wang, W.; Ye, Z.; Liu, S.; Yu, C.; Shi, W. Necroptosis, a novel form of caspase-independent cell death, contributes to renal epithelial cell damage in an ATP-depleted renal ischemia model. Mol. Med. Rep. 2014, 10, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Patschan, D.; Patschan, S.; Muller, G.A. Endothelial progenitor cells in acute ischemic kidney injury: Strategies for increasing the cells’ renoprotective competence. Int. J. Nephrol. 2011, 2011, 828369. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Cui, R.; Peng, L.; Ma, J.; Chen, X.; Xie, R.J.; Li, B. Mesenchymal stem cells, not conditioned medium, contribute to kidney repair after ischemia-reperfusion injury. Stem Cell Res. Ther. 2014, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.M. Vascular and circulating microRNAs in renal ischaemia-reperfusion injury. J. Physiol. 2015, 593, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Kim, J.; Traylor, A.M.; Sanders, P.W.; Balla, J.; Agarwal, A.; Curtis, L.M. Paracrine effects of mesenchymal stem cells in cisplatin-induced renal injury require heme oxygenase-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F254–F262. [Google Scholar] [CrossRef] [PubMed]
- Sumida, M.; Doi, K.; Ogasawara, E.; Yamashita, T.; Hamasaki, Y.; Kariya, T.; Takimoto, E.; Yahagi, N.; Nangaku, M.; Noiri, E. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J. Am. Soc. Nephrol. 2015, 26, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.X.; Wu, W.H.; Qiu, H.Y.; Bo, H.; Huang, S.M. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of DRP-1 translocation. J. Nephrol. 2013, 26, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.L.; Wang, L.T.; Huang, K.H.; Wang, C.C.; Chiang, C.K.; Liu, S.H. Quercetin attenuates renal ischemia/reperfusion injury via an activation of AMP-activated protein kinase-regulated autophagy pathway. J. Nutr. Biochem. 2014, 25, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Clatworthy, M.R. Immunotherapy for acute kidney injury. Immunotherapy 2012, 4, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, E.; Minambres, E.; Ruiz, J.C.; Ballesteros, A.; Pinera, C.; Quintanar, J.; Fernandez-Fresnedo, G.; Palomar, R.; Gomez-Alamillo, C.; Arias, M. Prediction of delayed graft function by means of a novel web-based calculator: A single-center experience. Am. J. Transplant. 2012, 12, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Chapal, M.; Le Borgne, F.; Legendre, C.; Kreis, H.; Mourad, G.; Garrigue, V.; Morelon, E.; Buron, F.; Rostaing, L.; Kamar, N.; et al. A useful scoring system for the prediction and management of delayed graft function following kidney transplantation from cadaveric donors. Kidney Int. 2014, 86, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duann, P.; Lianos, E.A.; Ma, J.; Lin, P.-H. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. Int. J. Mol. Sci. 2016, 17, 662. https://doi.org/10.3390/ijms17050662
Duann P, Lianos EA, Ma J, Lin P-H. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. International Journal of Molecular Sciences. 2016; 17(5):662. https://doi.org/10.3390/ijms17050662
Chicago/Turabian StyleDuann, Pu, Elias A. Lianos, Jianjie Ma, and Pei-Hui Lin. 2016. "Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury" International Journal of Molecular Sciences 17, no. 5: 662. https://doi.org/10.3390/ijms17050662