
Inhibition of Toll-Like Receptor 4 Signaling Mitigates Microvascular Loss but Not Fibrosis in a Model of Ischemic Acute Kidney Injury

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
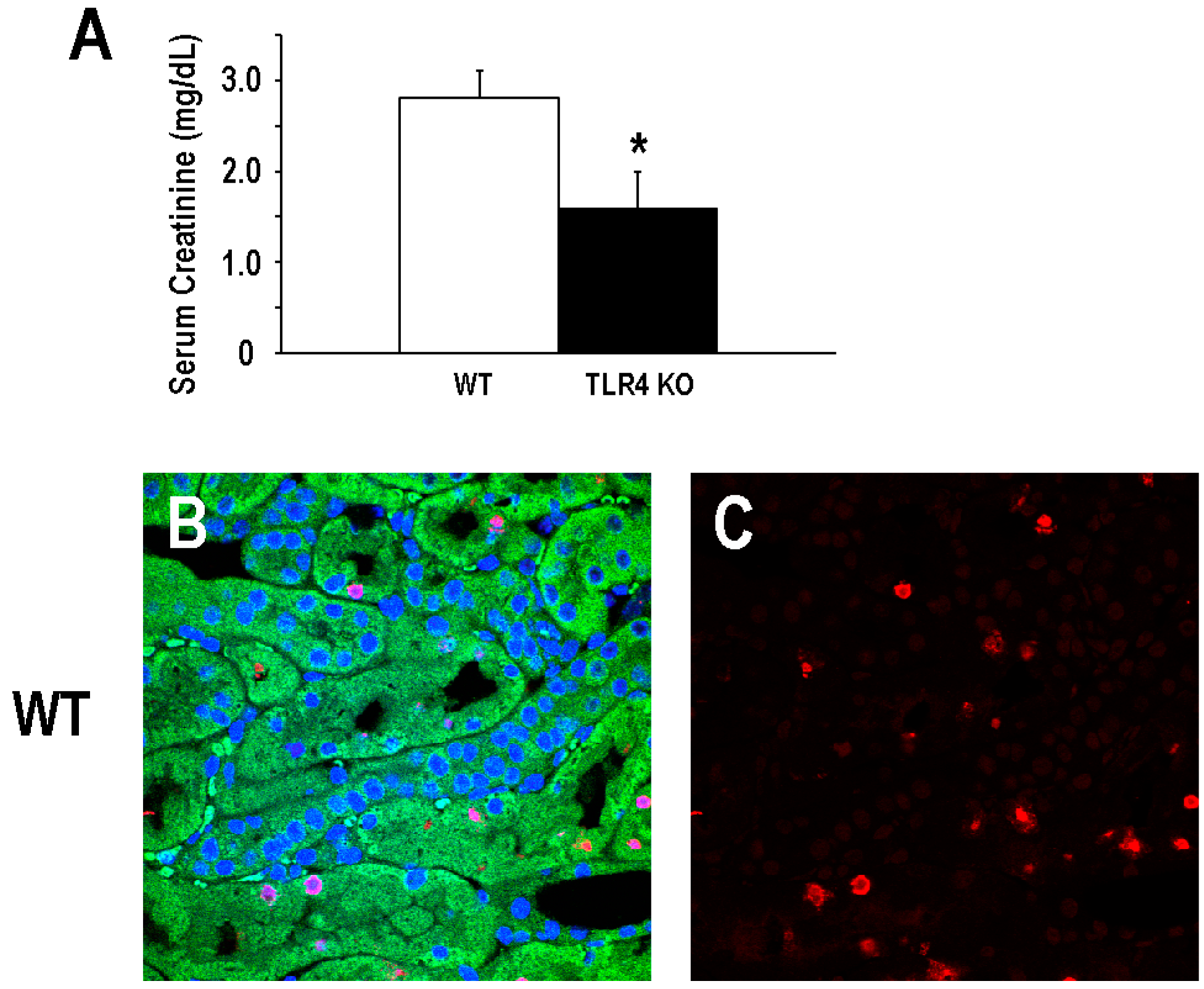
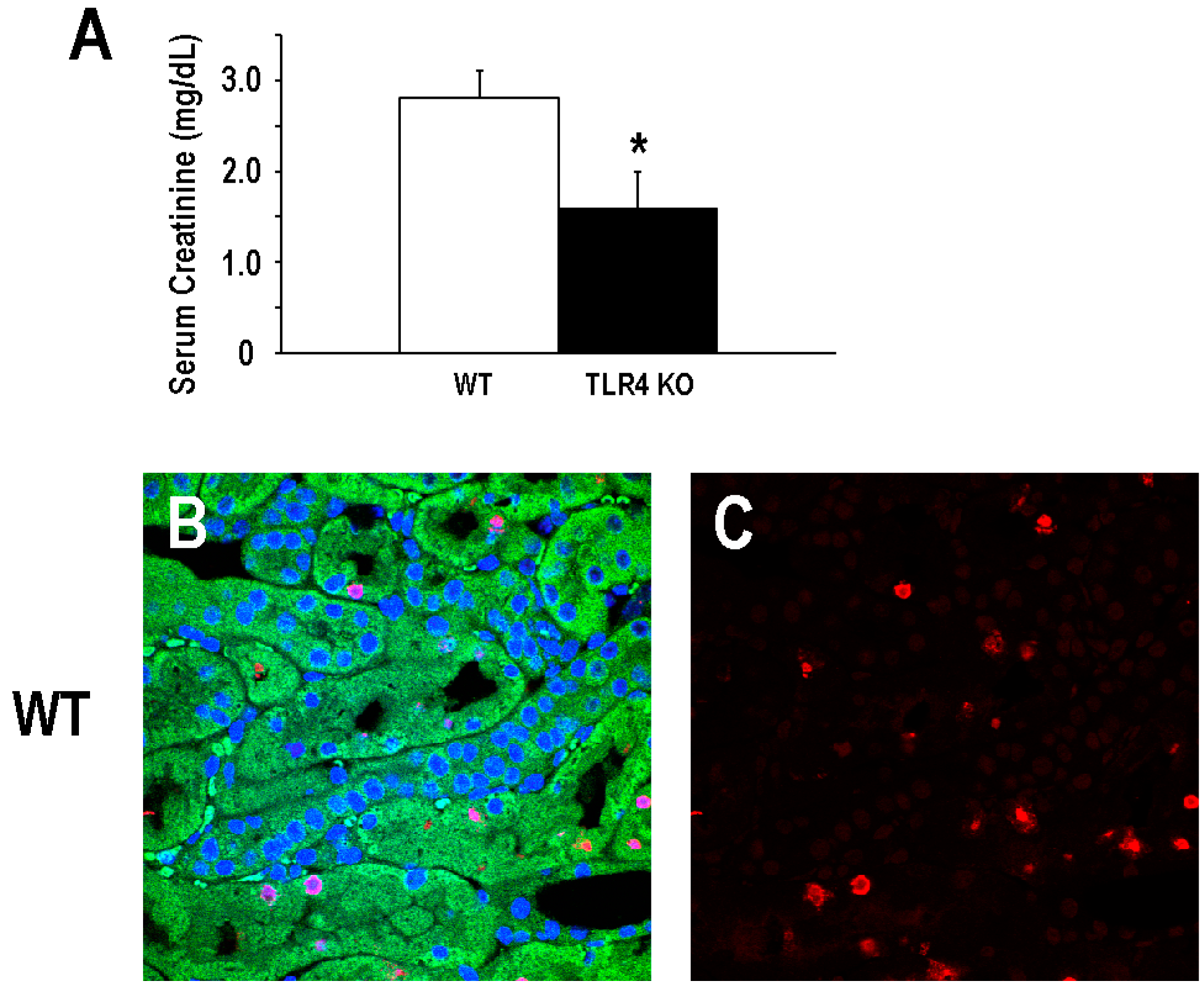
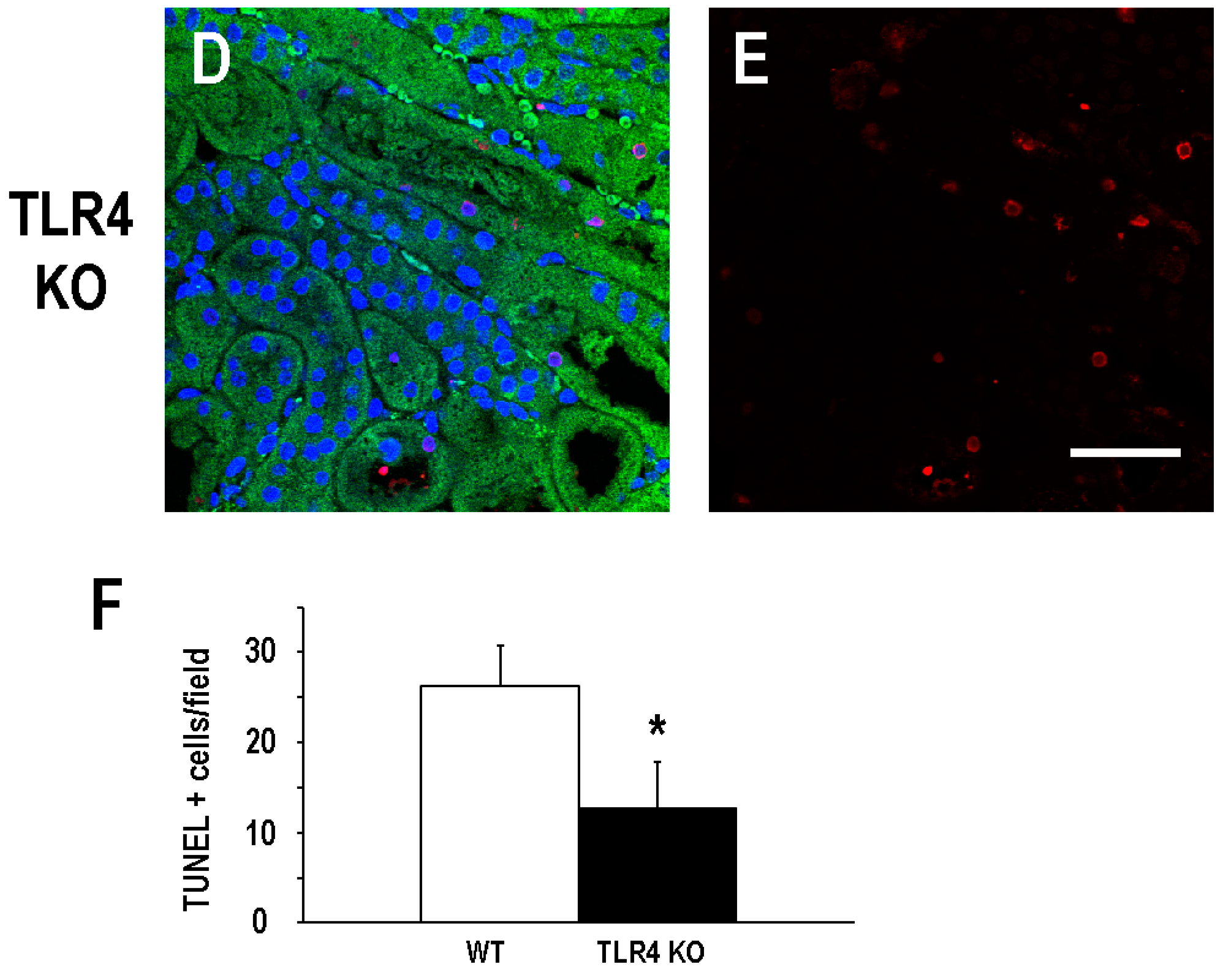
2.1. Functional Deletion of TLR4 Confers Acute Protection in a Model of Ischemic AKI
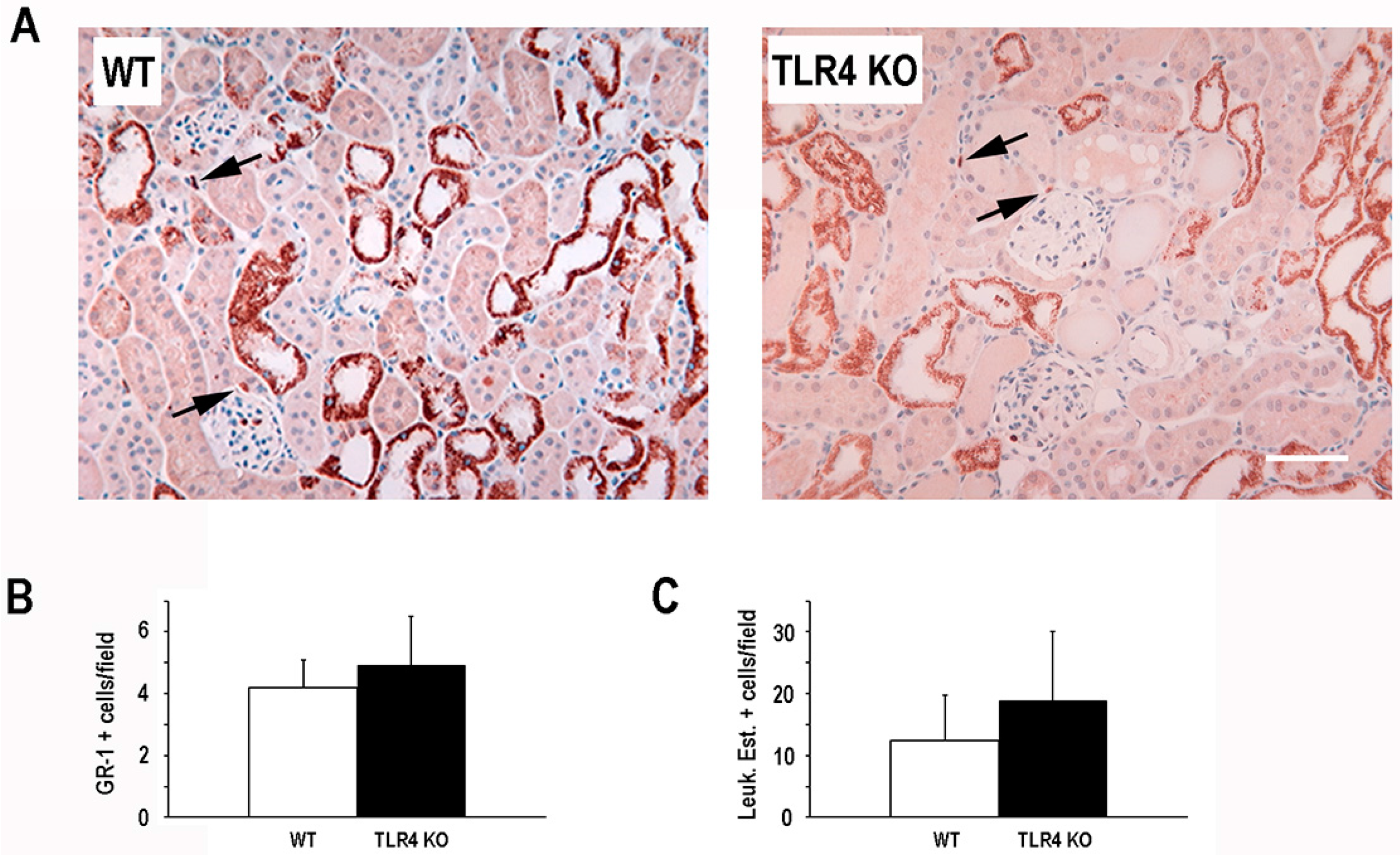
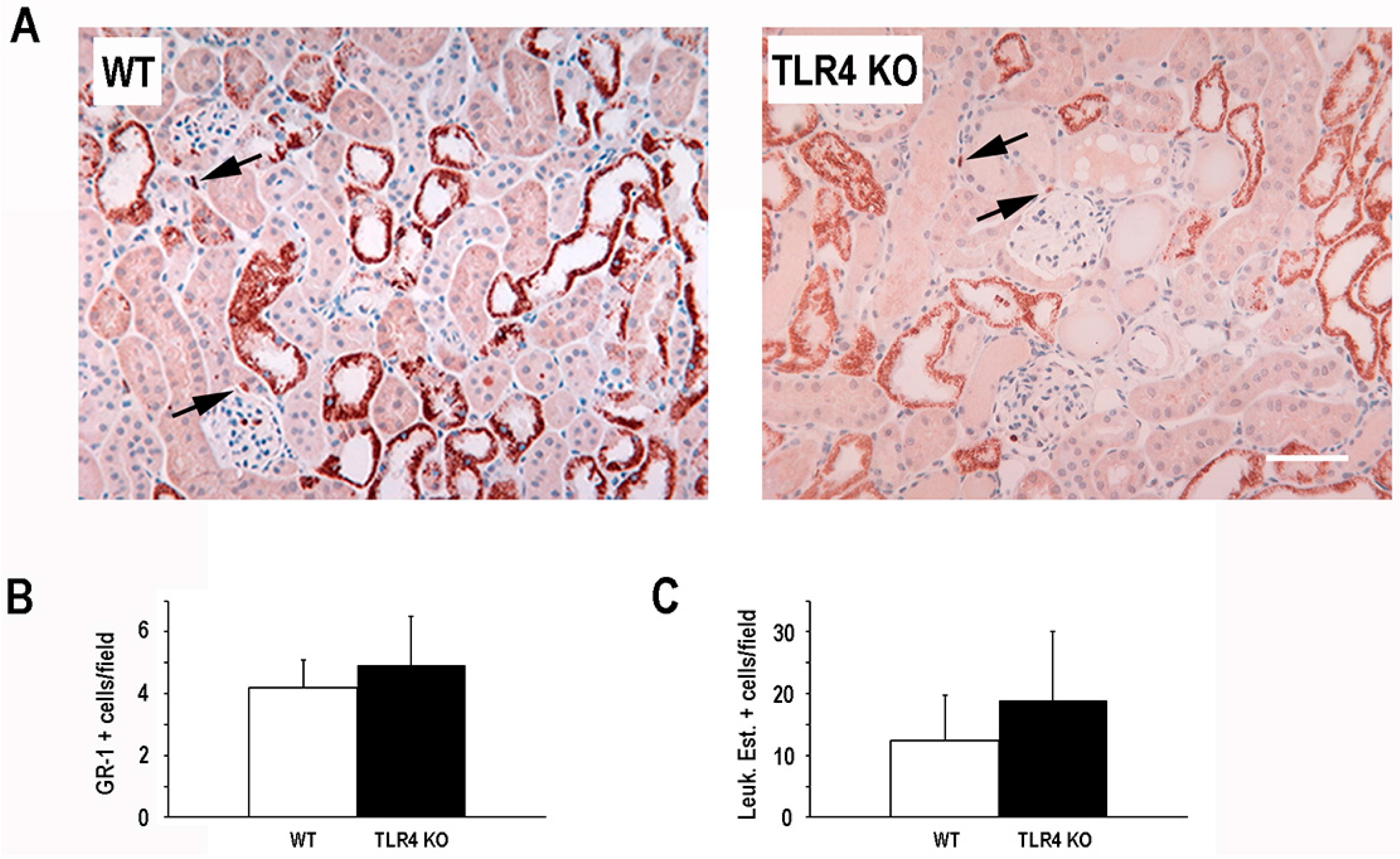
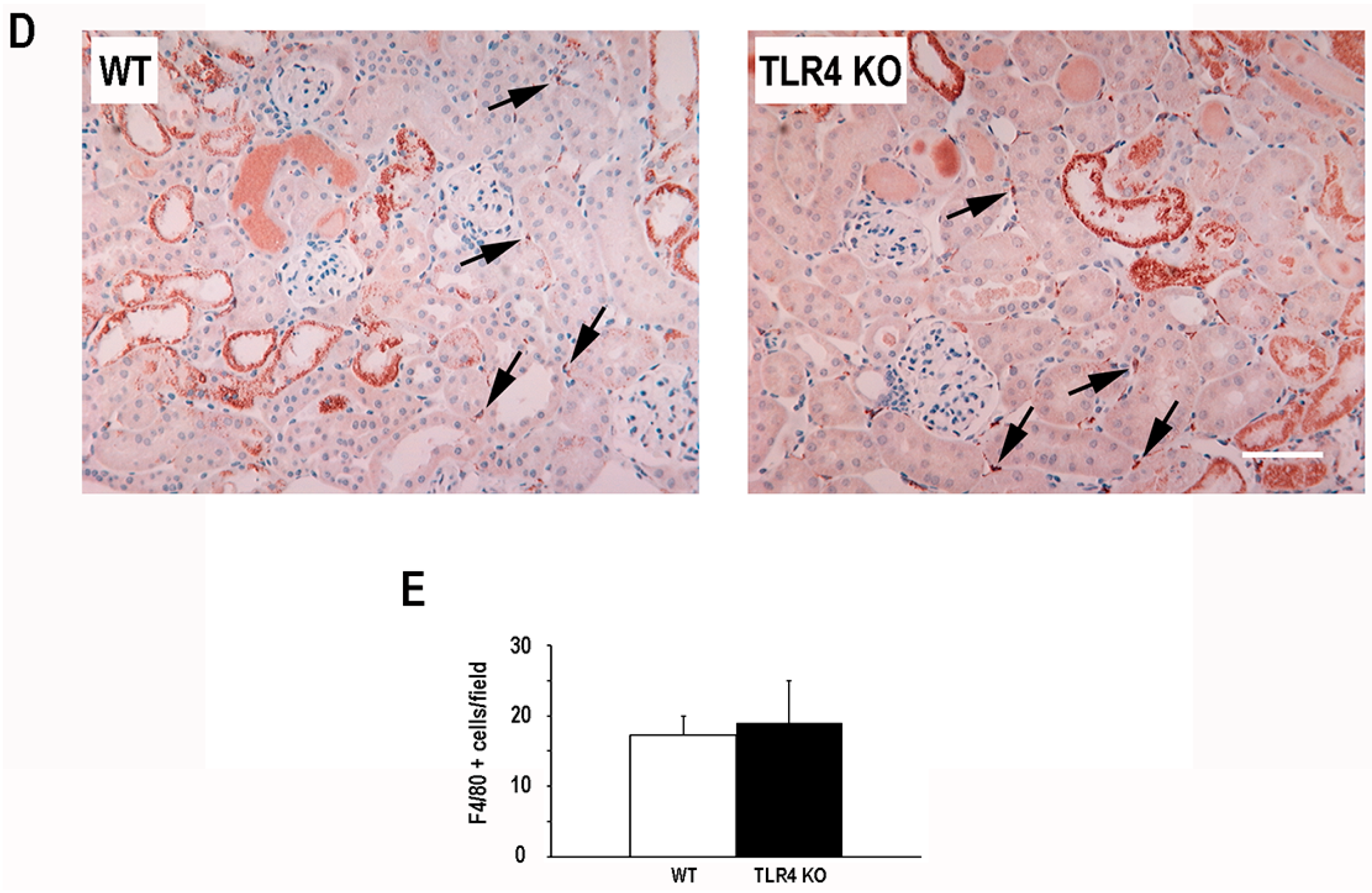
2.2. Functional Deletion of TLR4 Did Not Significantly Alter the Early Inflammatory Response in a Mouse Model of Ischemic AKI
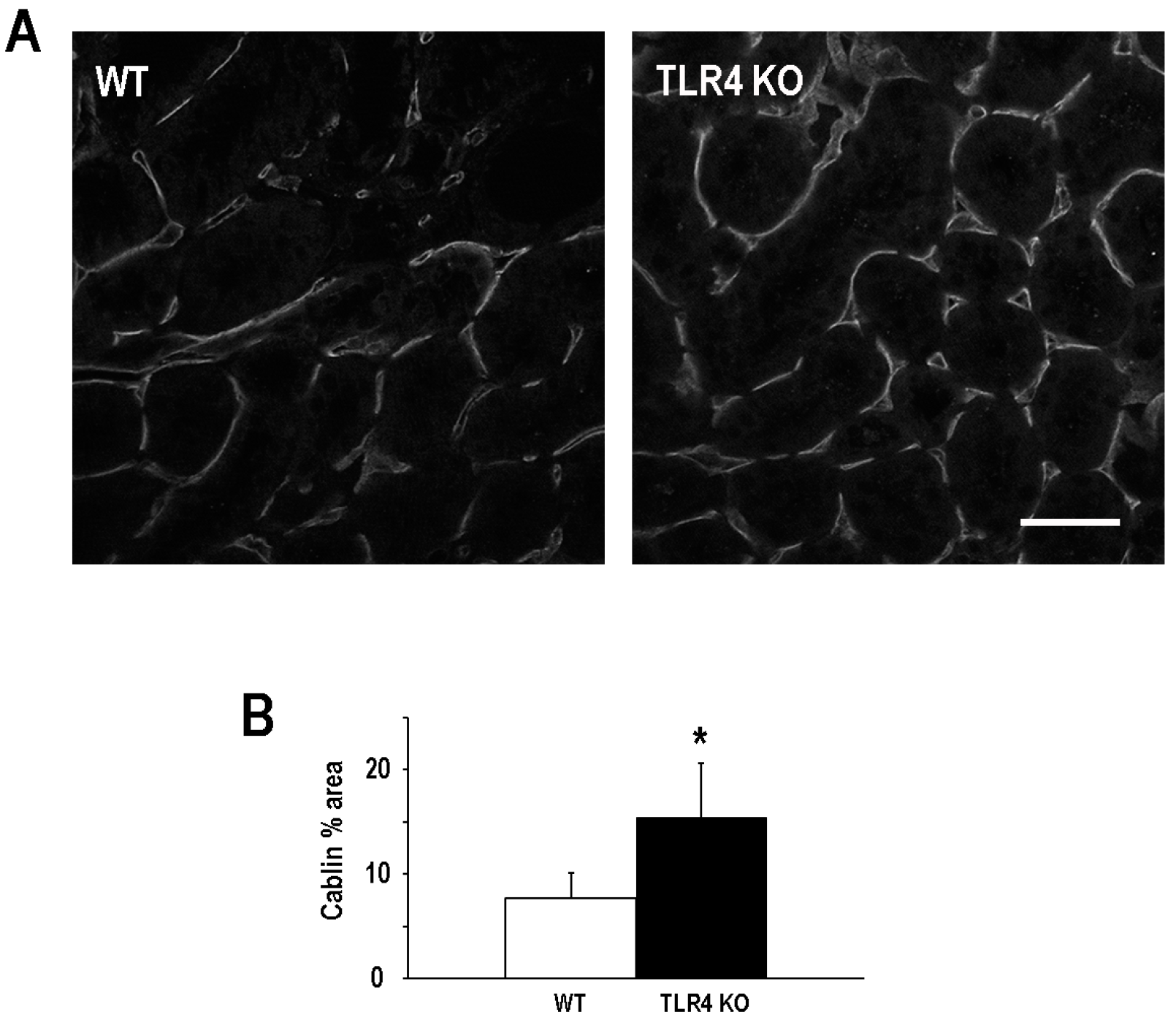
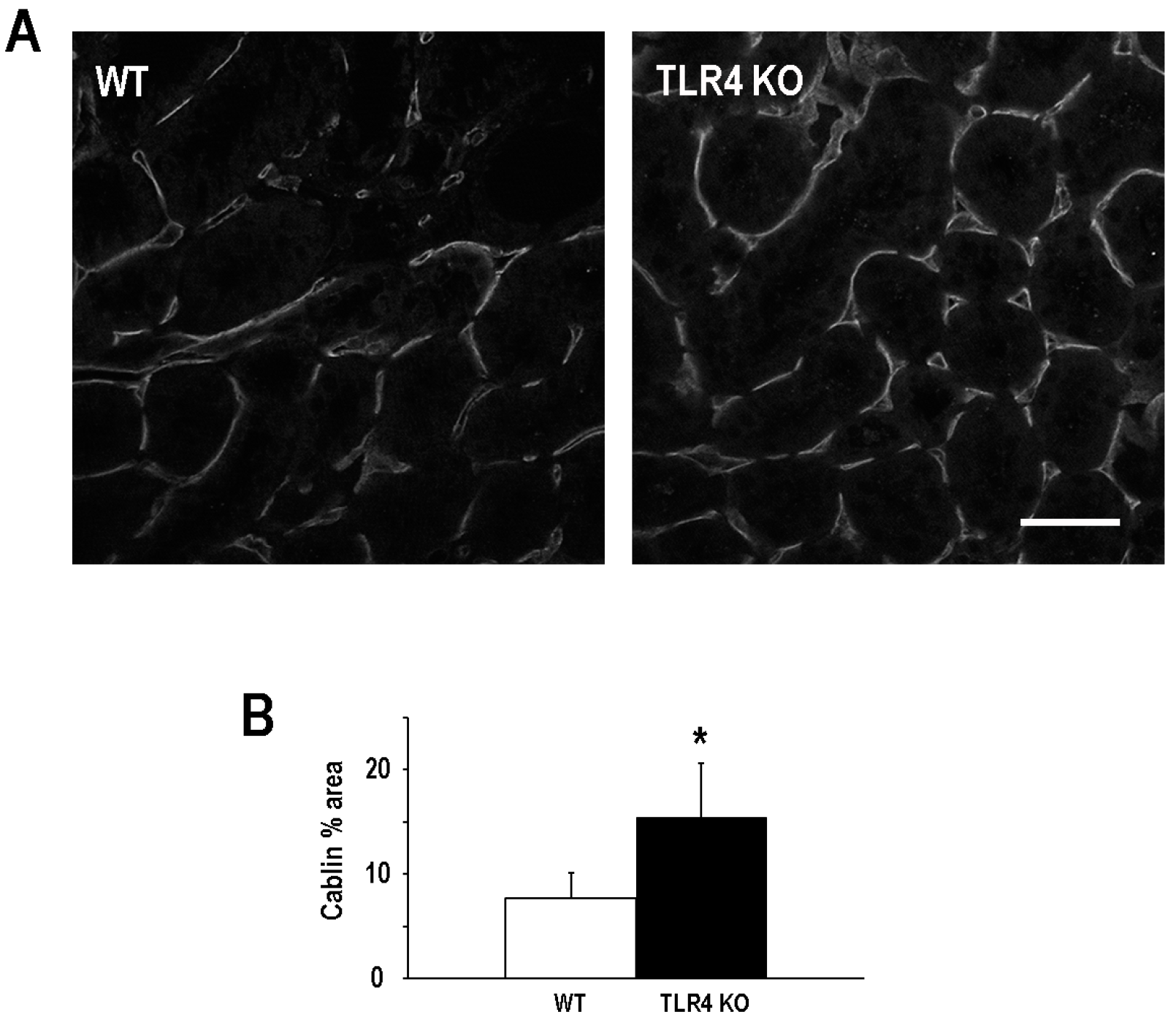
2.3. Functional Deletion of TLR4 Preserved Microvascular Density Following an Episode of Ischemic AKI
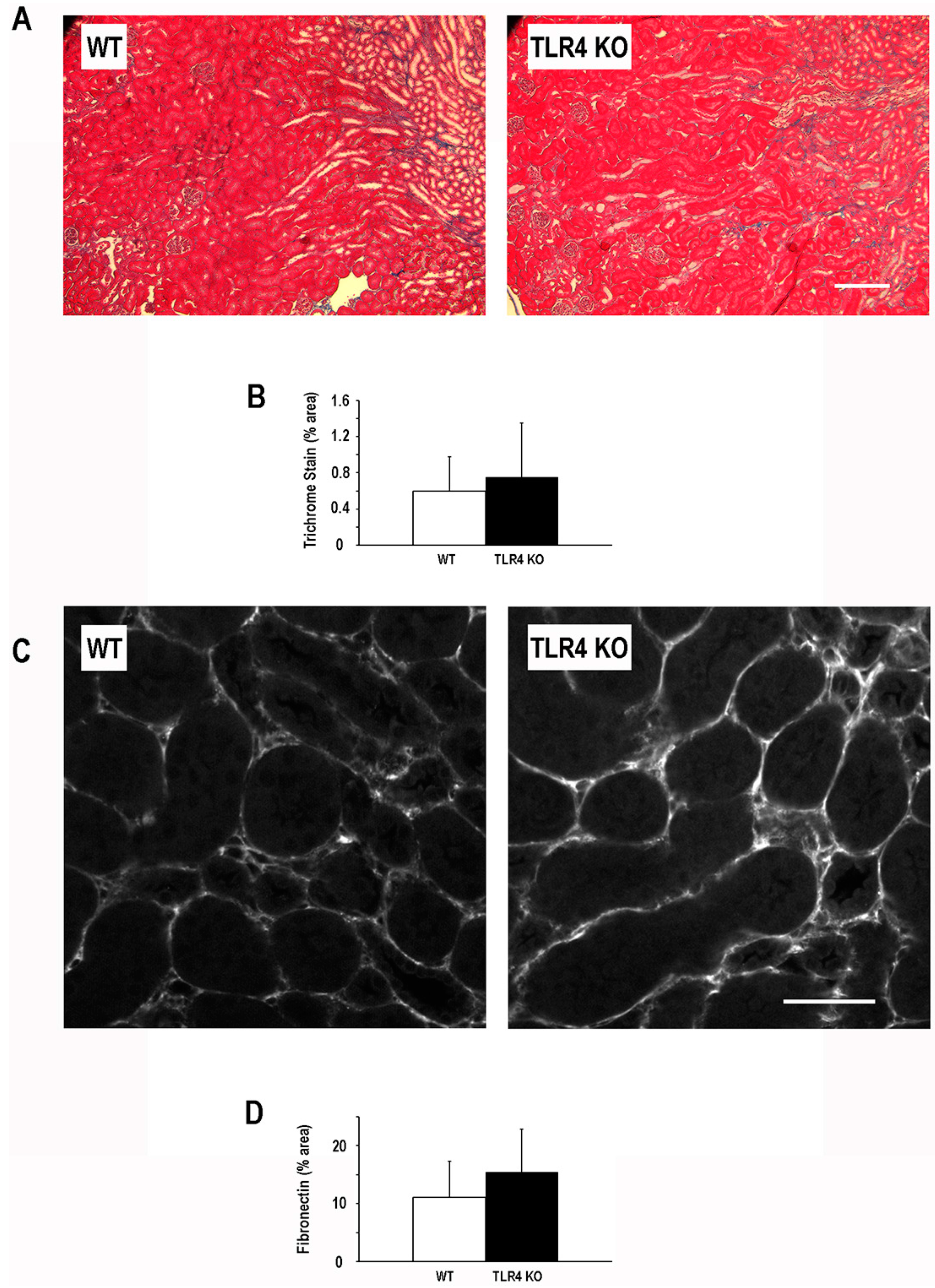
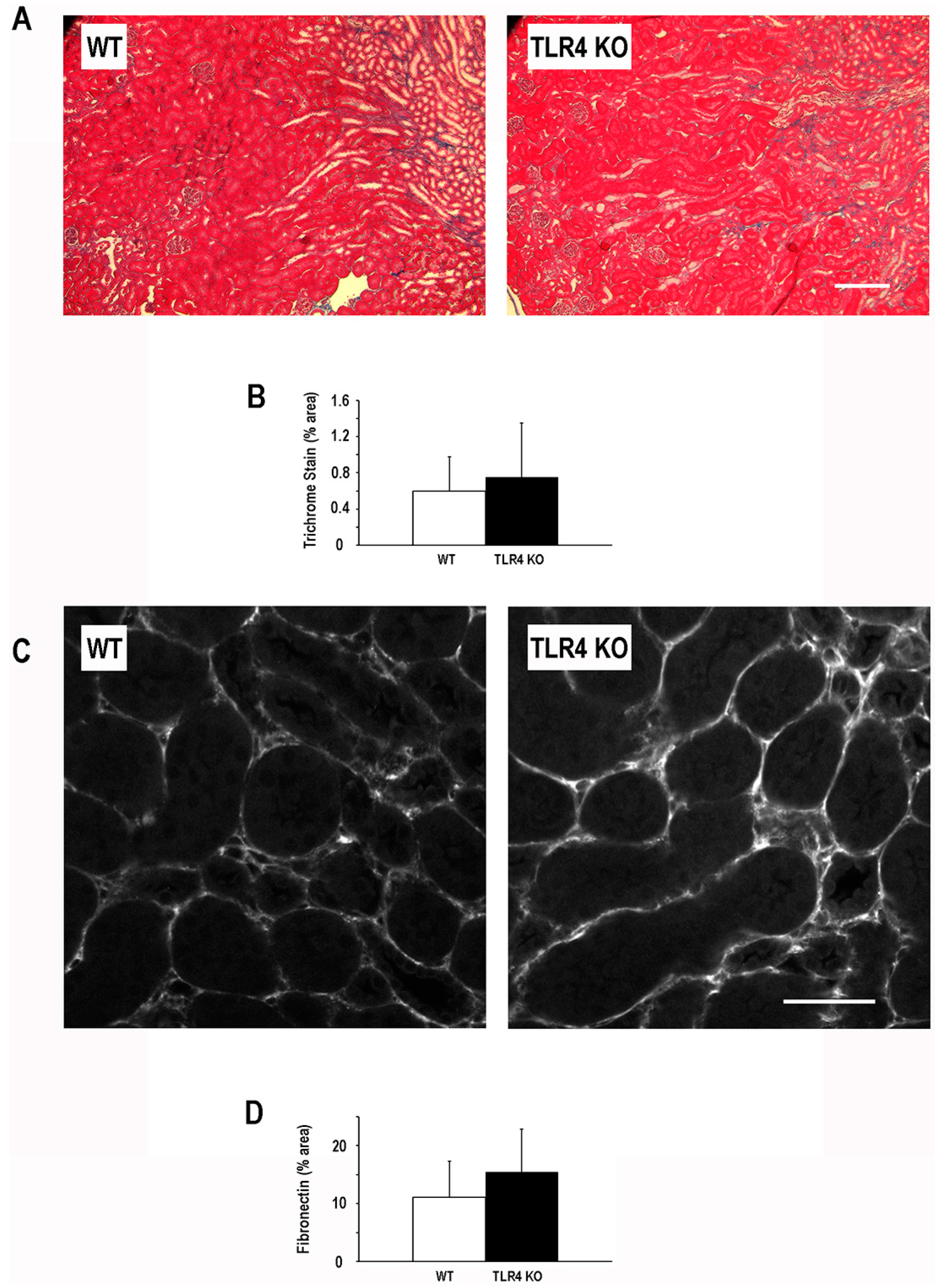
2.4. Functional Deletion of TLR4 Did Not Significantly Alter the Development of Fibrosis Following an Episode of Ischemic AKI
3. Discussion
4. Experimental Section
4.1. Animals and Experimental Models
4.2. Histopathology and Microscopy
4.3. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bagshaw, S.M.; Laupland, K.B.; Doig, C.J.; Mortis, G.; Fick, G.H.; Mucenski, M.; Godinez-Luna, T.; Svenson, L.W.; Rosenal, T. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: A population-based study. Crit. Care 2005, 9, R700–R709. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005, 16, 3365–3370. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.M.; Viscoli, C.M.; Horwitz, R.I. The effect of acute renal failure on mortality. A cohort analysis. JAMA 1996, 275, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.; Hafeez, A.; Hou, S. Hospital-acquired renal insufficiency. Am. J. Kidney Dis. 2002, 39, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Liangos, O.; Wald, R.; O’Bell, J.W.; Price, L.; Pereira, B.J.; Jaber, B.L. Epidemiology and outcomes of acute renal failure in hospitalized patients: A national survey. Clin. J. Am. Soc. Nephrol. 2006, 1, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Chawla, L.S.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. Outcomes following diagnosis of acute renal failure in US. Veterans: Focus on acute tubular necrosis. Kidney Int. 2009, 76, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of esrd among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Lo, L.J.; Go, A.S.; Chertow, G.M.; McCulloch, C.E.; Fan, D.; Ordonez, J.D.; Hsu, C.Y. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009, 76, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Wald, R.; Quinn, R.R.; Luo, J.; Li, P.; Scales, D.C.; Mamdani, M.M.; Ray, J.G. Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. JAMA 2009, 302, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- El-Achkar, T.M.; Dagher, P.C. Renal toll-like receptors: Recent advances and implications for disease. Nat. Clin. Pract. Nephrol. 2006, 2, 568–581. [Google Scholar] [CrossRef] [PubMed]
- El-Achkar, T.M.; Huang, X.; Plotkin, Z.; Sandoval, R.M.; Rhodes, G.J.; Dagher, P.C. Sepsis induces changes in the expression and distribution of toll-like receptor 4 in the rat kidney. Am. J. Physiol. Ren. Physiol. 2006, 290, F1034–F1043. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Pulskens, W.P.; Teske, G.J.; Butter, L.M.; Roelofs, J.J.; van der Poll, T.; Florquin, S.; Leemans, J.C. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS ONE 2008, 3, e3596. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ma, J.; Wang, P.; Corpuz, T.M.; Panchapakesan, U.; Wyburn, K.R.; Chadban, S.J. HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol. 2010, 21, 1878–1890. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; John, R.; Richardson, J.A.; Shelton, J.M.; Zhou, X.J.; Wang, Y.; Wu, Q.Q.; Hartono, J.R.; Winterberg, P.D.; Lu, C.Y. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 2011, 79, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Pulskens, W.P.; Rampanelli, E.; Teske, G.J.; Butter, L.M.; Claessen, N.; Luirink, I.K.; van der Poll, T.; Florquin, S.; Leemans, J.C. Tlr4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J. Am. Soc. Nephrol. 2010, 21, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.C.; Tsuji, T.; Baranova, I.N.; Bocharov, A.V.; Wilkins, K.J.; Street, J.M.; Alvarez-Prats, A.; Hu, X.; Eggerman, T.; Yuen, P.S.; et al. TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression. Physiol. Rep. 2015, 3, e12558. [Google Scholar] [CrossRef] [PubMed]
- Braga, T.T.; Correa-Costa, M.; Guise, Y.F.; Castoldi, A.; de Oliveira, C.D.; Hyane, M.I.; Cenedeze, M.A.; Teixeira, S.A.; Muscara, M.N.; Perez, K.R.; et al. MYD88 signaling pathway is involved in renal fibrosis by favoring a TH2 immune response and activating alternative M2 macrophages. Mol. Med. 2012, 18, 1231–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.T.; Hile, K.L.; Zhang, H.; Asanuma, H.; Vanderbrink, B.A.; Rink, R.R.; Meldrum, K.K. Toll-like receptor 4: A novel signaling pathway during renal fibrogenesis. J. Surg. Res. 2011, 168, e61–e69. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.A.; Hato, T.; Mai, E.; Yoshimoto, M.; Kuehl, S.; Anderson, M.; Mang, H.; Plotkin, Z.; Chan, R.J.; Dagher, P.C. P53 is renoprotective after ischemic kidney injury by reducing inflammation. J. Am. Soc. Nephrol. 2013, 24, 113–24. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Donohoe, D.; Roethe, K.; Osborn, J.L. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol. Ren. Physiol. 2001, 281, F887–F899. [Google Scholar] [CrossRef]
- Lee, S.Y.; Horbelt, M.; Mang, H.E.; Knipe, N.L.; Bacallao, R.L.; Sado, Y.; Sutton, T.A. MMP-9 gene deletion mitigates microvascular loss in a model of ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2011, 301, F101–F109. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.A.; Mang, H.E.; Campos, S.B.; Sandoval, R.M.; Yoder, M.C.; Molitoris, B.A. Injury of the renal microvascular endothelium alters barrier function after ischemia. Am. J. Physiol. Ren. Physiol. 2003, 285, F191–F198. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Friedrich, J.L.; Spahic, J.; Knipe, N.; Mang, H.; Leonard, E.C.; Changizi-Ashtiyani, S.; Bacallao, R.L.; Molitoris, B.A.; Sutton, T.A. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am. J. Physiol. Ren. Physiol. 2011, 300, F721–F733. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Donohoe, D.L.; Roethe, K.; Mattson, D.L. Chronic renal hypoxia after acute ischemic injury: Effects of l-arginine on hypoxia and secondary damage. Am. J. Physiol. Ren. Physiol. 2003, 284, F338–F348. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P. Rarefaction of peritubular capillaries following ischemic acute renal failure: A potential factor predisposing to progressive nephropathy. Curr. Opin. Nephrol. Hypertens. 2004, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, M.; Lee, S.Y.; Mang, H.E.; Knipe, N.L.; Sado, Y.; Kribben, A.; Sutton, T.A. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2007, 293, F688–F695. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.J.; Burns, J.; Dunnill, M.S.; McGee, J.O. Distribution of fibronectin in normal and diseased human kidneys. J. Clin. Pathol. 1980, 33, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.; Baelde, H.J.; Vleming, L.J.; de Heer, E.; Bruijn, J.A. Distribution of fibronectin isoforms in human renal disease. J. Pathol. 2001, 193, 256–262. [Google Scholar] [CrossRef]
- Zuk, A.; Bonventre, J.V.; Brown, D.; Matlin, K.S. Polarity, integrin, and extracellular matrix dynamics in the postischemic rat kidney. Am. J. Physiol. 1998, 275, C711–C731. [Google Scholar] [PubMed]
- Zuk, A.; Bonventre, J.V.; Matlin, K.S. Expression of fibronectin splice variants in the postischemic rat kidney. Am. J. Physiol. Ren. Physiol. 2001, 280, F1037–F1053. [Google Scholar]
- Chen, J.; Hartono, J.R.; John, R.; Bennett, M.; Zhou, X.J.; Wang, Y.; Wu, Q.; Winterberg, P.D.; Nagami, G.T.; Lu, C.Y. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int. 2011, 80, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Perez, J.S.; Lu, K.; George, A.J.; Ma, D. Role of toll-like receptor-4 in renal graft ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F801–F811. [Google Scholar] [CrossRef] [PubMed]
- Kalakeche, R.; Hato, T.; Rhodes, G.; Dunn, K.W.; El-Achkar, T.M.; Plotkin, Z.; Sandoval, R.M.; Dagher, P.C. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J. Am. Soc. Nephrol. 2011, 22, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramesh, G.; Uematsu, S.; Akira, S.; Reeves, W.B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol. 2008, 19, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Kanellis, J.; Hugo, C.; Truong, L.; Anderson, S.; Kerjaschki, D.; Schreiner, G.F.; Johnson, R.J. Role of the microvascular endothelium in progressive renal disease. J. Am. Soc. Nephrol. 2002, 13, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Sacks, S.H.; Sheerin, N.S. Endogenous ligands for TLR2 and TLR4 are not involved in renal injury following ureteric obstruction. Nephron Exp. Nephrol. 2010, 115, e122–e130. [Google Scholar] [CrossRef] [PubMed]
- Dagher, P.C.; Mai, E.M.; Hato, T.; Lee, S.Y.; Anderson, M.D.; Karozos, S.C.; Mang, H.E.; Knipe, N.L.; Plotkin, Z.; Sutton, T.A. The P53 inhibitor pifithrin-alpha can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2012, 302, F284–F291. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after aki leading to accelerated kidney ageing and ckd. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Charron, A.J.; Xu, W.; Bacallao, R.L.; Wandinger-Ness, A. Cablin: A novel protein of the capillary basal lamina. Am. J. Physiol. 1999, 277, H1985–H1996. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagher, P.C.; Hato, T.; Mang, H.E.; Plotkin, Z.; Richardson, Q.V.; Massad, M.; Mai, E.; Kuehl, S.E.; Graham, P.; Kumar, R.; et al. Inhibition of Toll-Like Receptor 4 Signaling Mitigates Microvascular Loss but Not Fibrosis in a Model of Ischemic Acute Kidney Injury. Int. J. Mol. Sci. 2016, 17, 647. https://doi.org/10.3390/ijms17050647
Dagher PC, Hato T, Mang HE, Plotkin Z, Richardson QV, Massad M, Mai E, Kuehl SE, Graham P, Kumar R, et al. Inhibition of Toll-Like Receptor 4 Signaling Mitigates Microvascular Loss but Not Fibrosis in a Model of Ischemic Acute Kidney Injury. International Journal of Molecular Sciences. 2016; 17(5):647. https://doi.org/10.3390/ijms17050647
Chicago/Turabian StyleDagher, Pierre C., Takashi Hato, Henry E. Mang, Zoya Plotkin, Quentin V. Richardson, Michael Massad, Erik Mai, Sarah E. Kuehl, Paige Graham, Rakesh Kumar, and et al. 2016. "Inhibition of Toll-Like Receptor 4 Signaling Mitigates Microvascular Loss but Not Fibrosis in a Model of Ischemic Acute Kidney Injury" International Journal of Molecular Sciences 17, no. 5: 647. https://doi.org/10.3390/ijms17050647