
Data Interoperability of Whole Exome Sequencing (WES) Based Mutational Burden Estimates from Different Laboratories

Abstract
:
1. Introduction
1.1. Immune Checkpoint and Cancer Mutational Burden
1.2. Current Status of Next Generation Sequencing (NGS) in Clinical Application
2. Interoperability of Whole Exome Sequencing (WES) Data from Different Labs
2.1. Mutation Data Reported by Vendors
2.2. Mutation Calls from Vendors’ Raw Data Using Unified Pipeline
3. Discussion
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T., Jr.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.F.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.L.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.D.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res. 2005, 571, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.W. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Goetz, L.; Bethel, K.; Topol, E.J. Rebooting cancer tissue handling in the sequencing era: Toward routine use of frozen tumor tissue. J. Am. Med. Assoc. 2013, 309, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Barroso-Sousa, R.; Jimenez, L.; Correa, B.R.; Sabbaga, J.; Hoff, P.M.; Reis, L.F.; Galante, P.A.; Camargo, A.A. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 2015, 6, 34221–34227. [Google Scholar] [PubMed]
- Jones, S.; Anagnostou, V.; Lytle, K.; Parpart-Li, S.; Nesselbush, M.; Riley, D.R.; Shukla, M.; Chesnick, B.; Kadan, M.; Papp, E.; et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Trans. Med. 2015, 7, 283ra53. [Google Scholar] [CrossRef] [PubMed]
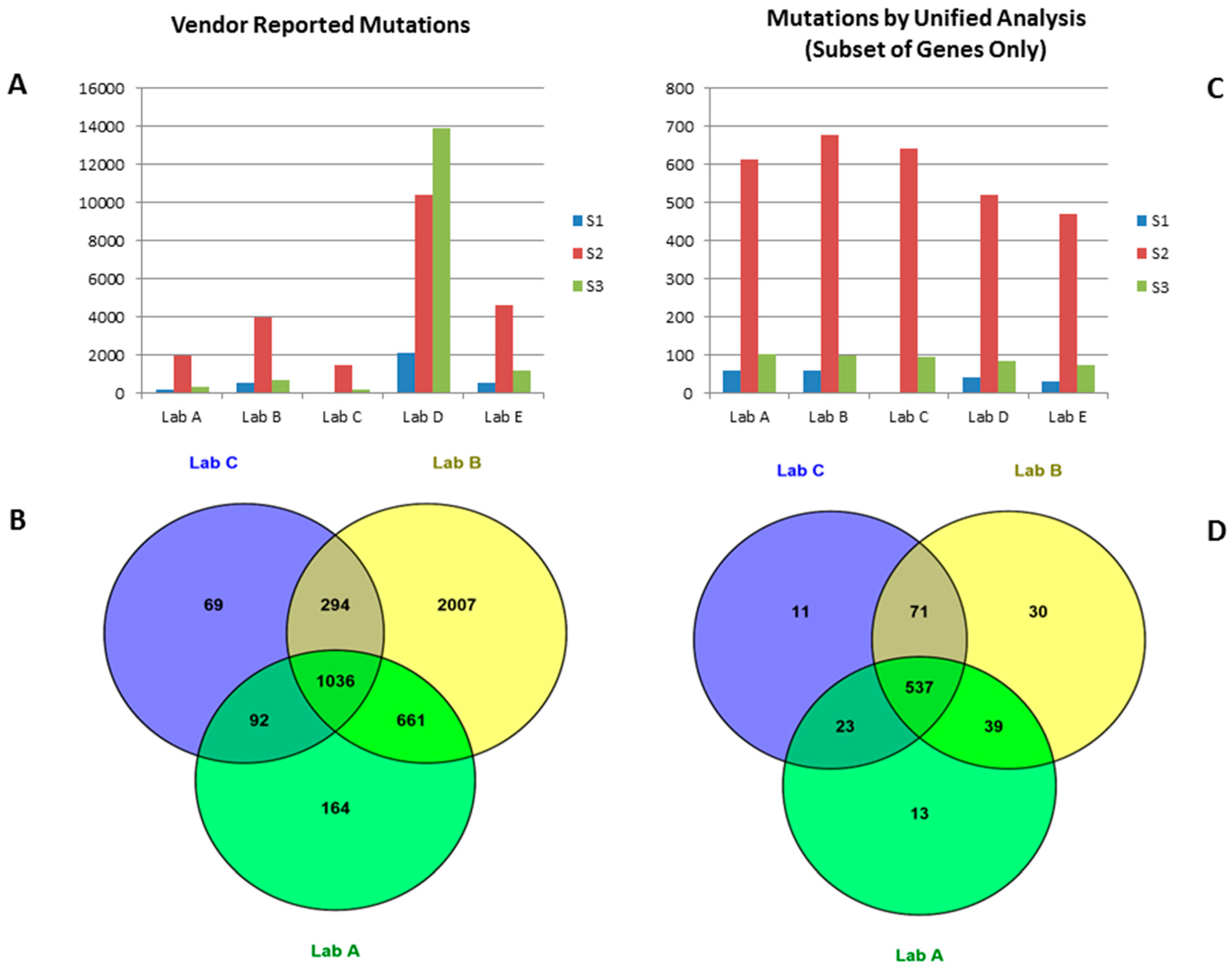

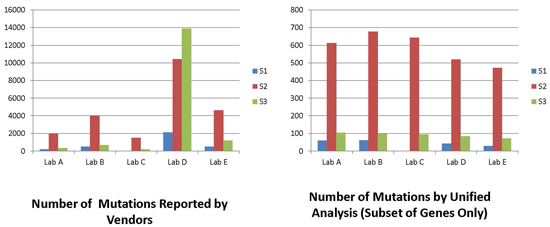{kind=link}
{kind=link}
| Vendor | Sample | Coverage | Mutations Reported by Each Vendor | Mutations Detected by Unified Analysis (for Genes in TruSight One Only) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Replicate 1 | Replicate 2 | Overlap | Concordance | Replicate 1 | Replicate 2 | Overlap | Concordance | |||
| Lab A | S1 | 181 | 200 | 200 | 178 | 89 | 60 | 62 | 54 | 88.52 |
| Lab B | S1 | 135 | 519 | 553 | 391 | 72.95 | 61 | 61 | 57 | 93.44 |
| Lab C | S1 | N.A. | ||||||||
| Lab D | S1 | 103 | 2113 | 2144 | 367 | 17.24 | 42 | 41 | 33 | 79.52 |
| Lab E | S1 | 66 | 510 | 622 | 327 | 57.77 | 29 | 38 | 25 | 74.63 |
| Lab A | S2 | 144 | 1953 | 1949 | 1634 | 83.75 | 612 | 627 | 555 | 89.59 |
| Lab B | S2 | 119 | 3998 | 3991 | 3470 | 86.87 | 677 | 656 | 623 | 93.47 |
| Lab C | S2 | 170 | 1491 | N.A. | N.A. | N.A. | 642 | N.A. | N.A. | N.A. |
| Lab D | S2 | 78 | 10,428 | 17,944 | 1968 | 13.87 | 519 | 573 | 466 | 85.35 |
| Lab E | S2 | 82 | 4619 | 5201 | 3788 | 77.15 | 472 | 568 | 442 | 85 |
| Lab A | S3 | 242 | 336 | 338 | 296 | 87.83 | 104 | 110 | 97 | 90.65 |
| Lab B | S3 | 133 | 871 | 823 | 611 | 72.14 | 98 | 103 | 90 | 89.55 |
| Lab C | S3 | 242 | 187 | N.A. | N.A. | N.A. | 94 | N.A. | N.A. | N.A. |
| Lab D | S3 | 67 | 13,892 | 23,410 | 573 | 3.07 | 84 | 90 | 77 | 88.51 |
| Lab E | S3 | 80 | 1214 | 1255 | 699 | 56.62 | 72 | 84 | 64 | 82.05 |
| Lab | Capture Kit Used | Total SNP Detected | Number of SNP Detected in COSMIC (Sensitivity of Detection in %) | ||
|---|---|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 1 | Replicate 2 | ||
| Lab A | Illumina Rapid Capture Exome Kit | 200 | 200 | 142 (80%) | 141 (79%) |
| Lab B | Proprietary Kit | 519 | 553 | 161 (90%) | 159 (89%) |
| Lab C | Agilent SureSelect v4 | N.A. | |||
| Lab D | Agilent SureSelect v4 | 2113 | 2144 | 104 (58%) | 98 (55%) |
| Lab E | Agilent SureSelect v5 + UTR | 510 | 622 | 116 (65%) | 142 (80%) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, P.; Pang, L.; Arreaza, G.; Maguire, M.; Chang, K.C.N.; Marton, M.J.; Levitan, D. Data Interoperability of Whole Exome Sequencing (WES) Based Mutational Burden Estimates from Different Laboratories. Int. J. Mol. Sci. 2016, 17, 651. https://doi.org/10.3390/ijms17050651
Qiu P, Pang L, Arreaza G, Maguire M, Chang KCN, Marton MJ, Levitan D. Data Interoperability of Whole Exome Sequencing (WES) Based Mutational Burden Estimates from Different Laboratories. International Journal of Molecular Sciences. 2016; 17(5):651. https://doi.org/10.3390/ijms17050651
Chicago/Turabian StyleQiu, Ping, Ling Pang, Gladys Arreaza, Maureen Maguire, Ken C. N. Chang, Matthew J. Marton, and Diane Levitan. 2016. "Data Interoperability of Whole Exome Sequencing (WES) Based Mutational Burden Estimates from Different Laboratories" International Journal of Molecular Sciences 17, no. 5: 651. https://doi.org/10.3390/ijms17050651