
Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice




Abstract
:
1. Introduction
2. Results
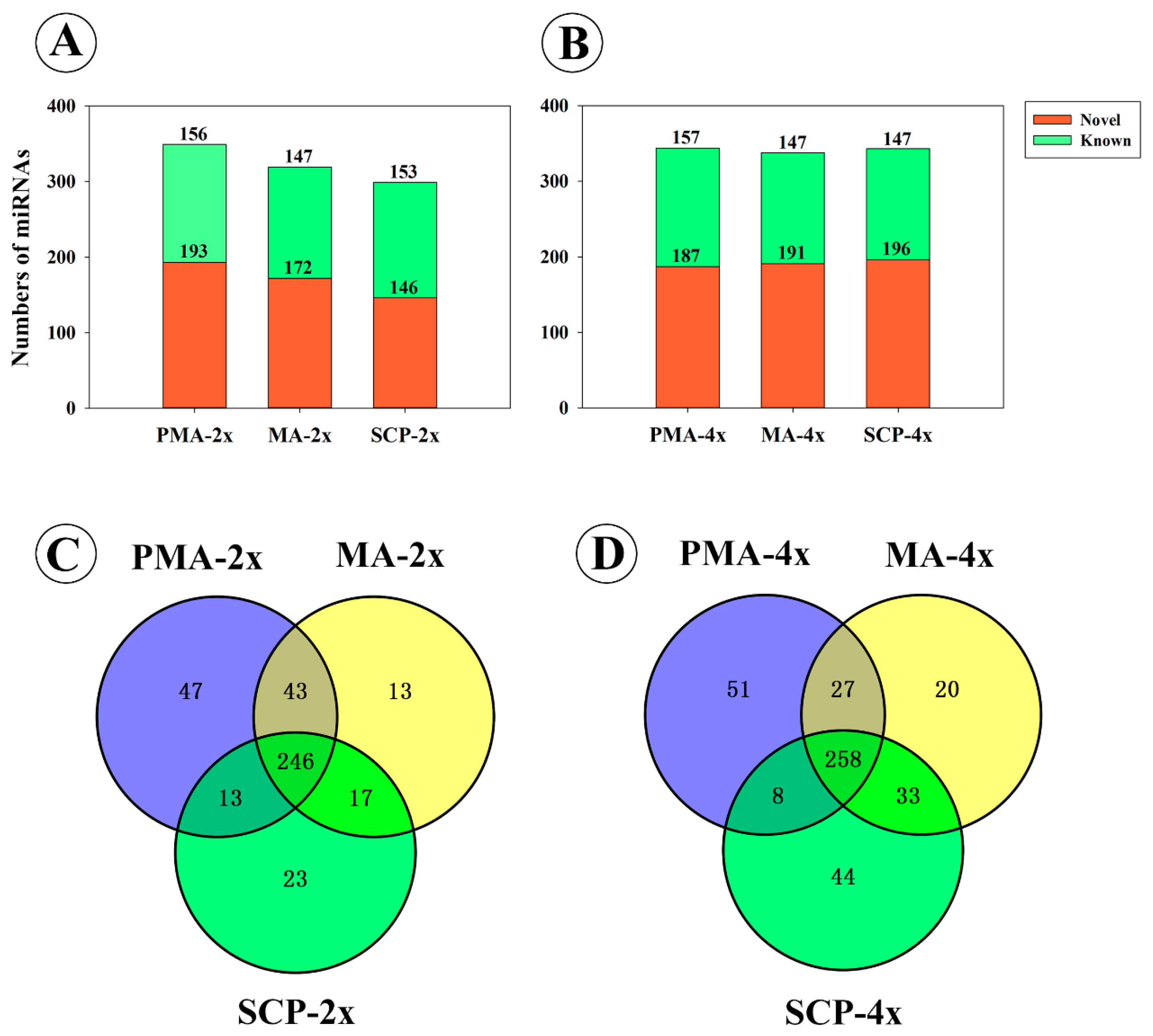
2.1. Overview of MicroRNAs (miRNAs) Sequencing Datasets in Developing Pollens of Rice
2.2. Association between miRNAs Expression Profiles and Pollen Development Stages in Autotetraploid and Diploid Rice
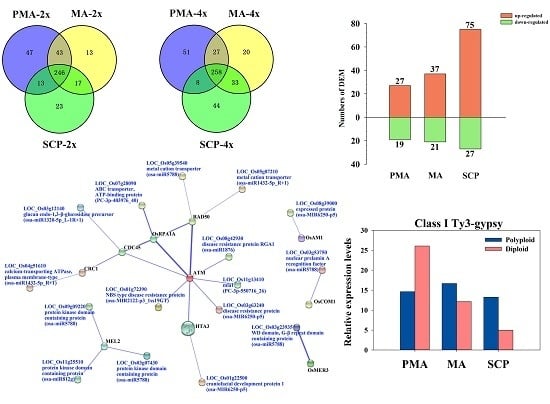
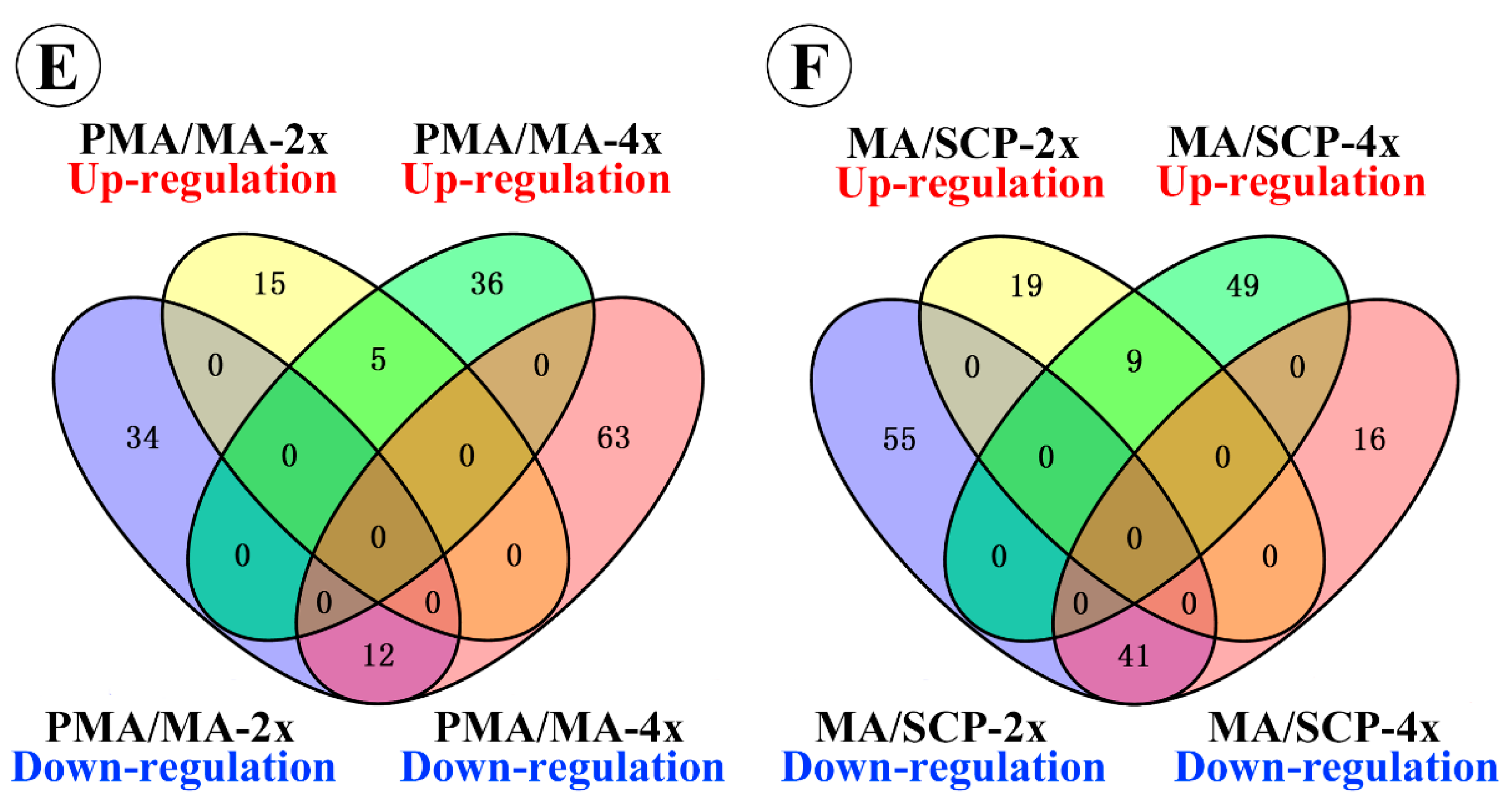
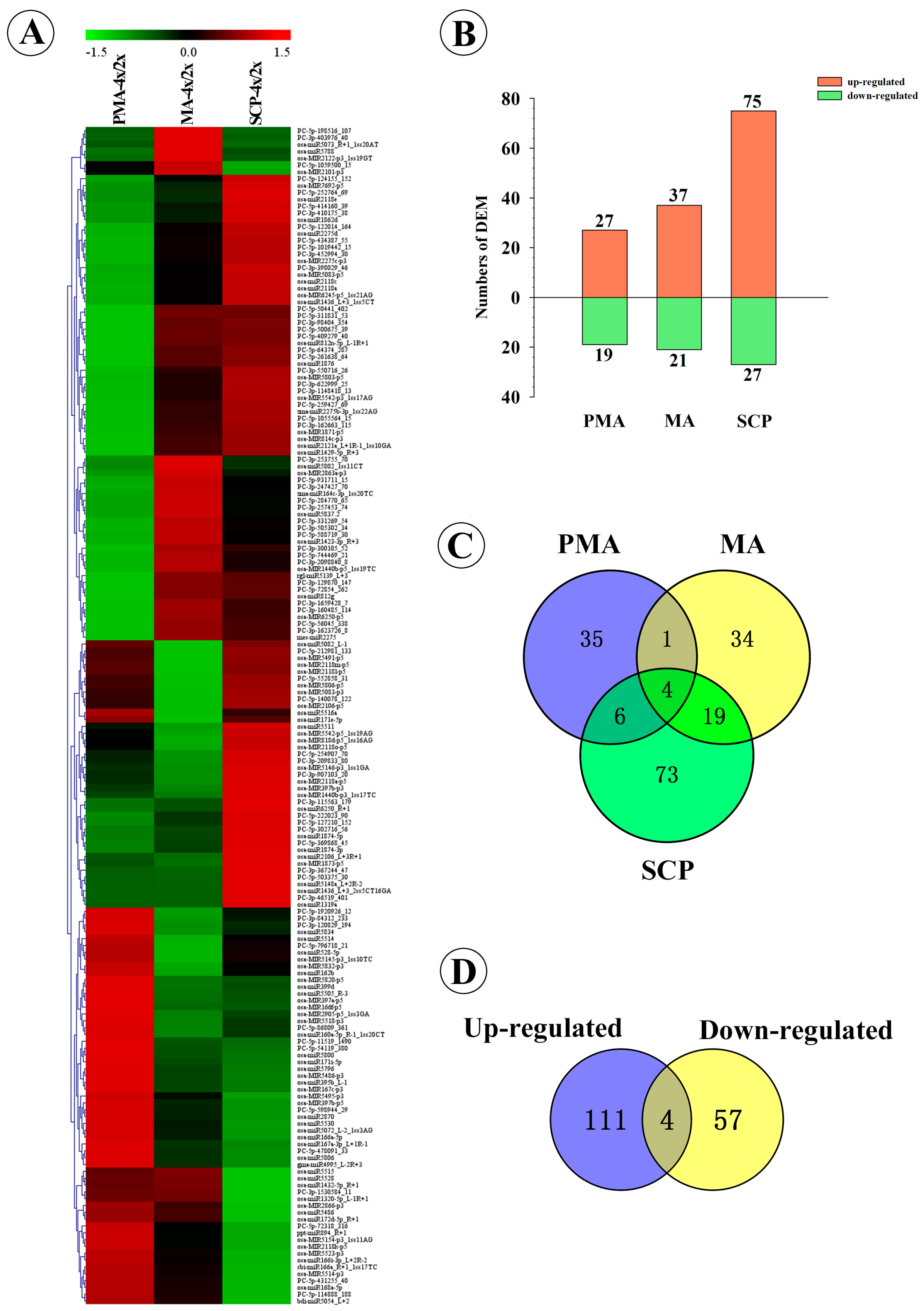
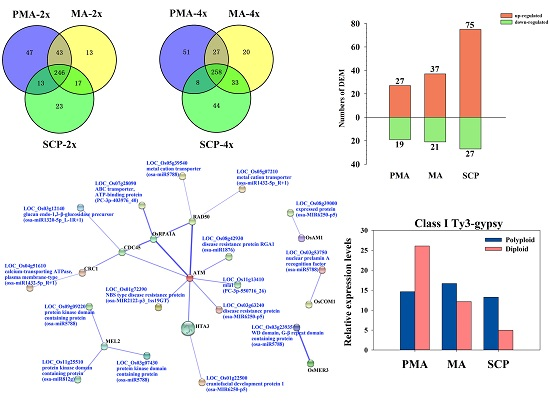
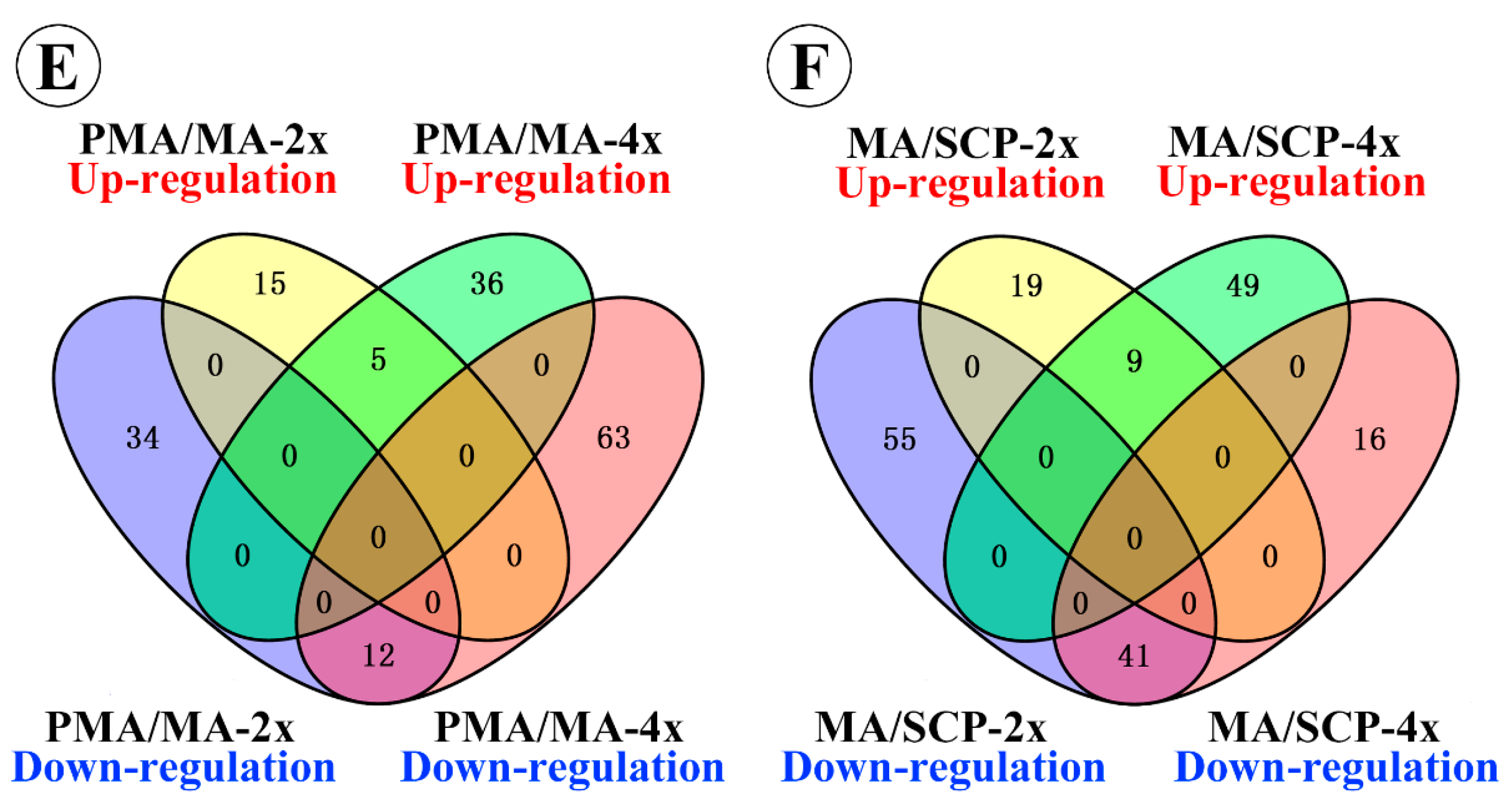
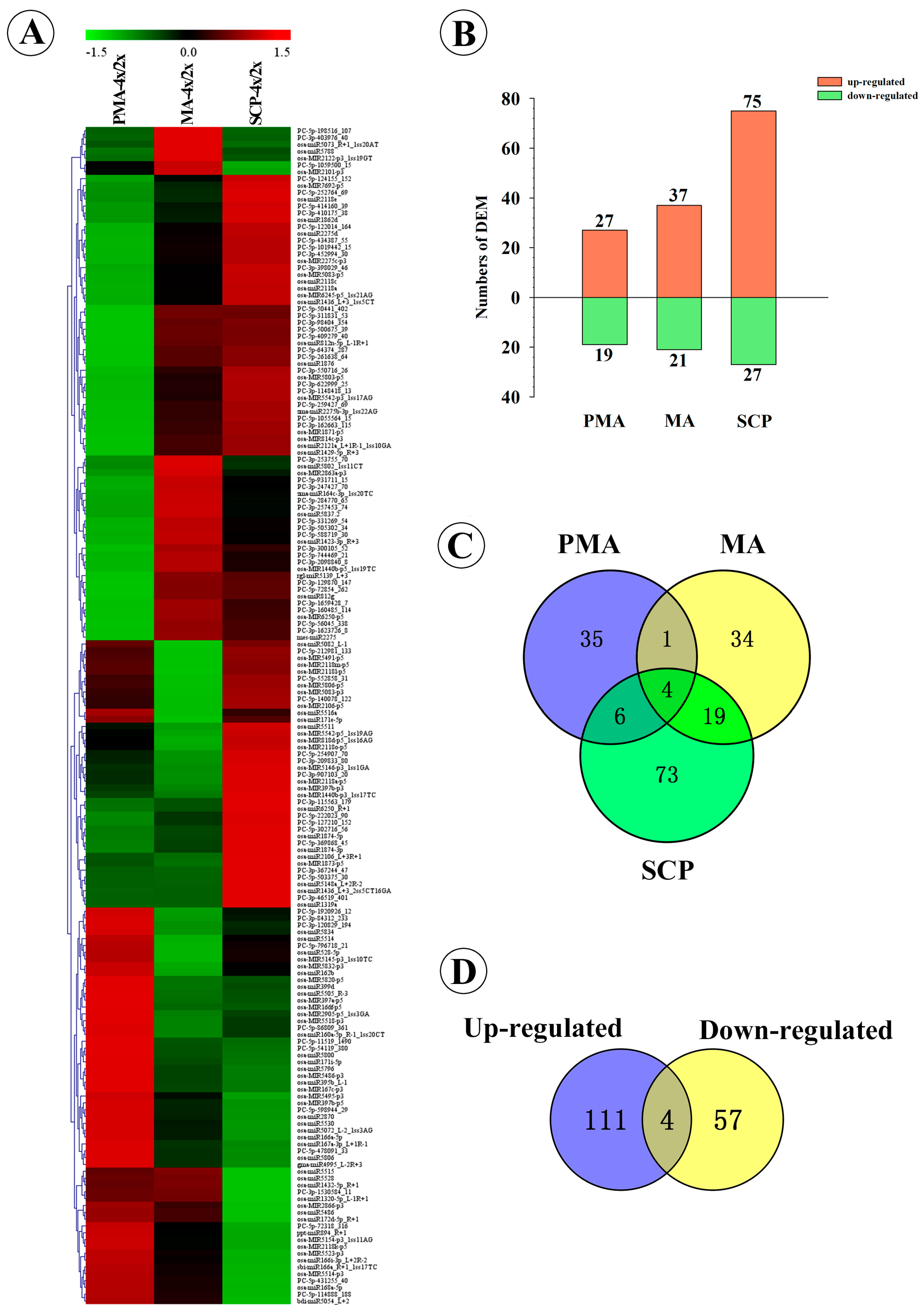
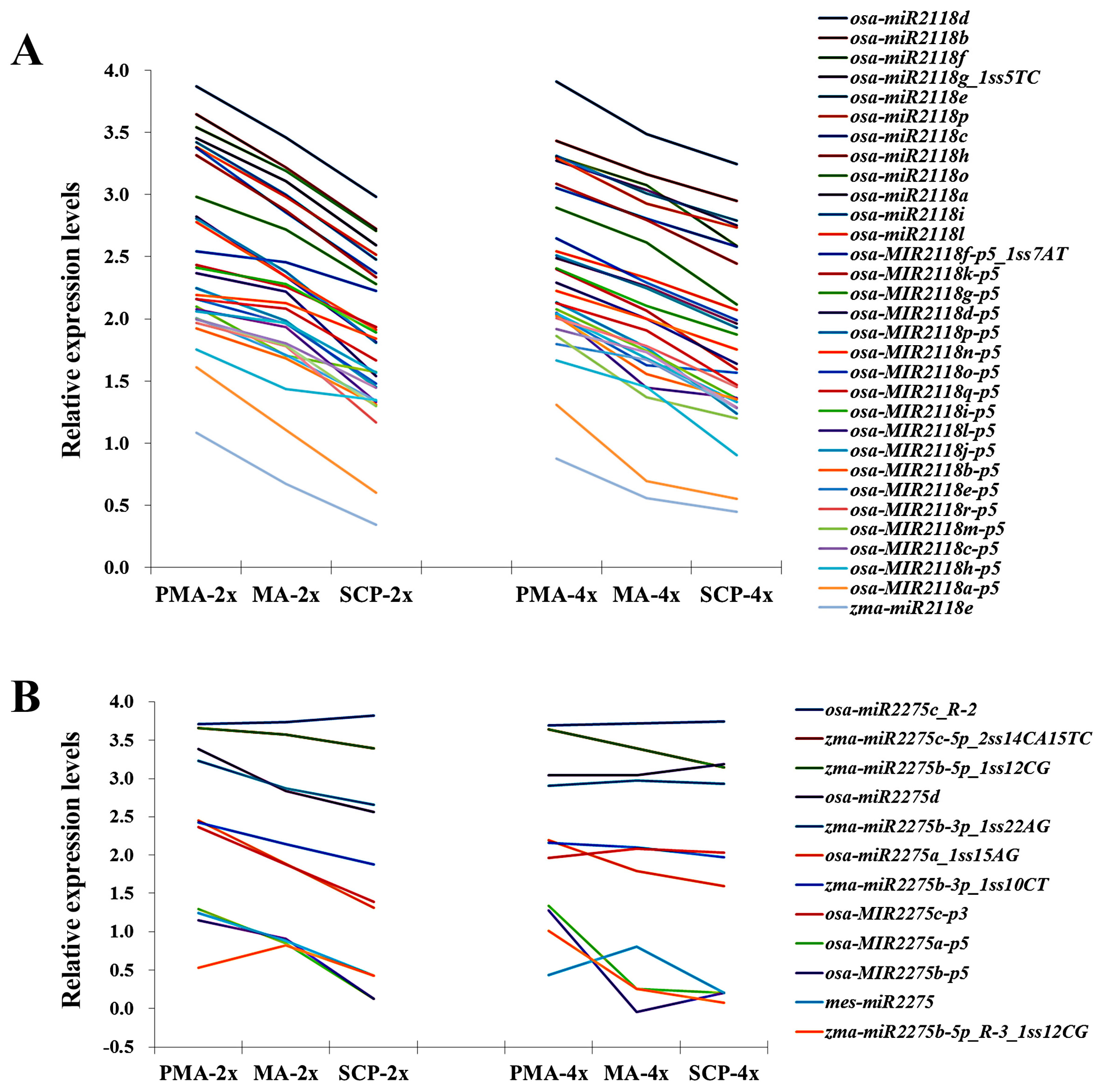
2.3. Differentially Expressed miRNAs during Pollen Development in Autotetraploid and Diploid Rice
2.4. Target Prediction of Differentially Expressed miRNAs (DEM) and Functional Classification
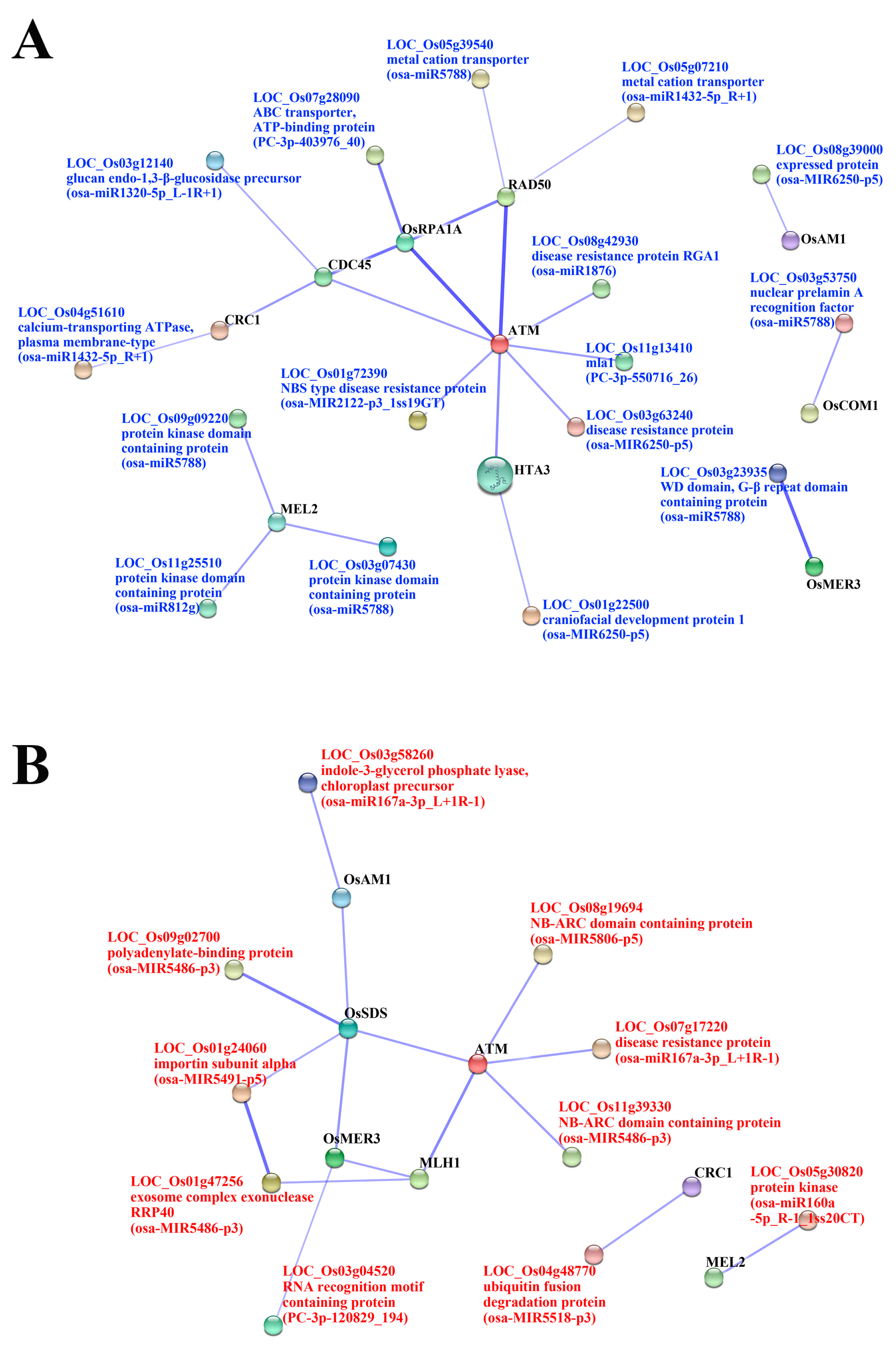
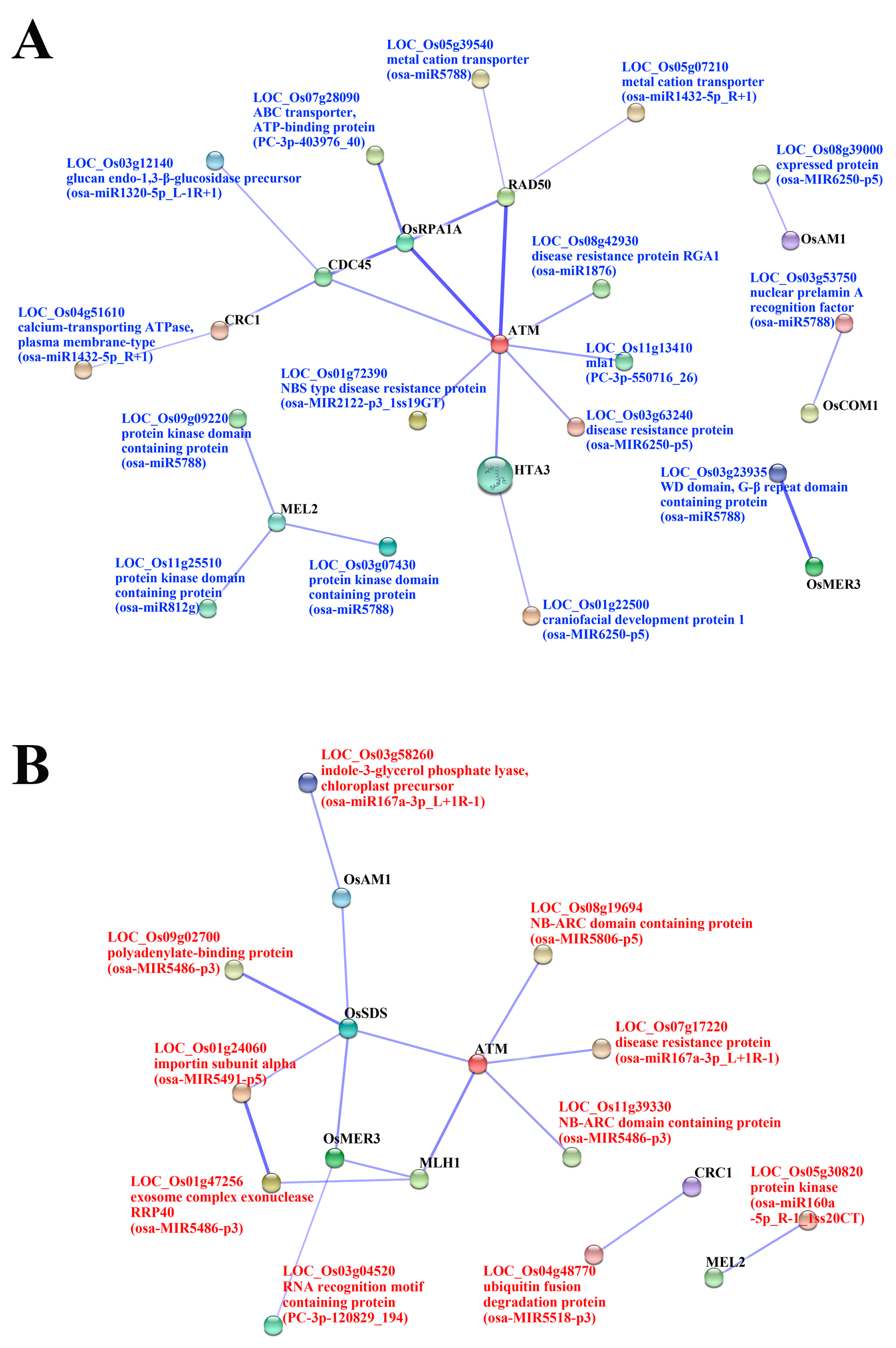
2.5. Functional Classification of Meiosis-Related Predicted Targets of Differentially Expressed miRNAs
2.6. Functional Classification of Pre-Meiotic Interphase (PMA) and Single Microspore Stage (SCP) Stage-Specific Targets Regulated by Differentially Expressed miRNAs
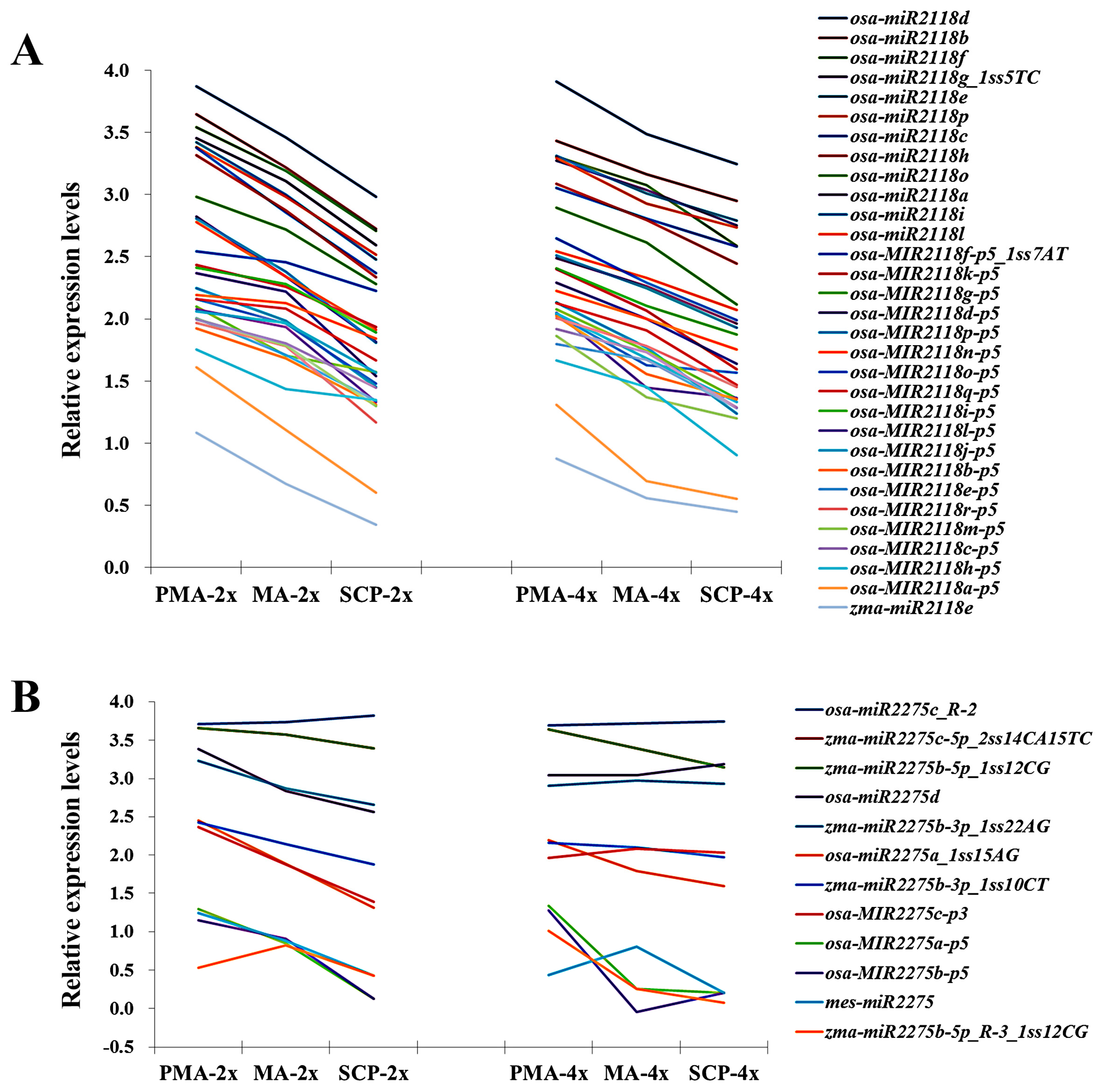
2.7. Correlation between miRNAs and Their Target Genes
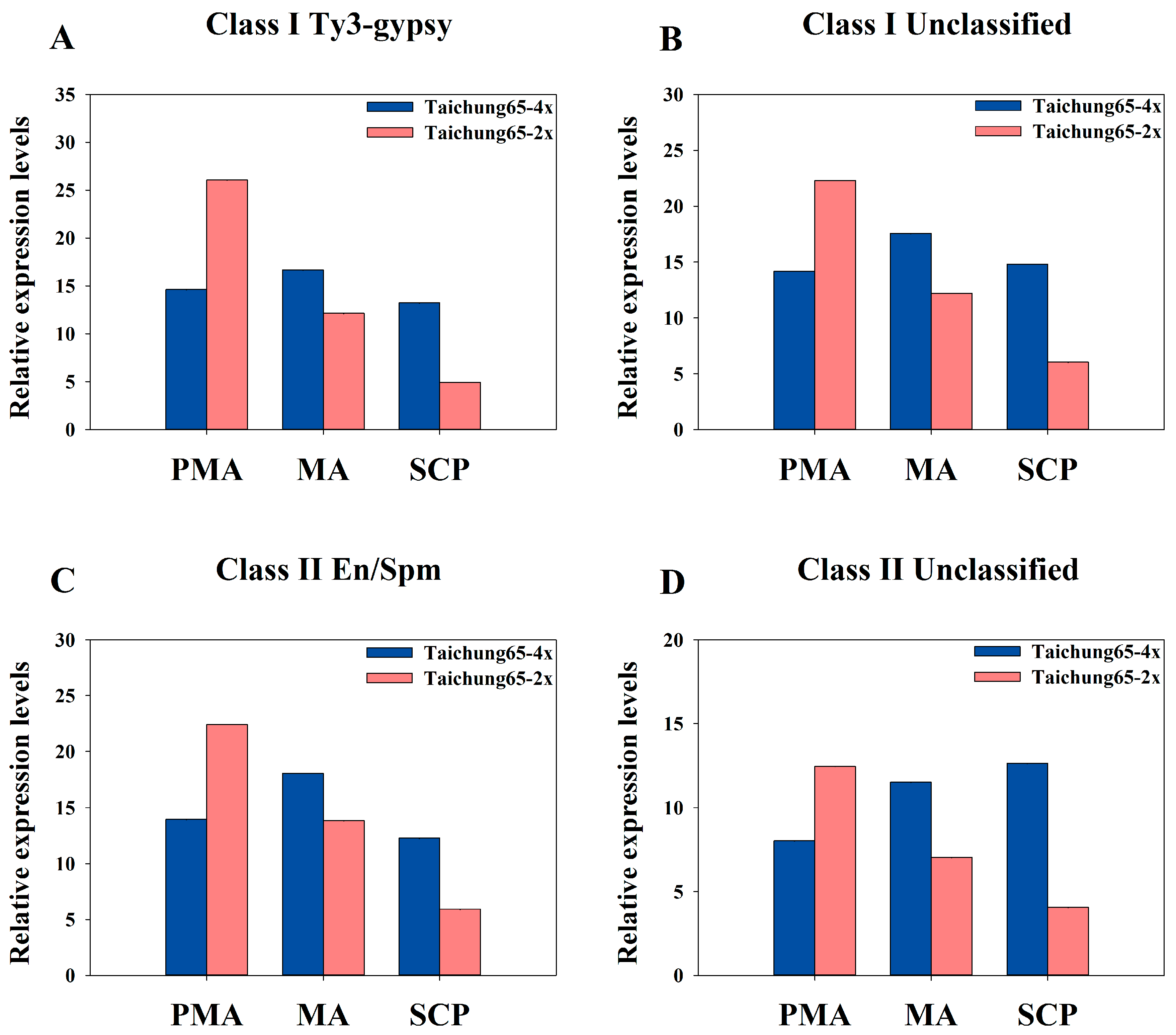
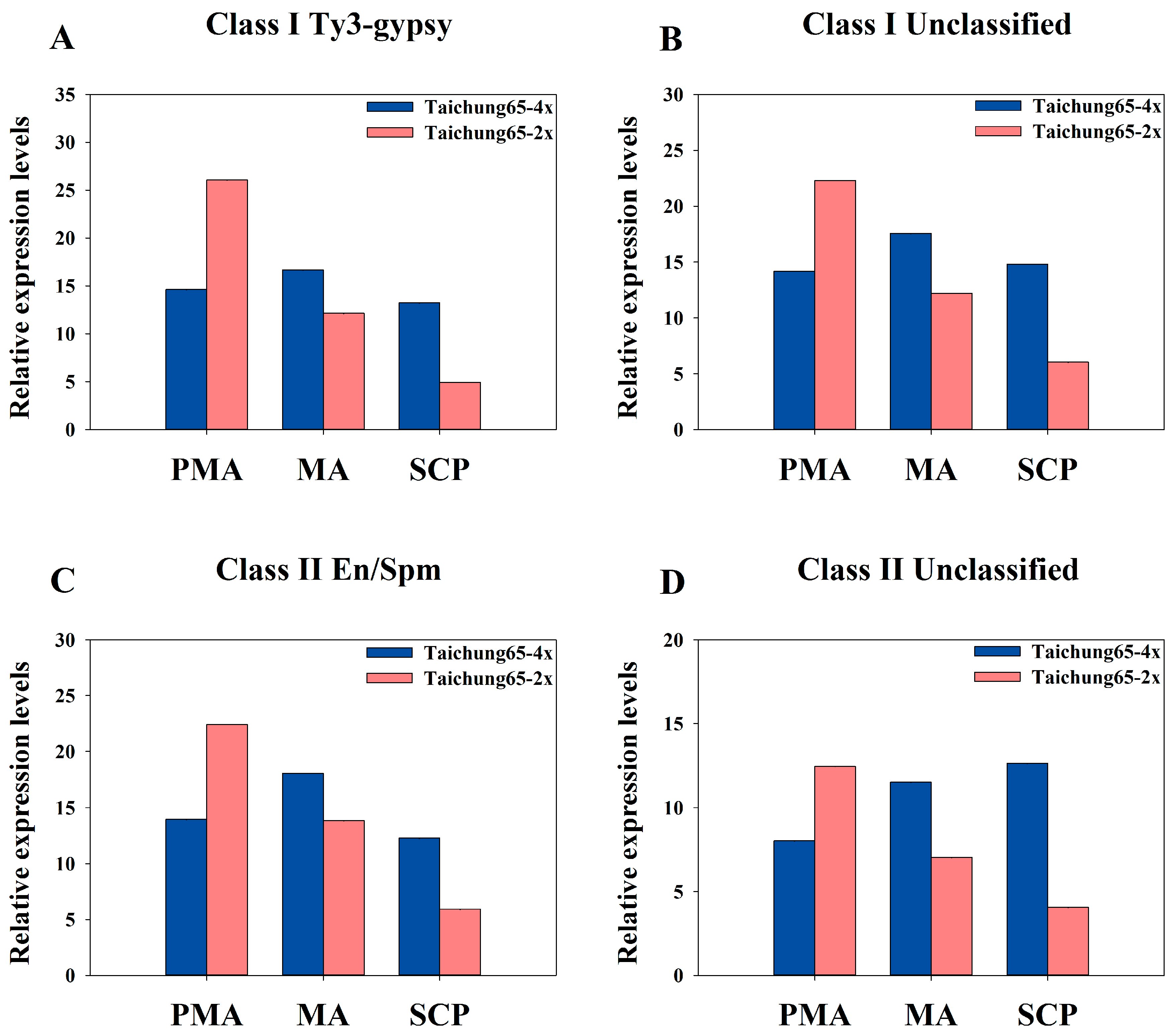
2.8. Differentially Expressed siRNAs in the Autopolyploid Pollen Associated with Transposable Elements
3. Discussion
3.1. Polyploidy Cause Changes in miRNA Expression Profiles during Pollen Development of Autotetraploid Rice
3.2. Specific miRNA Expression Patterns May Cause Meiosis Abnormalities in Autotetraploid Rice
3.3. Abundance of siRNAs Associated with Transposable Elements May Cause Meiosis Abnormalities in Autotetraploid Rice
4. Experimental Section
4.1. Rice Materials
4.2. Small RNA Library Construction, Sequencing and Data Processing
4.3. Analysis of Differentially Expressed miRNAs
4.4. The Prediction and Functional Analysis of Target Genes of miRNAs
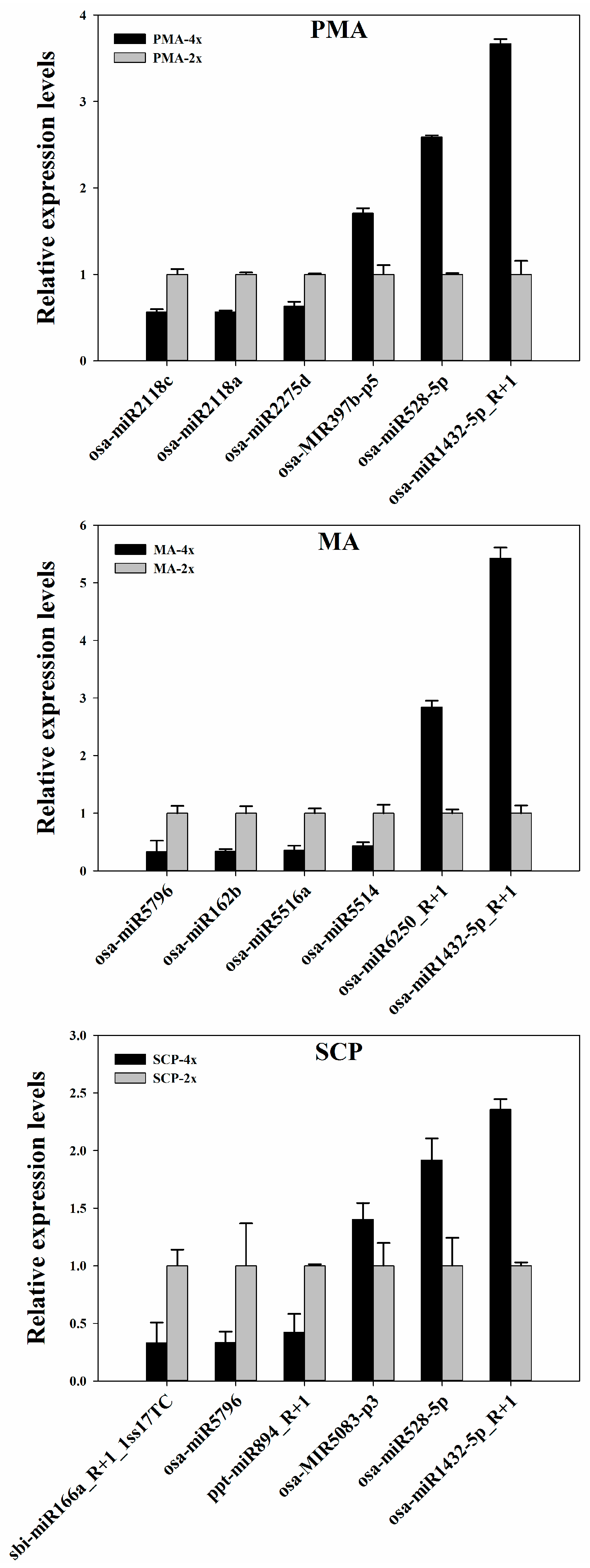
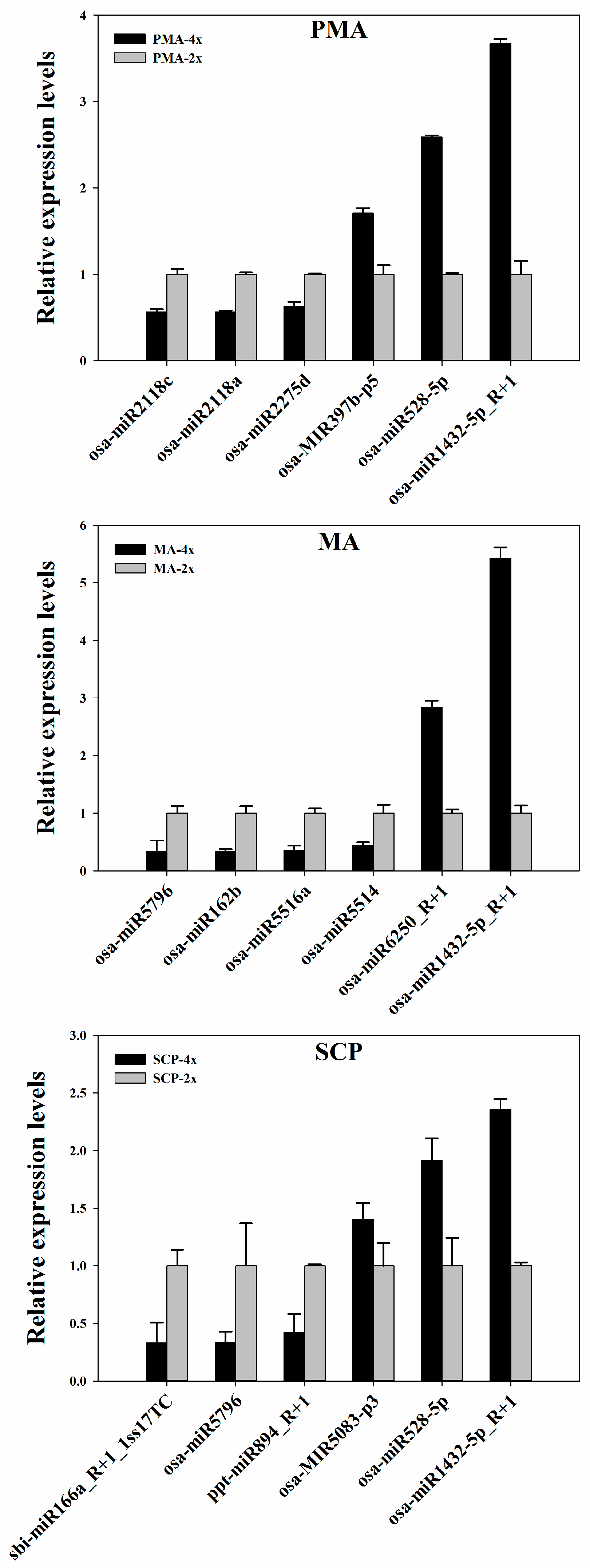
4.5. Quantitative Real-Time PCR (qRT-PCR) Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Soltis, D.E.; Bell, C.D.; Kim, S.; Soltis, P.S. Origin and early evolution of angiosperms. Ann. N. Y. Acad. Sci. 2008, 1133, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.D. Perspectives on polyploidy in plants—Ancient and neo. Biol. J. Linn. Soc. 2004, 82, 411–423. [Google Scholar] [CrossRef]
- Shahid, M.Q.; Li, Y.J.; Saleem, M.F.; Naeem, M.; Wei, C.M.; Liu, X.D. Yield and yield components in autotetraploid and diploid rice genotypes (indica and japonica) sown in early and late seasons. Aust. J. Crop Sci. 2013, 7, 632–641. [Google Scholar]
- Osborn, T.C.; Pires, J.C.; Birchler, J.A.; Auger, D.L.; Chen, Z.J.; Lee, H.S.; Comai, L.; Madlung, A.; Doerge, R.W.; Colot, V.; et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003, 19, 141–147. [Google Scholar] [CrossRef]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Haage, K.; Streit, V.E.; Gierl, A.; Ruiz, R.A. A large number of tetraploid Arabidopsis thaliana lines, generated by a rapid strategy, reveal high stability of neo-tetraploids during consecutive generations. Theor. Appl. Genet. 2009, 118, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.Q.; Liu, G.; Li, J.; Naeem, M.; Liu, X. Heterosis and gene action study of agronomic traits in diploid and autotetraploid rice. Acta Agric. Scand. Sect. B Soil Plant Sci. 2011, 61, 23–32. [Google Scholar] [CrossRef]
- Shahid, M.Q.; Xu, H.; Lin, S.; Chen, Z.; Naeem, M.; Li, Y.; Liu, X. Genetic analysis and hybrid vigor study of grain yield and other quantitative traits in autotetraploid rice. Pak. J. Bot. 2012, 44, 237–246. [Google Scholar]
- Tu, S.; Luan, L.; Liu, Y.; Long, W.; Kong, F.; He, T.; Xu, Q.; Yan, W.; Yu, M. Production and heterosis analysis of rice autotetraploid hybrids. Crop Sci. 2007, 47, 2356–2363. [Google Scholar] [CrossRef]
- He, J.H.; Shahid, M.Q.; Chen, Z.X.; Chen, X.A.; Liu, X.D.; Lu, Y.G. Abnormal PMC microtubule distribution pattern and chromosome behavior resulted in low pollen fertility of an intersubspecific autotetraploid rice hybrid. Plant Syst. Evol. 2011, 291, 257–265. [Google Scholar] [CrossRef]
- He, J.H.; Shahid, M.Q.; Li, Y.J.; Guo, H.B.; Cheng, X.A.; Liu, X.D.; Lu, Y.G. Allelic interaction of F1 pollen sterility loci and abnormal chromosome behaviour caused pollen sterility in intersubspecific autotetraploid rice hybrids. J. Exp. Bot. 2011, 62, 4433–4445. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.Q.; Sun, J.F.; Wei, C.M.; Zhang, P.; Liu, X.D. Studies on the abnormality of embryo sac and pollen fertility in autotetraploid rice during different growing seasons. Pak. J. Bot. 2010, 42, 7–19. [Google Scholar]
- Wu, J.; Shahid, M.Q.; Guo, H.; Yin, W.; Chen, Z.; Wang, L.; Liu, X.; Lu, Y. Comparative cytological and transcriptomic analysis of pollen development in autotetraploid and diploid rice. Plant Reprod. 2014, 27, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shahid, M.Q.; Chen, L.; Chen, Z.; Wang, L.; Liu, X.; Lu, Y. Polyploidy enhances F1 pollen sterility loci interactions that increase meiosis abnormalities and pollen sterility in autotetraploid rice. Plant Physiol. 2015, 169, 2700–2717. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Green, P.J. The role of rice microRNAs in abiotic stress responses. J. Plant Biol. 2013, 56, 187–197. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, M.; Xu, X.; Li, X.; Li, C.; Ding, Z. System analysis of microRNAs in the development and aluminium stress responses of the maize root system. Plant Biotechnol. J. 2014, 12, 1108–1121. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Shao, C.; Wang, H.; Ma, X.; Chen, M. Construction of gene regulatory networks mediated by vegetative and reproductive stage-specific small RNAs in rice (Oryza sativa). New Phytol. 2013, 197, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Mao, D.; Xu, L.; Li, D.; Song, S.; Chen, C. Integrated analysis of miRNA and mRNA expression profiles in response to Cd exposure in rice seedlings. BMC Genom. 2014, 15, 835. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, T.; Kaneko, F.; Kazama, T.; Suwabe, K.; Suzuki, G.; Makino, A.; Mae, T.; Endo, M.; Kawagishi-Kobayashi, M.; Watanabe, M. Identification of small RNAs in late developmental stage of rice anthers. Genes Genet. Syst. 2008, 83, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Chun, J.; Ai, T.B.; Tong, Y.A.; Zhang, R.; Zhao, M.M.; Chen, F.; Wang, S.H. MicroRNA profiles and their control of male gametophyte development in rice. Plant Mol. Biol. 2012, 80, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.Q.; Yan, L.F.; Wang, T. Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol. 2011, 12, R53. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Horiuchi, Y.; Ueda, Y.; Mizuta, Y.; Kubo, T.; Yano, K.; Yamaki, S.; Tsuda, K.; Nagata, T.; Niihama, M.; et al. Rice expression atlas in reproductive development. Plant Cell Physiol. 2010, 51, 2060–2081. [Google Scholar] [CrossRef] [PubMed]
- Deveshwar, P.; Bovill, W.D.; Sharma, R.; Able, J.A.; Kapoor, S. Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice. BMC Plant Biol. 2011, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Yant, L.; Hollister, J.D.; Wright, K.M.; Arnold, B.J.; Higgins, J.D.; Franklin, F.C.; Bomblies, K. Meiotic adaptation to genome duplication in Arabidopsis arenosa. Curr. Biol. 2013, 23, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Arnold, B.; Xue, K.; Surinova, M.; O’Connell, J.; Bomblies, K. Selection on meiosis genes in diploid and tetraploid Arabidopsis arenosa. Mol. Biol. Evol. 2015, 32, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yang, H.; Wei, Z.; Ma, H.; Ge, X. Rice male development under drought stress: Phenotypic changes and stage-dependent transcriptomic reprogramming. Mol. Plant 2013, 6, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.K.; Poulsen, L.R.; Schulz, A.; Fleurat-Lessard, P.; Moller, A.; Husted, S.; Schiott, M.; Amtmann, A.; Palmgren, M.G. Pollen development and fertilization in Arabidopsis is dependent on the MALE GAMETOGENESIS IMPAIRED ANTHERS gene encoding a type V P-type ATPase. Genes Dev. 2005, 19, 2757–2769. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.X.; Liu, Y.G. Molecular control of male reproductive development and pollen fertility in rice. J. Integr. Plant Biol. 2012, 54, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Li, Y.; Shen, Y.; Cheng, Z. Ten years of gene discovery for meiotic event control in rice. J. Genet. Genom. 2014, 41, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Komiya, R.; Ohyanagi, H.; Niihama, M.; Watanabe, T.; Nakano, M.; Kurata, N.; Nonomura, K. Rice germline-specific Argonaute MEL1 protein binds to phasiRNAs generated from more than 700 lincRNAs. Plant J. 2014, 78, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Zhang, H.; Arikit, S.; Huang, K.; Nan, G.L.; Walbot, V.; Meyers, B.C. Spatiotemporally dynamic, cell-type-dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc. Natl. Acad. Sci. USA 2015, 112, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- De Storme, N.; Geelen, D. The Arabidopsis mutant jason produces unreduced first division restitution male gametes through a parallel/fused spindle mechanism in meiosis II. Plant Physiol. 2011, 155, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Uanschou, C.; Ronceret, A.; Von Harder, M.; de Muyt, A.; Vezon, D.; Pereira, L.; Chelysheva, L.; Kobayashi, W.; Kurumizaka, H.; Schlogelhofer, P.; et al. Sufficient amounts of functional HOP2/MND1 complex promote interhomolog DNA repair but are dispensable for intersister DNA repair during meiosis in Arabidopsis. Plant Cell 2013, 25, 4924–4940. [Google Scholar] [CrossRef] [PubMed]
- Merkle, T.; Grasser, K.D. Unexpected mobility of plant chromatin-associated HMGB proteins. Plant Signal Behav. 2011, 6, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Block-Schmidt, A.S.; Dukowic-Schulze, S.; Wanieck, K.; Reidt, W.; Puchta, H. BRCC36A is epistatic to BRCA1 in DNA crosslink repair and homologous recombination in Arabidopsis thaliana. Nucleic Acids Res. 2011, 39, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.A.; Meyers, B.C. Small RNA-mediated epigenetic modifications in plants. Curr. Opin. Plant Biol. 2011, 14, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Xia, E.H.; Yao, Q.Y.; Liu, X.D.; Gao, L.Z. Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, E7022–E7029. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, Z.; Lin, H.; Liu, H.; Chen, J.; Peng, H.; Cao, M.; Rong, T.; Pan, G. Cytoplasmic male sterility-regulated novel microRNAs from maize. Funct. Integr. Genom. 2011, 11, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Wei, H.; Wu, M.; Song, M.; Zhang, J.; Yu, J.; Fan, S.; Yu, S. Comparative expression profiling of miRNA during anther development in genetic male sterile and wild type cotton. BMC Plant Biol. 2013, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Xu, B.; Zhao, N.; Yang, X.; Zhang, M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genom. 2013, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Sun, H.; Du, Y.; Zhang, J.; Li, J.; Liu, Y.; Zhao, Y.; Zhao, Q. Characterization and expression patterns of microRNAs involved in rice grain filling. PLoS ONE 2013, 8, e54148. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Zhu, Z.; Hu, J.; Qian, Q.; Dai, J.; Ding, Y. Identification and expression analysis of microRNAs at the grain filling stage in rice (Oryza sativa L.) via deep sequencing. PLoS ONE 2013, 8, e57863. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Salinas, M.; Hohmann, S.; Berndtgen, R.; Huijser, P. miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 2010, 22, 3935–3950. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.F.; Tian, Q.; Reed, J.W. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 2006, 133, 4211–4218. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Zhang, B. microRNA evolution and expression analysis in polyploidized cotton genome. Plant Biotechnol. J. 2015, 13, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Zhai, X.; Niu, S.; Ren, Y. Dynamic expression of novel and conserved microRNAs and their targets in diploid and tetraploid of Paulownia tomentosa. Biochimie 2014, 102, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Lu, J.; Tian, L.; Ramachandran, V.; Kasschau, K.D.; Chapman, E.J.; Carrington, J.C.; Chen, X.; Wang, X.J.; Chen, Z.J. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl. Acad. Sci. USA 2009, 106, 17835–17840. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, K.; Morohoshi, A.; Nakano, M.; Eiguchi, M.; Miyao, A.; Hirochika, H.; Kurata, N. A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell 2007, 19, 2583–2594. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, P.; Zhai, J.; Zhou, M.; Ma, L.; Liu, B.; Jeong, D.H.; Nakano, M.; Cao, S.; Liu, C.; et al. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 2012, 69, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Gong, L.; Yuan, W.; Li, X.; Chen, G.; Li, X.; Zhang, Q.; Wu, C. Replication protein A (RPA1a) is required for meiotic and somatic DNA repair but is dispensable for DNA replication and homologous recombination in rice. Plant Physiol. 2009, 151, 2162–2173. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Tang, D.; Wang, M.; Lu, J.; Yu, H.; Liu, J.; Qian, B.; Gong, Z.; Wang, X.; Chen, J.; et al. MER3 is required for normal meiotic crossover formation, but not for presynaptic alignment in rice. J. Cell Sci. 2009, 122, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Vespa, L.; Warrington, R.T.; Mokros, P.; Siroky, J.; Shippen, D.E. ATM regulates the length of individual telomere tracts in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 18145–18150. [Google Scholar] [CrossRef] [PubMed]
- Vespa, L.; Couvillion, M.; Spangler, E.; Shippen, D.E. ATM and ATR make distinct contributions to chromosome end protection and the maintenance of telomeric DNA in Arabidopsis. Genes Dev. 2005, 19, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Friesner, J.D.; Liu, B.; Culligan, K.; Britt, A.B. Ionizing radiation-dependent γ-H2AX focus formation requires ataxia telangiectasia mutated and ataxia telangiectasia mutated and Rad3-related. Mol. Biol. Cell 2005, 16, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Clerici, M.; Mantiero, D.; Lucchini, G.; Longhese, M.P. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J. Biol. Chem. 2005, 280, 38631–38638. [Google Scholar] [CrossRef] [PubMed]
- Gallego, M.E.; Jeanneau, M.; Granier, F.; Bouchez, D.; Bechtold, N.; White, C.I. Disruption of the Arabidopsis RAD50 gene leads to plant sterility and MMS sensitivity. Plant J. 2001, 25, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, H.; Zheng, Y.; Ding, Y. Comparative expression profiling of miRNAs between the cytoplasmic male sterile line MeixiangA and its maintainer line MeixiangB during rice anther development. Planta 2015, 241, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2, E104. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Liu, H.L.; Daxinger, L.; Pontes, O.; He, X.; Qian, W.; Lin, H.; Xie, M.; Lorkovic, Z.J.; Zhang, S.; et al. An RNA polymerase II- and AGO4-associated protein acts in RNA-directed DNA methylation. Nature 2010, 465, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Ausin, I.; Johnson, L.M.; Vashisht, A.A.; Zhu, J.K.; Wohlschlegel, J.A.; Jacobsen, S.E. A protein complex required for polymerase V transcripts and RNA-directed DNA methylation in Arabidopsis. Curr. Biol. 2010, 20, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yin, H.; Song, X.; Zhang, Y.; Liu, M.; Sang, J.; Jiang, J.; Li, J.; Zhuo, R. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol. J. 2016. [Google Scholar] [CrossRef]
- miRBase 20.0. Available online: ftp://mirbase.org/pub/mirbase/CURRENT/ (accessed on 20 July 2015).
- Rice Genomes. Available online: ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v9.0/Osativa (accessed on 15 July 2015).
- RNAfold Software. Available online: http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi (accessed on 10 August 2015).
- Cer, R.Z.; Herrera-Galeano, J.E.; Anderson, J.J.; Bishop-Lilly, K.A.; Mokashi, V.P. miRNA Temporal Analyzer (miRNATA): A bioinformatics tool for identifying differentially expressed microRNAs in temporal studies using normal quantile transformation. Gigascience 2014, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Carrington, J.C. miRNA target prediction in plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar] [PubMed]
- AgriGO. Available online: http://bioinfo.cau.edu.cn/agriGO/ (accessed on 25 July 2015).
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi-Gasic, E.; Wu, R.; Wood, M.; Walton, E.F.; Hellens, R.P. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 2007, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Description/Stage-Specific Expression | Up-Regulated | Down-Regulated | Total |
|---|---|---|---|---|
| Taichung65-2x | DEM in MA compared to PMA | 20 | 46 | 66 |
| DEM in SCP compared to MA | 28 | 96 | 124 | |
| Taichung65-4x | DEM in MA compared to PMA | 41 | 75 | 116 |
| DEM in SCP compared to MA | 58 | 57 | 115 | |
| Taichung65-4x vs. Taichung65-2x | DEM in PMA | 27 | 19 | 46 |
| DEM in MA | 37 | 21 | 58 | |
| DEM in SCP | 75 | 27 | 102 | |
| DEM specifically in MA compared to PMA of Taichung65-2x | 15 | 34 | 49 | |
| DEM specifically in SCP compared to MA of Taichung65-2x | 19 | 55 | 74 | |
| DEM specifically in MA compared to PMA of Taichung65-4x | 36 | 63 | 99 | |
| DEM specifically in SCP compared to MA of Taichung65-4x | 49 | 16 | 65 |
| miRNA | Targets | Genes Annotation | PMA | MA | SCP | Specific in MA | Meiosis-Related Genes |
|---|---|---|---|---|---|---|---|
| osa-miR1320-5p_L-1R+1 | LOC_Os03g12140 | glucan endo-1,3-β-glucosidase precursor, putative, expressed | up | up | up | LOC_Os11g03430 (CDC45) | |
| osa-miR1432-5p_R+1 | LOC_Os04g51610 | calcium-transporting ATPase, plasma membrane-type, putative, expressed | up | up | up | LOC_Os04g40290 (CRC1) | |
| LOC_Os05g07210 | metal cation transporter, putative, expressed | up | up | up | LOC_Os02g29464 (RAD50) | ||
| osa-miR1876 | LOC_Os08g42930 | disease resistance protein RGA1, putative, expressed | ns | up | up | LOC_Os01g01689 (ATM) | |
| osa-MIR2122-p3_1ss19GT | LOC_Os01g72390 | NBS type disease resistance protein, putative, expressed | ns | up | ns | osa-MIR2122-p3_1ss19GT | LOC_Os01g01689 (ATM) |
| osa-miR5788 | LOC_Os03g07430 | protein kinase domain containing protein, expressed | ns | up | ns | osa-miR5788 | LOC_Os12g38460 (MEL2) |
| LOC_Os03g23935 | WD domain, G-β repeat domain containing protein, expressed | ns | up | ns | osa-miR5788 | LOC_Os02g40450 (OsMER3) | |
| LOC_Os03g53750 | nuclear prelamin A recognition factor, putative, expressed | ns | up | ns | osa-miR5788 | LOC_Os06g41050 (OsCOM1) | |
| LOC_Os05g39540 | metal cation transporter, putative, expressed | ns | up | ns | osa-miR5788 | LOC_Os02g29464 (RAD50) | |
| LOC_Os09g09220 | protein kinase domain containing protein, expressed | ns | up | ns | osa-miR5788 | LOC_Os12g38460 (MEL2) | |
| osa-MIR6250-p5 | LOC_Os01g22500 | craniofacial development protein 1, putative, expressed | ns | up | up | LOC_Os12g34510 (HTA3) | |
| LOC_Os03g63240 | disease resistance protein, putative, expressed | ns | up | up | LOC_Os01g01689 (ATM) | ||
| LOC_Os08g39000 | expressed protein | ns | up | up | LOC_Os03g44760 (OsAM1) | ||
| osa-miR812g | LOC_Os11g25510 | protein kinase domain containing protein, expressed | ns | up | up | LOC_Os12g38460 (MEL2) | |
| PC-3p-403976_40 | LOC_Os07g28090 | ABC transporter, ATP-binding protein, putative, expressed | ns | up | up | LOC_Os02g53680 (OsRPA1A) | |
| PC-3p-550716_26 | LOC_Os11g13410 | mla1, putative, expressed | ns | up | ns | PC-3p-550716_26 | LOC_Os01g01689 (ATM) |
| osa-miR160a-5p_R-1_1ss20CT | LOC_Os05g30820 | protein kinase, putative, expressed | ns | down | ns | osa-miR160a-5p_R-1_1ss20CT | LOC_Os12g38460 (MEL2) |
| osa-miR167a-3p_L+1R-1 | LOC_Os03g58260 | indole-3-glycerol phosphate lyase, chloroplast precursor, putative, expressed | ns | down | down | LOC_Os03g44760 (OsAM1) | |
| LOC_Os07g17220 | disease resistance protein, putative, expressed | ns | down | down | LOC_Os01g01689 (ATM) | ||
| osa-MIR5486-p3 | LOC_Os01g47256 | exosome complex exonuclease RRP40, putative, expressed | ns | down | down | LOC_Os01g72880 (MLH1) | |
| LOC_Os09g02700 | polyadenylate-binding protein, putative, expressed | ns | down | down | LOC_Os03g12414 (OsSDS) | ||
| LOC_Os11g39330 | NB-ARC domain containing protein, putative, expressed | ns | down | down | LOC_Os01g01689 (ATM) | ||
| osa-MIR5491-p5 | LOC_Os01g24060 | importin subunit α, putative, expressed | ns | down | ns | osa-MIR5491-p5 | LOC_Os03g12414 (OsSDS) |
| osa-MIR5518-p3 | LOC_Os04g48770 | ubiquitin fusion degradation protein, putative, expressed | ns | down | ns | osa-MIR5518-p3 | LOC_Os04g40290 (CRC1) |
| osa-MIR5806-p5 | LOC_Os08g19694 | NB-ARC domain containing protein, expressed | ns | down | ns | osa-MIR5806-p5 | LOC_Os01g01689 (ATM) |
| PC-3p-120829_194 | LOC_Os03g04520 | RNA recognition motif containing protein, putative, expressed | ns | down | ns | PC-3p-120829_194 | LOC_Os02g40450 (OsMER3) |
| MicroRNA_Family | miRNA | Sequence (5’ to 3’) | PMA | MA | SCP |
|---|---|---|---|---|---|
| miR2118 | osa-MIR2118k-p5 | TAAGCTCTATTGTCCCCTCTA | ns | ns | down |
| osa-miR2118e | TTCCCAATGCCTCCCATGCCTA | ns | ns | up | |
| osa-MIR2118l-p5 | TTAGGAAGAGGAAGAAATTGA | ns | down | ns | |
| osa-MIR2118m-p5 | GGAATGGGAACATGAAGGAAAG | ns | down | ns | |
| osa-MIR2118o-p5 | GGCATGGGGACATGAAGGAATG | ns | down | ns | |
| osa-miR2118a | TTCTCGATGCCTCCCATTCCTA | down | ns | ns | |
| osa-miR2118c | TTCCCGATGCCTCCTATTCCTA | down | ns | ns | |
| osa-MIR2118a-p5 | GGACTGGGAACATATGAGAAAG | down | ns | ns | |
| miR2275 | osa-miR2275d | CTTGTTTTTCTCCAATATCTCA | down | ns | up |
| osa-MIR2275c-p3 | ATTGTTTTTCTCCAATATCTCA | down | ns | up | |
| mes-miR2275 | TTTGGTTTCCTCCAATATCTTA | down | ns | ns | |
| zma-miR2275b-3p_1ss22AG | TTCAGTTTCCTCTAATATCTCG | down | ns | ns |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Shahid, M.Q.; Wu, J.; Wang, L.; Liu, X.; Lu, Y. Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice. Int. J. Mol. Sci. 2016, 17, 499. https://doi.org/10.3390/ijms17040499
Li X, Shahid MQ, Wu J, Wang L, Liu X, Lu Y. Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice. International Journal of Molecular Sciences. 2016; 17(4):499. https://doi.org/10.3390/ijms17040499
Chicago/Turabian StyleLi, Xiang, Muhammad Qasim Shahid, Jinwen Wu, Lan Wang, Xiangdong Liu, and Yonggen Lu. 2016. "Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice" International Journal of Molecular Sciences 17, no. 4: 499. https://doi.org/10.3390/ijms17040499
APA StyleLi, X., Shahid, M. Q., Wu, J., Wang, L., Liu, X., & Lu, Y. (2016). Comparative Small RNA Analysis of Pollen Development in Autotetraploid and Diploid Rice. International Journal of Molecular Sciences, 17(4), 499. https://doi.org/10.3390/ijms17040499