
The Protective Role of the TOPK/PBK Pathway in Myocardial Ischemia/Reperfusion and H2O2-Induced Injury in H9C2 Cardiomyocytes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
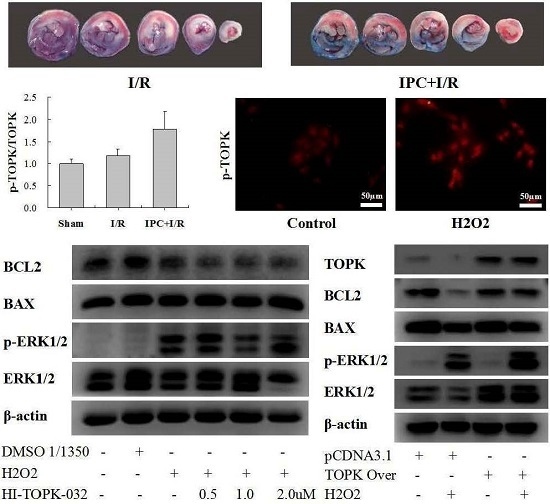
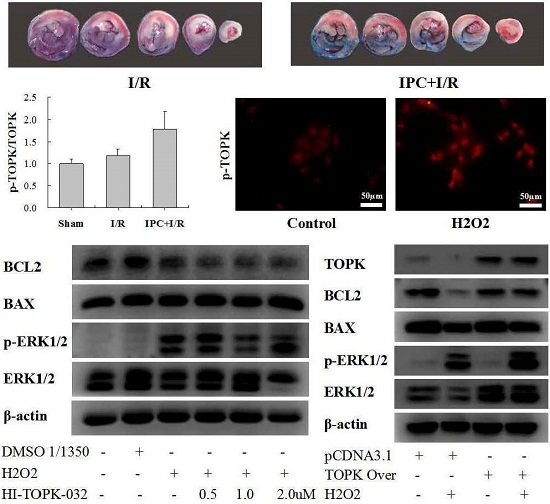
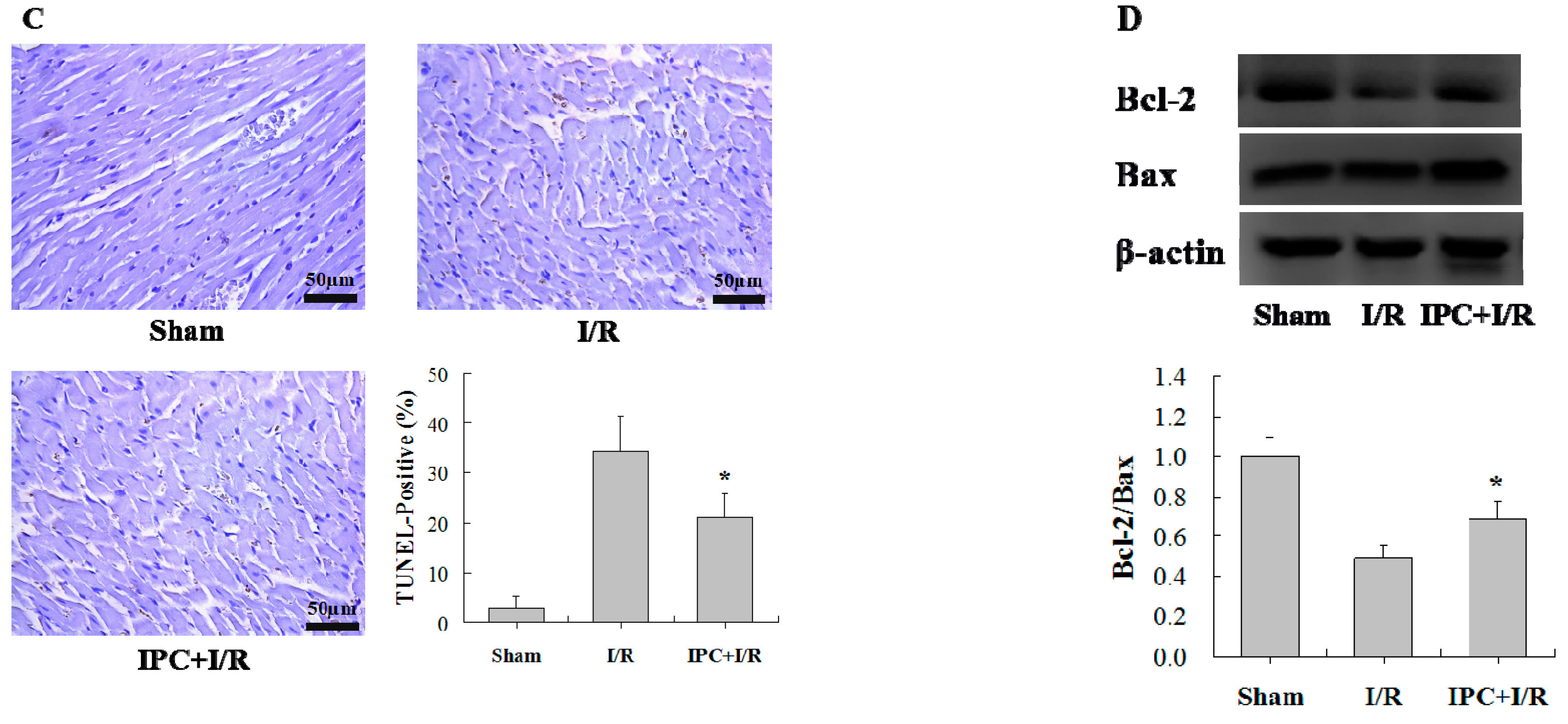
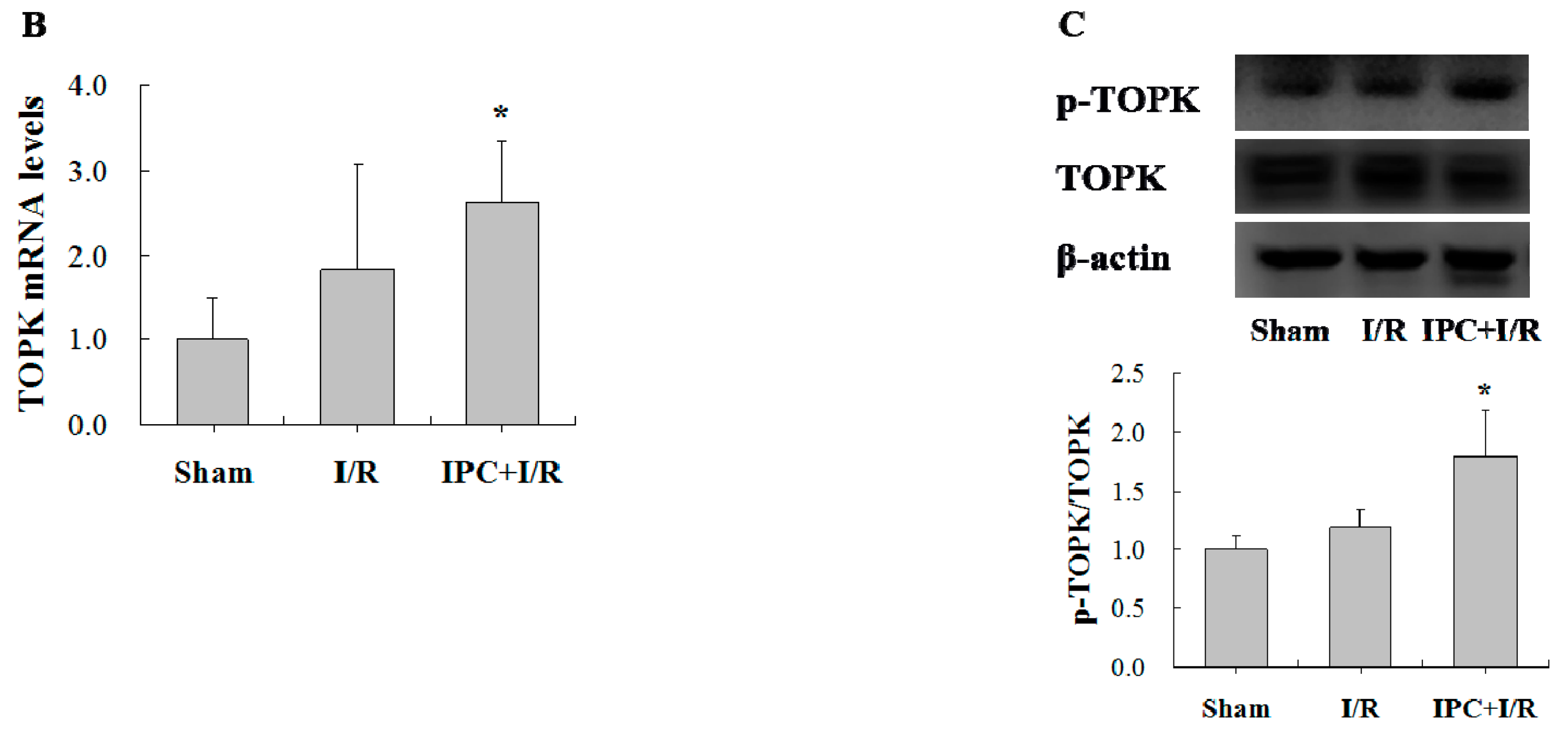
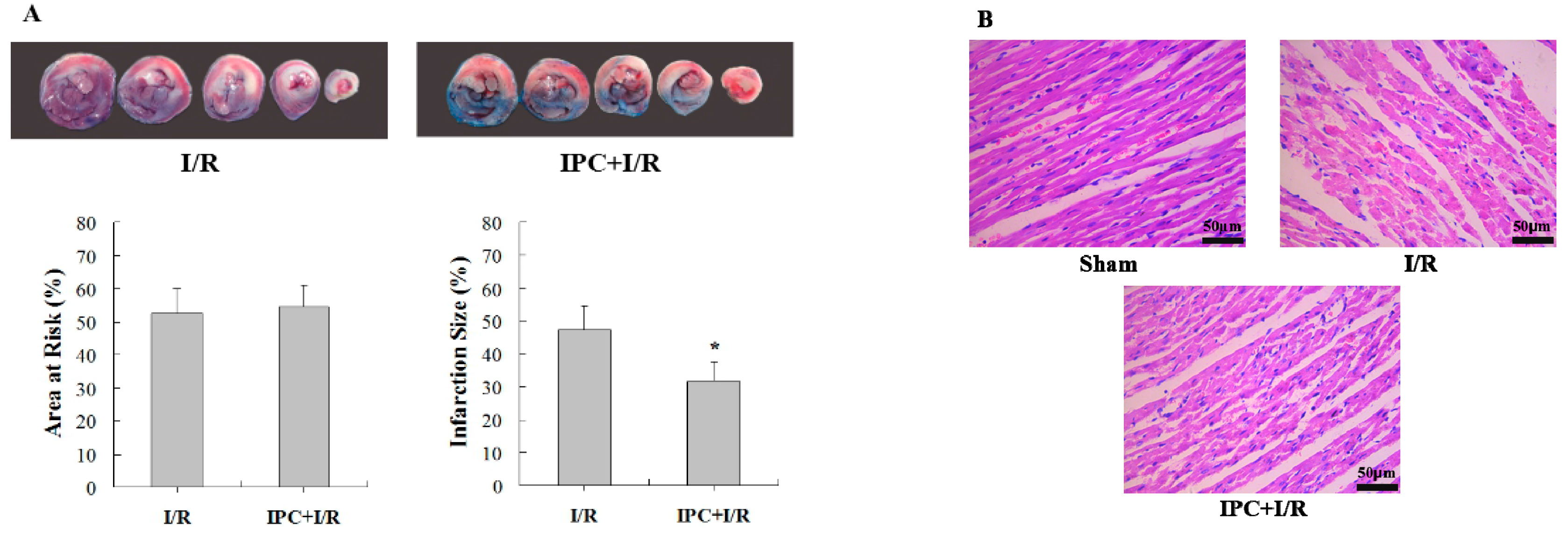
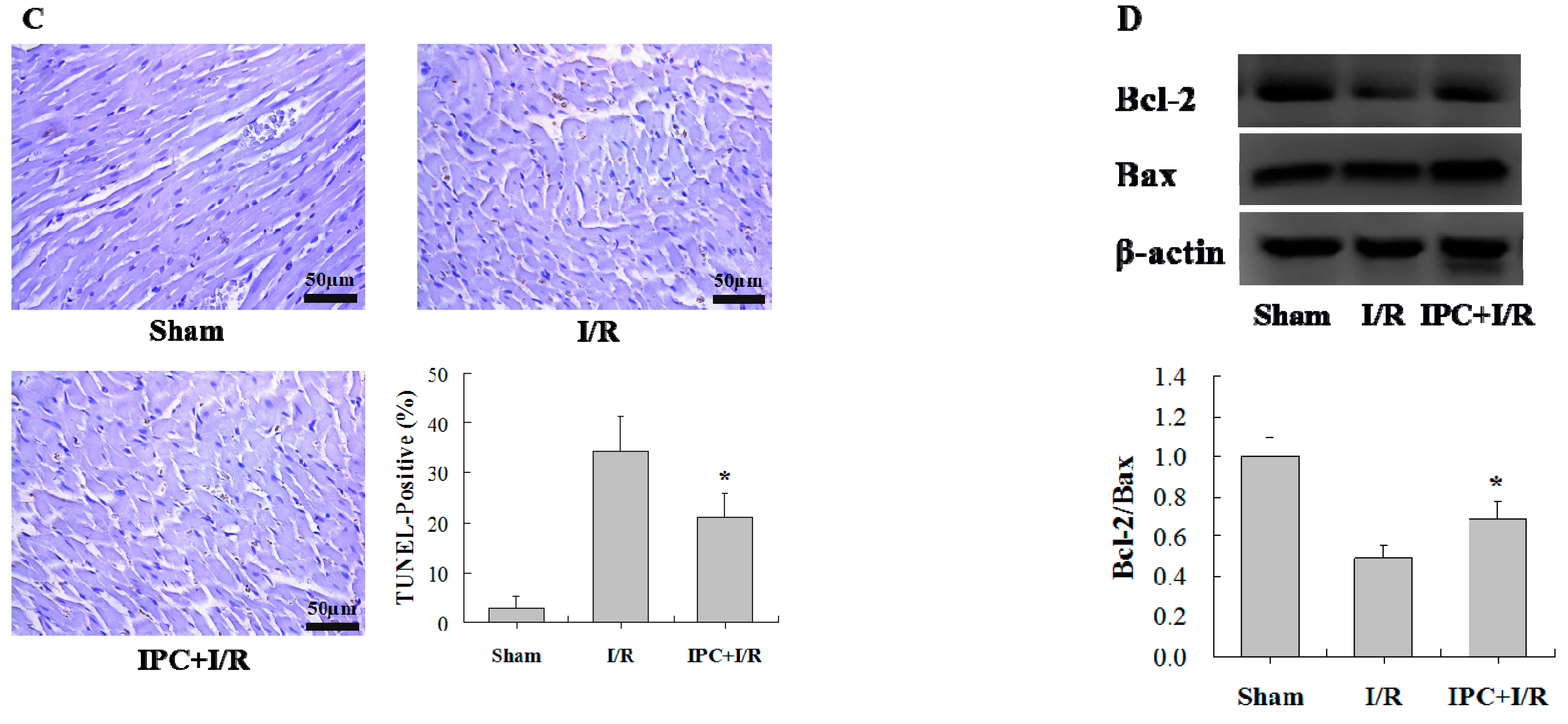
2.1. IPC Conferred Protection against Myocardial I/R Injury
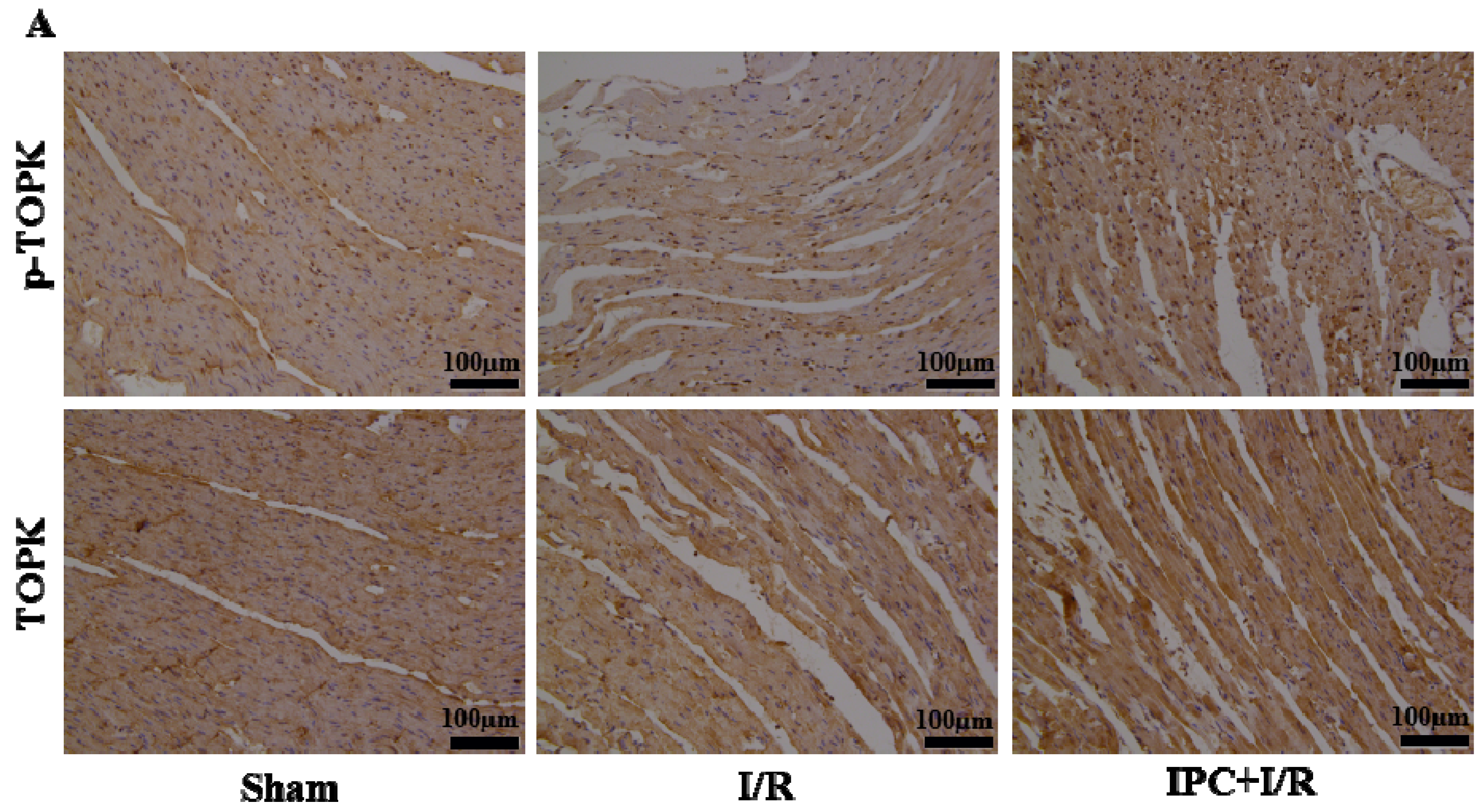
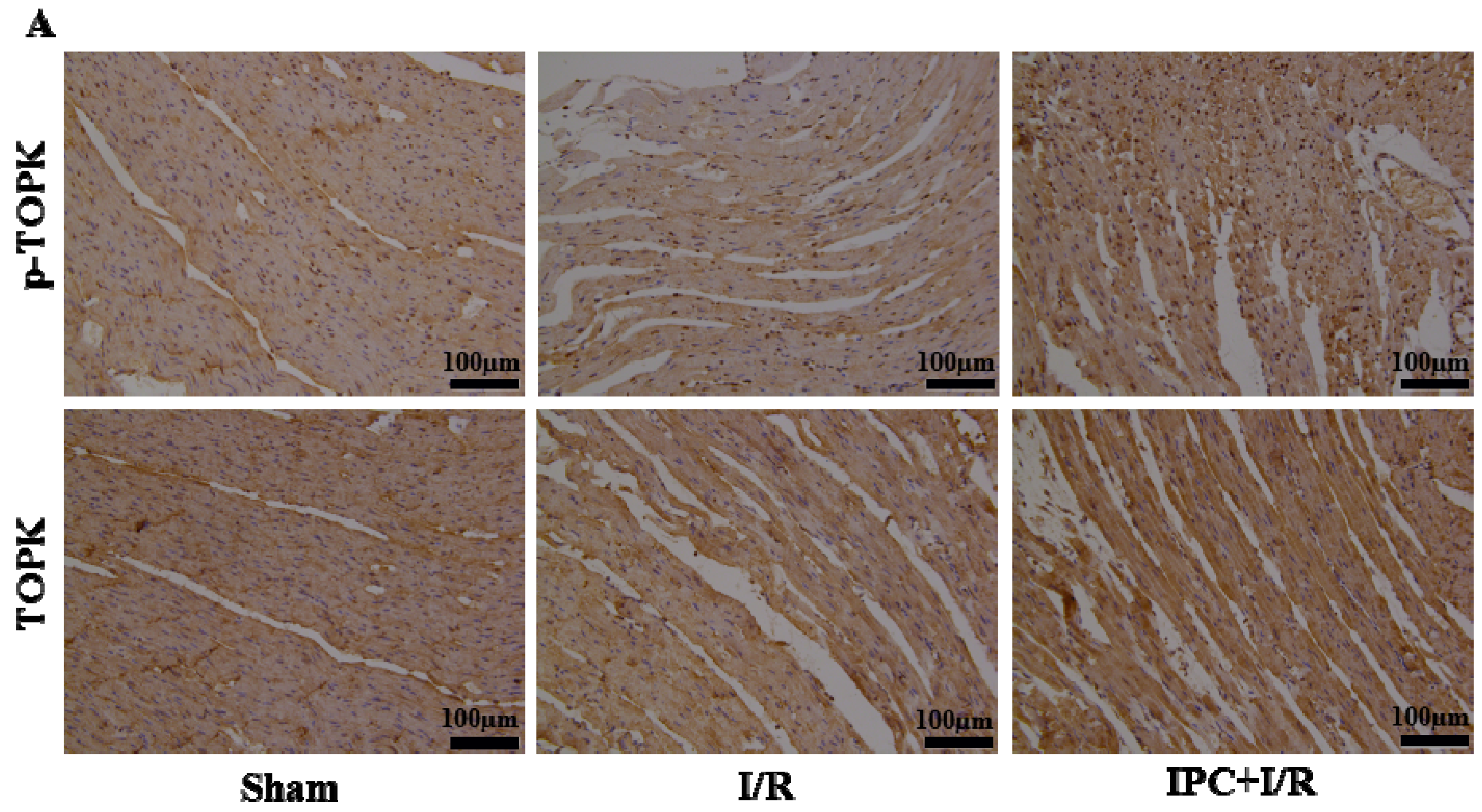
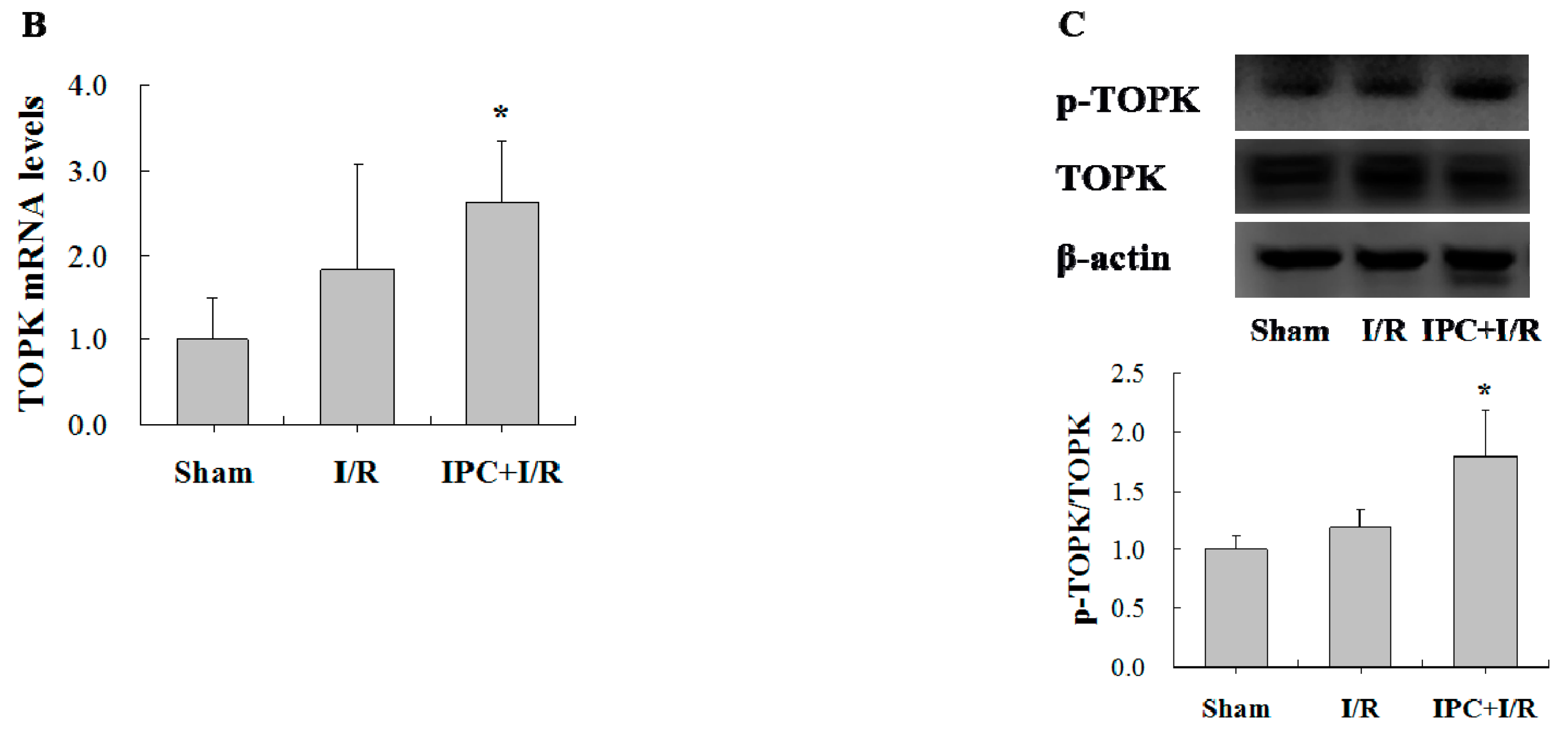
2.2. IPC Activated TOPK Signaling Pathway
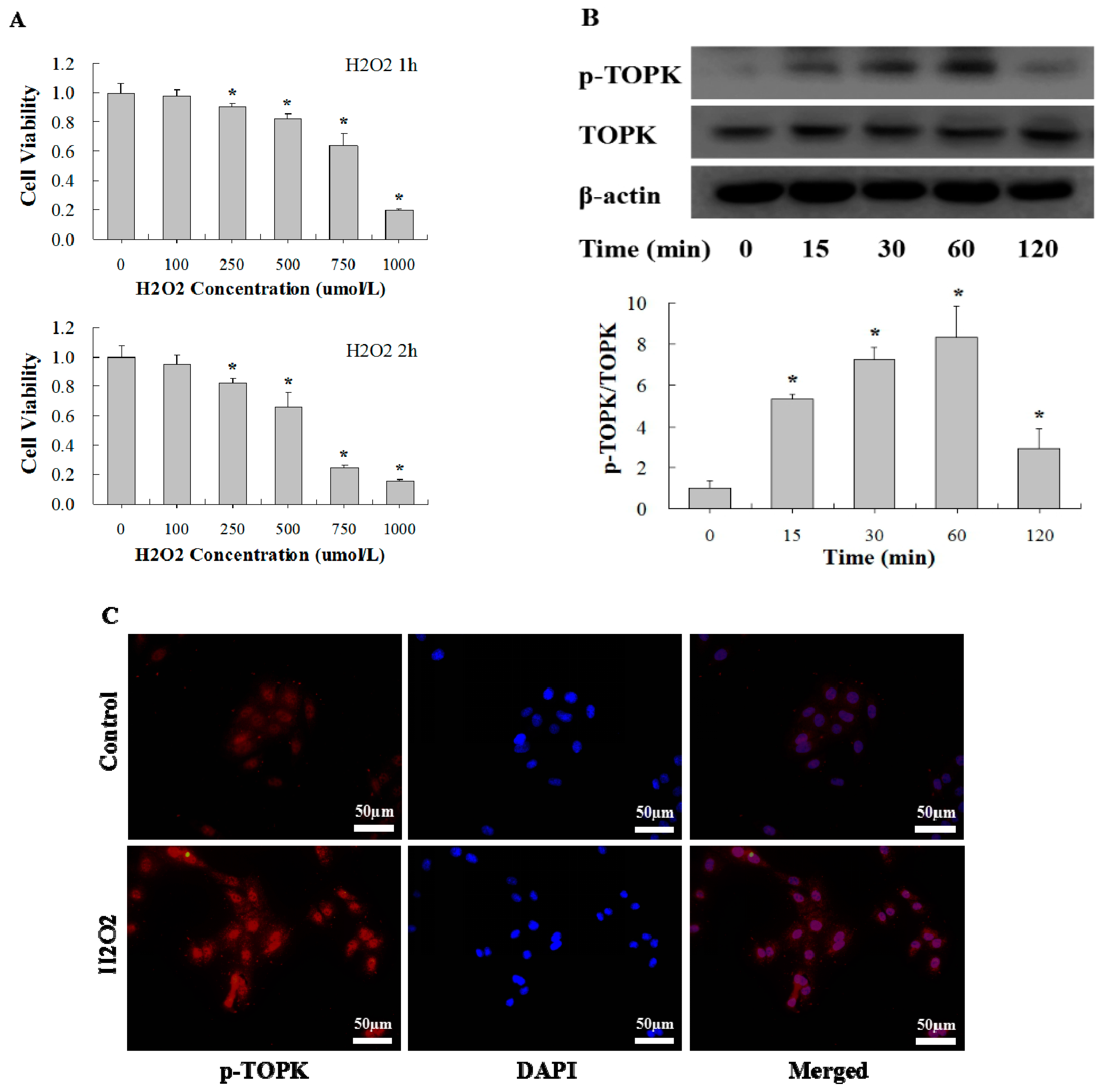
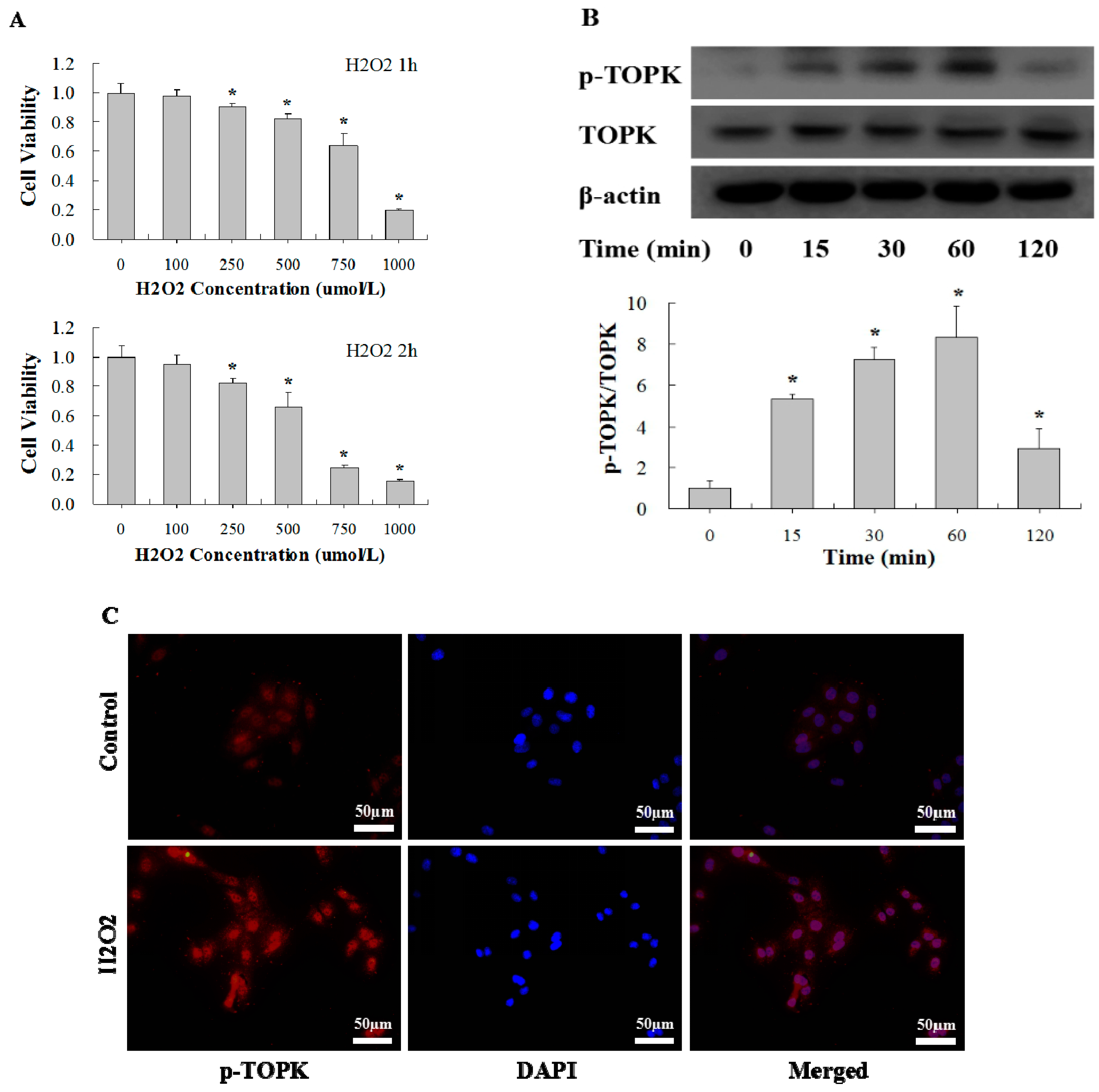
2.3. H2O2 Activates TOPK in Cardiomyocytes in Time-Dependent Manner
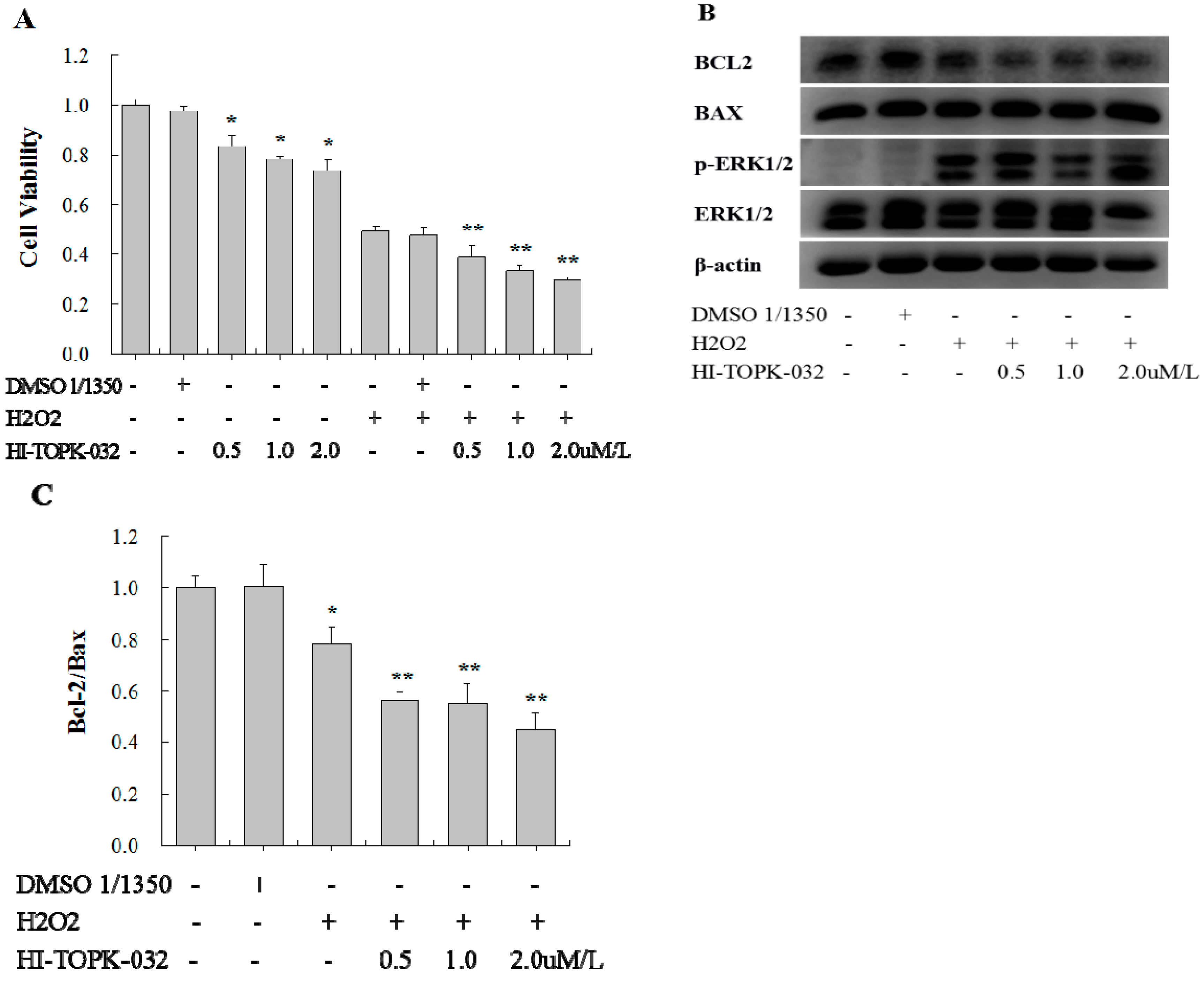
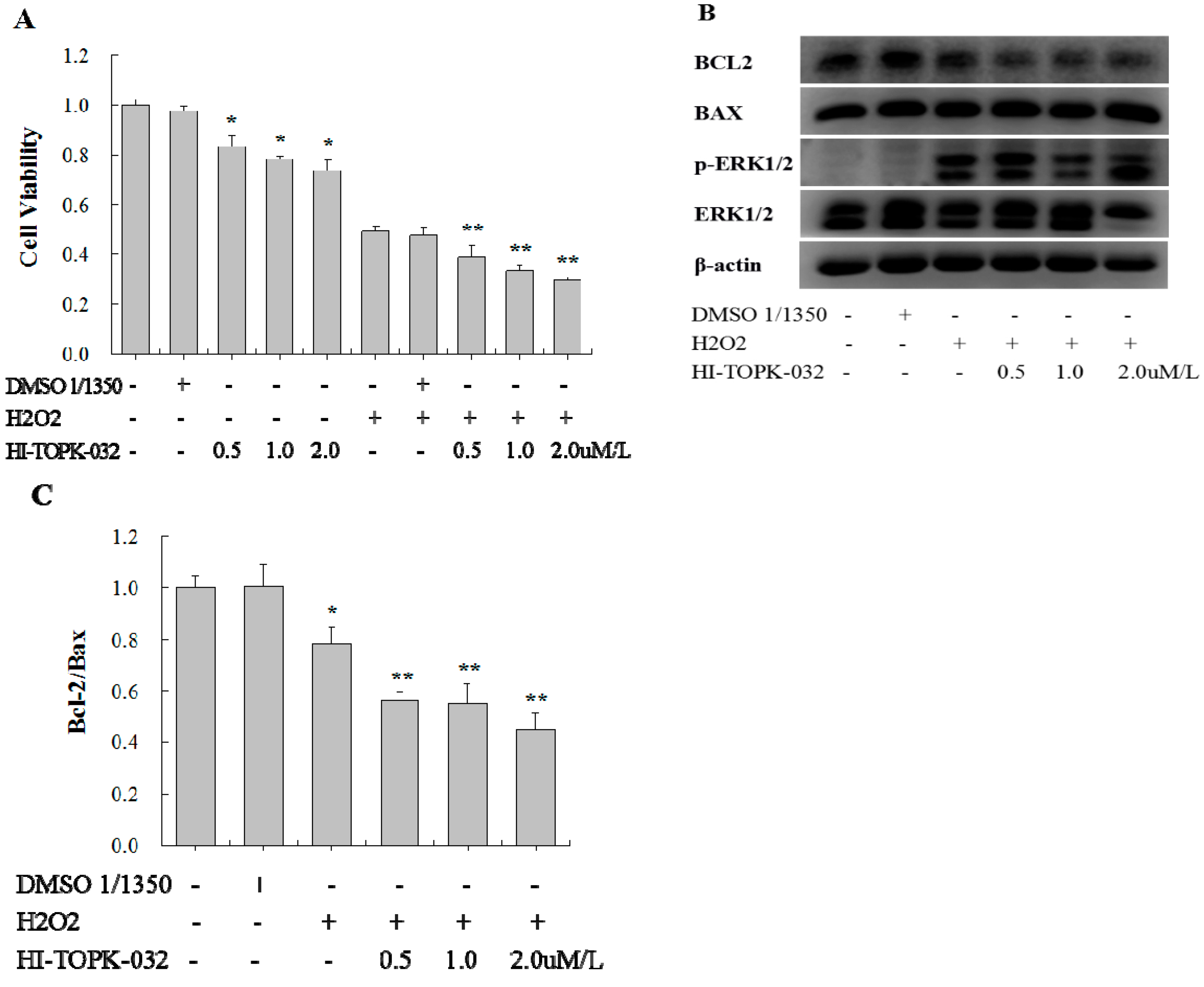
2.4. TOPK Inhibitor Aggravated the Oxidative Stress Injury in H9C2 Cardiomyocytes via the ERK Signaling Pathway
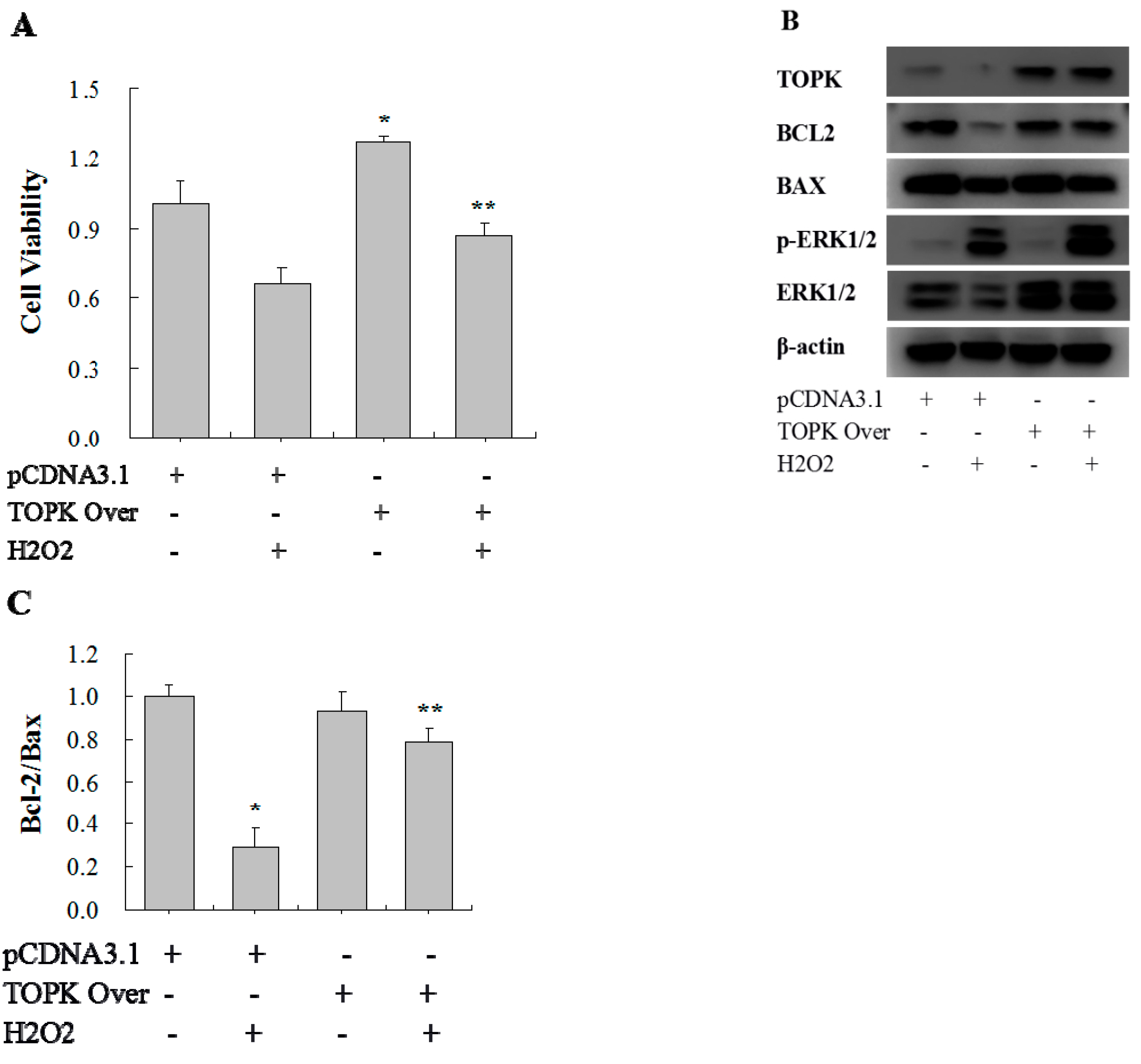
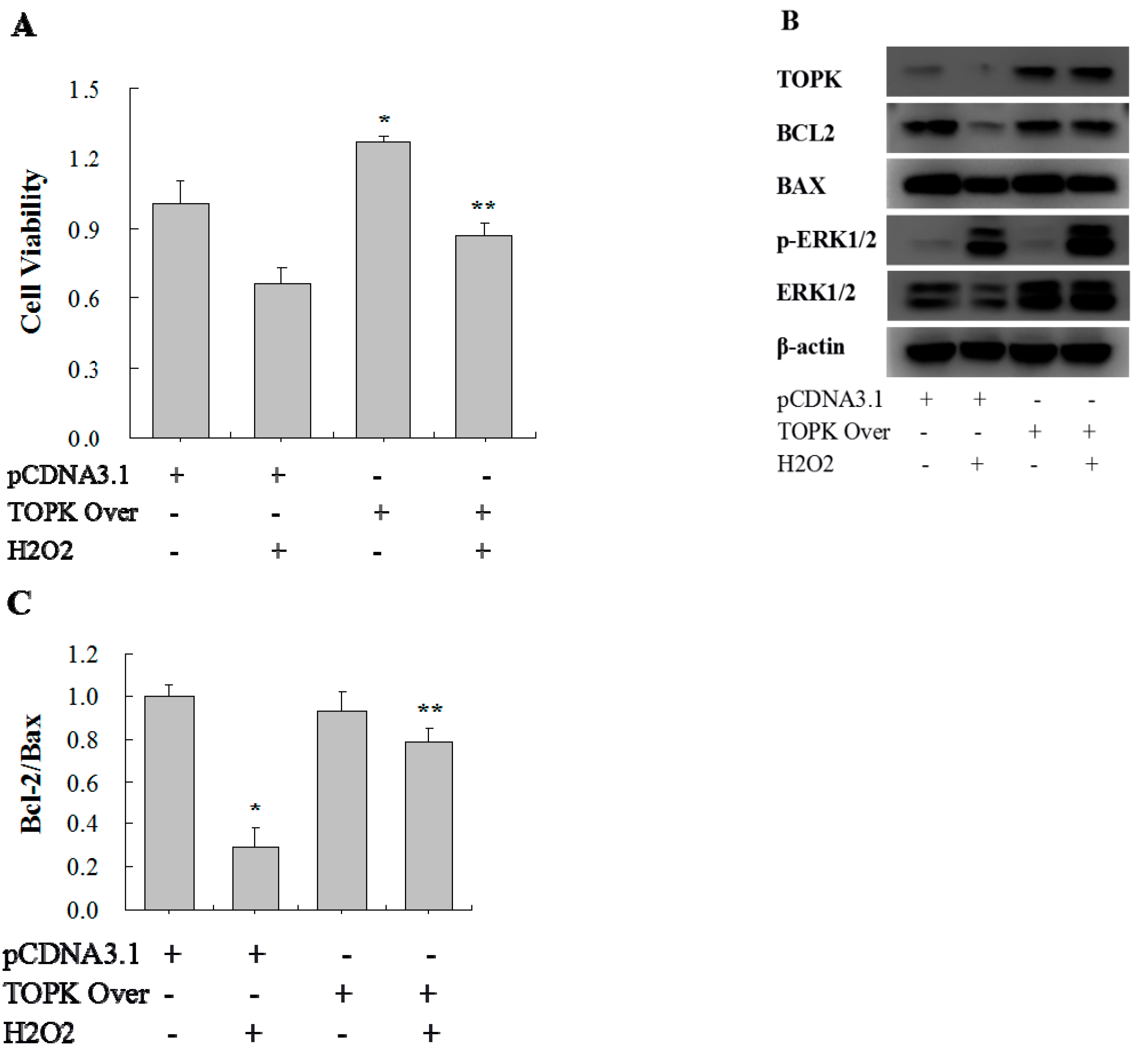
2.5. TOPK Overexpression Protected H9C2 Cardiomyocytes from Oxidative Stress Injury via the ERK Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. In Vivo Experimental Protocols
4.3. Cell Culture and in Vitro Experimental Protocols
4.4. Determination of Myocardial Infarct Size
4.5. In-Situ Detection of Apoptosis in Heart Tissue
4.6. HE and Immunohistochemical Staining
4.7. Quantification of Cell Viability by MTS Test
4.8. Immunofluorescence Analysis
4.9. Real-Time Quantitative PCR
4.10. Western Blot Analysis
4.11. Statistical Analysis
Author Contributions
Conflicts of Interest
Abbreviations
| AMI: acute myocardial infarction; |
| DMSO: dimethyl sulfoxide; |
| ECG: electrocardiogram; |
| ECL: enhanced chemiluminescence; |
| ERK: extracellular signal-regulated kinases; |
| H2O2: hydrogen peroxide; |
| HE: hematoxylin and eosin; |
| I/R: ischemia/reperfusion; |
| IPC: ischemic preconditioning; |
| LAD: left anterior descending coronary artery; |
| LV: left ventricular; |
| MAPK: mitogen-activated protein kinase; |
| MAPKK: mitogen-activated protein kinase kinase; |
| ROS: reactive oxygen species; |
| TOPK: T-LAK-cell-originated protein kinase; |
| TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling. |
References
- Hearse, D.J.; Bolli, R. Reperfusion induced injury: Manifestations, mechanisms, and clinical relevance. Cardiovasc. Res. 1992, 26, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Otani, H.; Tanaka, H.; Inoue, T.; Umemoto, M.; Omoto, K.; Tanaka, K.; Sato, T.; Osako, T.; Masuda, A.; Nonoyama, A.; et al. In vitro study on contribution of oxidative metabolism of isolated rabbit heart mitochondria to myocardial reperfusion injury. Circ. Res. 1984, 55, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Duchen, M.R.; Yellon, D.M. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc. Res. 2003, 60, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, R.; Komuro, I.; Yamazaki, T.; Zou, Y.; Kudoh, S.; Tanaka, M.; Shiojima, I.; Hiroi, Y.; Yazaki, Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J. Clin. Investig. 1997, 100, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Duan, W.; Jin, Z.; Yi, W.; Yan, J.; Zhang, S.; Wang, N.; Liang, Z.; Li, Y.; Chen, W.; et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J. Pineal Res. 2013, 55, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, V.; O’Connor, R. PBK/TOPK promotes tumour cell proliferation through p38 MAPK activity and regulation of the DNA damage response. Oncogene 2007, 26, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Gartenhaus, R.B.; Eichberg, D.; Liu, Z.; Fang, H.B.; Rapoport, A.P. PBK/TOPK interacts with the DBD domain of tumor suppressor p53 and modulates expression of transcriptional targets including p21. Oncogene 2010, 29, 5464–5474. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zykova, T.A.; Kang, B.S.; Wang, Z.; Ebeling, M.C.; Abe, Y.; Ma, W.Y.; Bode, A.M.; Dong, Z. Bidirectional signals transduced by TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells. Gastroenterology 2007, 133, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Zykova, T.A.; Zhu, F.; Lu, C.; Higgins, L.; Tatsumi, Y.; Abe, Y.; Bode, A.M.; Dong, Z. Lymphokine-activated killer T-cell-originated protein kinase phosphorylation of histone H2AX prevents arsenite-induced apoptosis in RPMI7951 melanoma cells. Clin. Cancer Res. 2006, 12, 6884–6893. [Google Scholar] [CrossRef] [PubMed]
- Zykova, T.A.; Zhu, F.; Vakorina, T.I.; Zhang, J.; Higgins, L.A.; Urusova, D.V.; Bode, A.M.; Dong, Z. T-LAK cell-originated protein kinase (TOPK) phosphorylation of Prx1 at Ser-32 prevents UVB-induced apoptosis in RPMI7951 melanoma cells through the regulation of Prx1 peroxidase activity. J. Biol. Chem. 2010, 285, 29138–29146. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Nishidate, T.; Nakamura, Y.; Katagiri, T. Critical roles of T-LAK cell-originated protein kinase in cytokinesis. Cancer Sci. 2010, 101, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Matsumoto, S.; Kito, K.; Ueda, N. Cloning and expression of a novel MAPKK-like protein kinase, lymphokine-activated killer T-cell-originated protein kinase, specifically expressed in the testis and activated lymphoid cells. J. Biol. Chem. 2000, 275, 21525–21531. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Yan, Q.; Fan, L.; Liu, Y.; Cui, J.; Wang, J.; Wang, L.; Wang, Y.; Wang, Z.; Guo, Y.; et al. PBK/TOPK in the differential diagnosis of cholangiocarcinoma from hepatocellular carcinoma and its involvement in prognosis of human cholangiocarcinoma. Hum. Pathol. 2010, 41, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Srivastava, A.K.; Dalela, D.; Rath, S.K.; Goel, M.M.; Bhatt, M.L. Expression of PDZ-binding kinase/T-LAK cell-originated protein kinase (PBK/TOPK) in human urinary bladder transitional cell carcinoma. Immunobiology 2014, 219, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Simons-Evelyn, M.; Bailey-Dell, K.; Toretsky, J.A.; Ross, D.D.; Fenton, R.; Kalvakolanu, D.; Rapoport, A.P. PBK/TOPK is a novel mitotic kinase which is upregulated in Burkitt’s lymphoma and other highly proliferative malignant cells. Blood Cells Mol. Dis. 2001, 27, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Li, Y.; Reddy, K.; Lee, M.H.; Kim, M.O.; Cho, Y.Y.; Lee, S.Y.; Kim, J.E.; Bode, A.M.; Dong, Z. Novel TOPK inhibitor HI-TOPK-032 effectively suppresses colon cancer growth. Cancer Res. 2012, 72, 3060–3068. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, S.; Branton, D.; Lue, R.A. Characterization of PDZ-binding kinase, a mitotic kinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5167–5172. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.A.; Martini, J.F.; Bui, S.K.; Wolfe, C.L. Ischemic preconditioning attenuates ischemia/reperfusion-induced activation of caspases and subsequent cleavage of poly(ADP-ribose) polymerase in rat hearts in vivo. Cardiovasc. Res. 1999, 44, 536–542. [Google Scholar] [CrossRef]
- Miller, D.L.; van Winkle, D.M. Ischemic preconditioning limits infarct size following regional ischemia-reperfusion in in situ mouse hearts. Cardiovasc. Res. 1999, 42, 680–684. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Otani, H. Ischemic preconditioning: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 207–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Li, R.; Wang, C.; Cao, L.; Wang, Y.; Yu, L. T-LAK cell-originated protein kinase is essential for the proliferation of hepatocellular carcinoma SMMC-7721 cells. Cell Biochem. Funct. 2013, 31, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, R.; Tao, Z.; Gao, L.; Yan, F.; Gao, Z.; Liu, X.; Ji, X.; Luo, Y. Ischemic postconditioning relieves cerebral ischemia and reperfusion injury through activating T-LAK cell-originated protein kinase/protein kinase B pathway in rats. Stroke 2014, 45, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Libuy, M.; Feliu, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Markers 2013, 35, 773–790. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Zhu, H.; Wei, X.; Zhang, C.; Jiang, L.; Liu, Y.; Luo, Q.; Xiao, X. Oxidative stress-mediated up-regulation of myocardial ischemic preconditioning up-regulated protein 1 gene expression in H9c2 cardiomyocytes is regulated by cyclic AMP-response element binding protein. Free Radic. Biol. Med. 2010, 49, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Vanden Hoek, T.; Becker, L.B.; Shao, Z.H.; Li, C.Q.; Schumacker, P.T. Preconditioning in cardiomyocytes protects by attenuating oxidant stress at reperfusion. Circ. Res. 2000, 86, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Tritto, I.; D’Andrea, D.; Eramo, N.; Scognamiglio, A.; de Simone, C.; Violante, A.; Esposito, A.; Chiariello, M.; Ambrosio, G. Oxygen radicals can induce preconditioning in rabbit hearts. Circ. Res. 1997, 80, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res. Cardiol. 2006, 101, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Farmakis, D.; Kremastinos, D.T. To prevent, protect and save the ischemic heart: Antioxidants revisited. Expert Opin. Ther. Targets 2009, 13, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fujiwara, H.; Yamasaki, K.; Sasayama, S. Superoxide dismutase and N-2-mercaptopropionyl glycine attenuate infarct size limitation effect of ischaemic preconditioning in the rabbit. Cardiovasc. Res. 1994, 28, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Schulz, R.; Gres, P.; Korth, H.G.; Heusch, G. Attenuation of ischemic preconditioning in pigs by scavenging of free oxyradicals with ascorbic acid. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H698–H703. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Tsovolas, K.; Aggeli, I.K.; Zoga, A.; Gaitanaki, C.; Paraskevaidis, I.A.; Markantonis, S.L.; Beis, I.; Kremastinos, D.T. Acute administration of vitamin E triggers preconditioning via K(ATP) channels and cyclic-GMP without inhibiting lipid peroxidation. Free Radic. Biol. Med. 2006, 41, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, X.M.; Iliodromitis, E.K.; Kremastinos, D.T.; Dost, T.; Cohen, M.V.; Downey, J.M. Redox signaling at reperfusion is required for protection from ischemic preconditioning but not from a direct PKC activator. Basic Res. Cardiol. 2008, 103, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, L.; Shi, C.; Hu, P.; Yan, W.; Wang, Z.; Duan, Q.; Lu, F.; Qin, L.; Lu, T.; et al. TOPK is highly expressed in circulating tumor cells, enabling metastasis of prostate cancer. Oncotarget 2015, 6, 12392–12404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, H.; Wang, R.; Tao, Z.; Yan, F.; Gao, L.; Liu, X.; Wang, N.; Min, L.; Jia, Y.; Zhao, Y.; et al. Activation of T-LAK-cell-originated protein kinase-mediated antioxidation protects against focal cerebral ischemia-reperfusion injury. FEBS J. 2014, 281, 4411–4420. [Google Scholar] [CrossRef] [PubMed]
- Shih, M.C.; Chen, J.Y.; Wu, Y.C.; Jan, Y.H.; Yang, B.M.; Lu, P.J.; Cheng, H.C.; Huang, M.S.; Yang, C.J.; Hsiao, M.; et al. TOPK/PBK promotes cell migration via modulation of the PI3K/PTEN/AKT pathway and is associated with poor prognosis in lung cancer. Oncogene 2012, 31, 2389–2400. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.M.; Zhu, F.; Cho, Y.Y.; Lee, K.W.; Kang, B.S.; Kim, H.G.; Zykova, T.; Bode, A.M.; Dong, Z. T-lymphokine-activated killer cell-originated protein kinase functions as a positive regulator of c-Jun-NH2-kinase 1 signaling and H-Ras-induced cell transformation. Cancer Res. 2007, 67, 5186–5194. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Tao, L.; Liu, H.; Christopher, T.A.; Lopez, B.L.; Ma, X.L. Role of ERK1/2 in the anti-apoptotic and cardioprotective effects of nitric oxide after myocardial ischemia and reperfusion. Apoptosis 2006, 11, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.A.; Padmanaban, D.; Ursell, P.C.; Sievers, R.E.; Wolfe, C.L. Ischemic preconditioning decreases apoptosis in rat hearts in vivo. Circulation 1997, 96, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, G.; Ye, N.; Dai, D.; Chen, Y.; Li, C.; Sun, Y. The Protective Role of the TOPK/PBK Pathway in Myocardial Ischemia/Reperfusion and H2O2-Induced Injury in H9C2 Cardiomyocytes. Int. J. Mol. Sci. 2016, 17, 267. https://doi.org/10.3390/ijms17030267
Sun G, Ye N, Dai D, Chen Y, Li C, Sun Y. The Protective Role of the TOPK/PBK Pathway in Myocardial Ischemia/Reperfusion and H2O2-Induced Injury in H9C2 Cardiomyocytes. International Journal of Molecular Sciences. 2016; 17(3):267. https://doi.org/10.3390/ijms17030267
Chicago/Turabian StyleSun, Guozhe, Ning Ye, Dongxue Dai, Yintao Chen, Chao Li, and Yingxian Sun. 2016. "The Protective Role of the TOPK/PBK Pathway in Myocardial Ischemia/Reperfusion and H2O2-Induced Injury in H9C2 Cardiomyocytes" International Journal of Molecular Sciences 17, no. 3: 267. https://doi.org/10.3390/ijms17030267
APA StyleSun, G., Ye, N., Dai, D., Chen, Y., Li, C., & Sun, Y. (2016). The Protective Role of the TOPK/PBK Pathway in Myocardial Ischemia/Reperfusion and H2O2-Induced Injury in H9C2 Cardiomyocytes. International Journal of Molecular Sciences, 17(3), 267. https://doi.org/10.3390/ijms17030267