
Modulators of Macrophage Polarization Influence Healing of the Infarcted Myocardium

 ,
,
Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Macrophage Polarization
3. Modulators of Macrophage Polarization after AMI
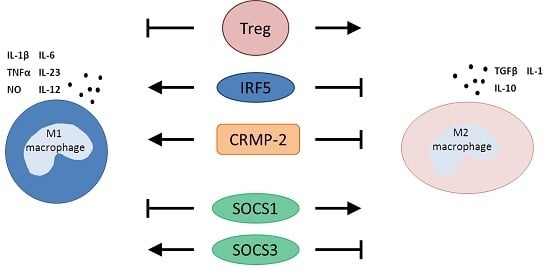
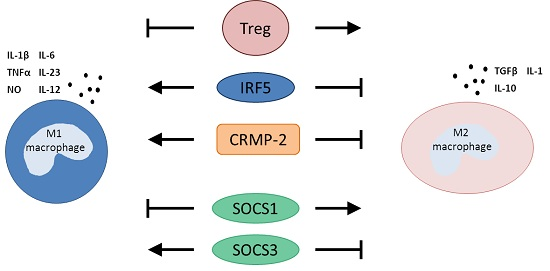
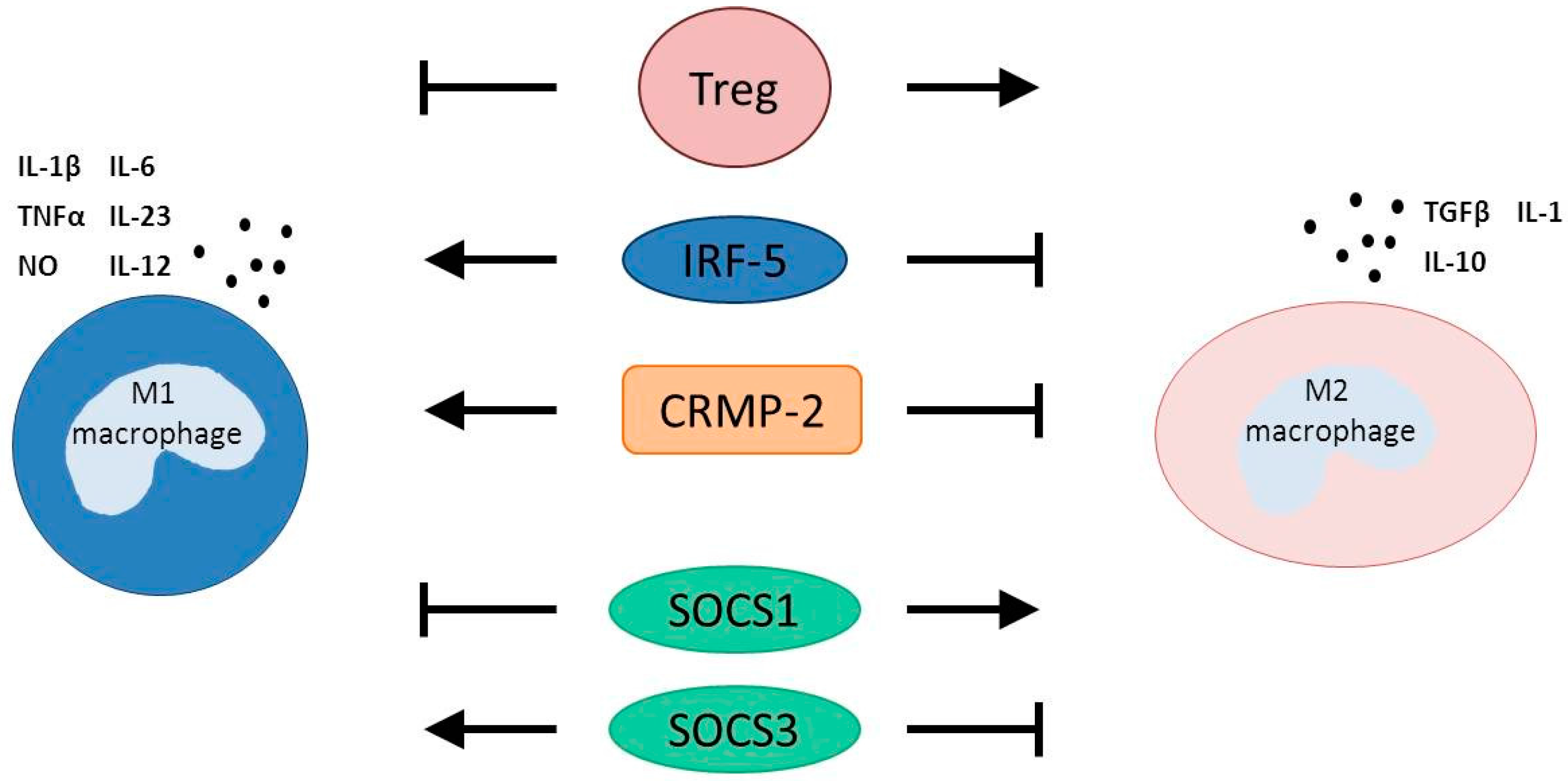
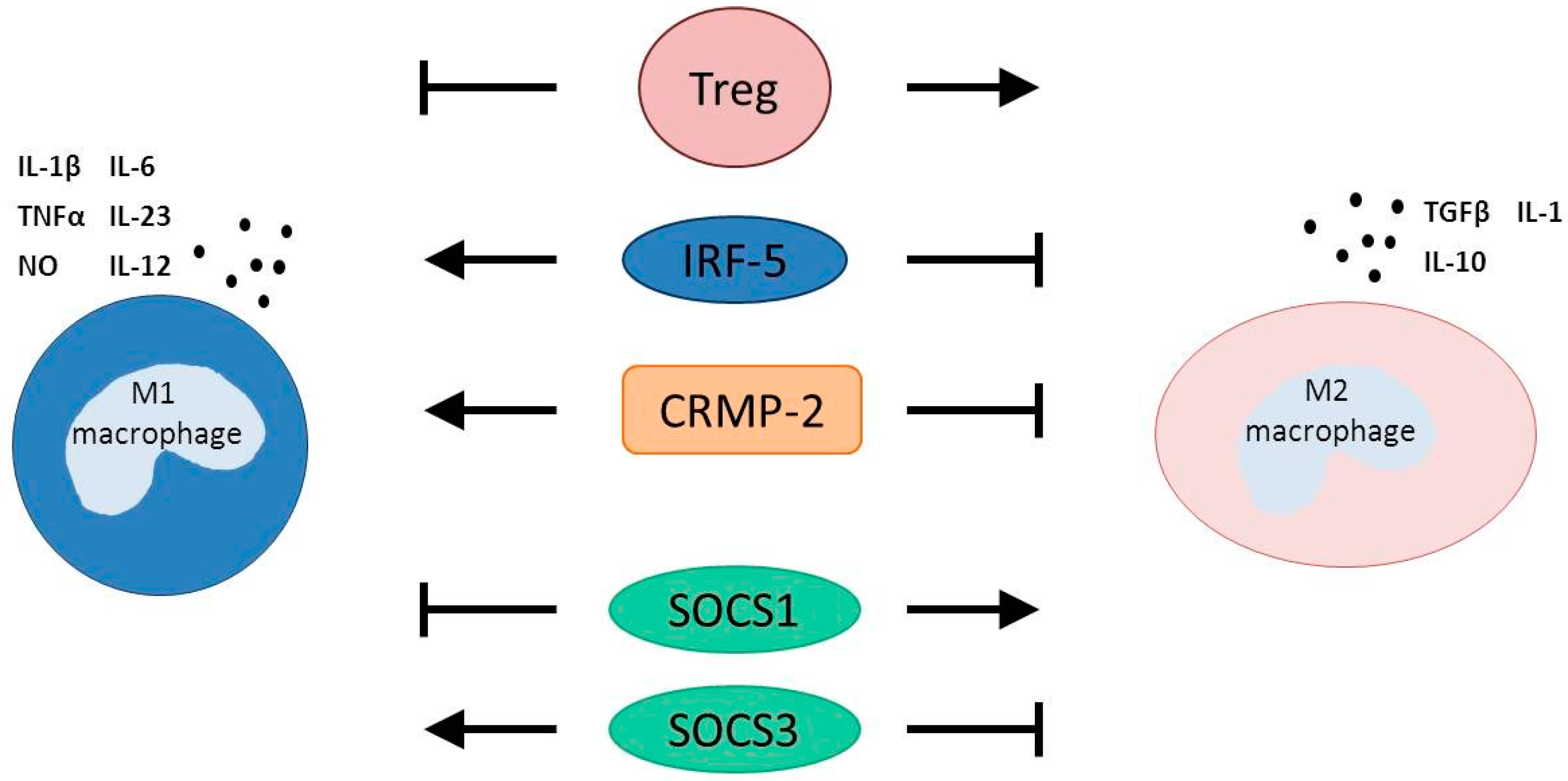
3.1. T Regulatory Cells
3.2. Interferon Regulatory Factor 5
3.3. Collapsin Response Mediator Protein-2
3.4. Suppressors of Cytokine Signaling
4. Targeting Monocytes in Clinical Studies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dutta, P.; Nahrendorf, M. Monocytes in myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Van der Laan, A.M.; Ter Horst, E.N.; Delewi, R.; Begieneman, M.P.; Krijnen, P.A.; Hirsch, A.; Lavaei, M.; Nahrendorf, M.; Horrevoets, A.J.; Niessen, H.W.; et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur. Heart J. 2014, 35, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Inflammation in cardiac injury, repair and regeneration. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic sirna silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Harel-Adar, T.; Ben Mordechai, T.; Amsalem, Y.; Feinberg, M.S.; Leor, J.; Cohen, S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl. Acad. Sci. USA 2011, 108, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Courties, G.; Heidt, T.; Sebas, M.; Iwamoto, Y.; Jeon, D.; Truelove, J.; Tricot, B.; Wojtkiewicz, G.; Dutta, P.; Sager, H.B.; et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J. Am. Coll. Cardiol. 2014, 63, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.S.; Zhao, G.L.; Liu, Q.; Jiang, S.C.; Wang, Y.; Zhang, D.M. Silencing collapsin response mediator protein-2 reprograms macrophage phenotype and improves infarct healing in experimental myocardial infarction model. J. Inflamm. (Lond.) 2015, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Somasuntharam, I.; Boopathy, A.V.; Khan, R.S.; Martinez, M.D.; Brown, M.E.; Murthy, N.; Davis, M.E. Delivery of NOX2-NADPH oxidase sirna with polyketal nanoparticles for improving cardiac function following myocardial infarction. Biomaterials 2013, 34, 7790–7798. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.H.; Bebien, M.; Didierlaurent, A.; Nebauer, R.; Hussell, T.; Broide, D.; Karin, M.; Lawrence, T. An antiinflammatory role for IKKβ through the inhibition of “classical” macrophage activation. J. Exp. Med. 2008, 205, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Soler, C.; Felipe, A.; Garcia-Manteiga, J.; Serra, M.; Guillen-Gomez, E.; Casado, F.J.; MacLeod, C.; Modolell, M.; Pastor-Anglada, M.; Celada, A. Interferon-γ regulates nucleoside transport systems in macrophages through signal transduction and activator of transduction factor 1 (STAT1)-dependent and -independent signalling pathways. Biochem. J. 2003, 375, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhu, J.; Smith, S.; Foldi, J.; Zhao, B.; Chung, A.Y.; Outtz, H.; Kitajewski, J.; Shi, C.; Weber, S.; et al. Notch-RBP-j signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol. 2012, 13, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.T.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Kamper, P.; Bendix, K.; Hamilton-Dutoit, S.; Honore, B.; Nyengaard, J.R.; d’Amore, F. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica 2011, 96, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.H.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage polarisation: An immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.J.; Ni Choileain, N.; Zang, Y.; Mannick, J.A.; Lederer, J.A. CD4+CD25+ regulatory T cells control innate immune reactivity after injury. J. Immunol. 2005, 174, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Marson, A.; Kretschmer, K.; Frampton, G.M.; Jacobsen, E.S.; Polansky, J.K.; MacIsaac, K.D.; Levine, S.S.; Fraenkel, E.; von Boehmer, H.; Young, R.A. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 2007, 445, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Aikawa, E.; Figueiredo, J.L.; Stangenberg, L.; van den Borne, S.W.; Blankesteijn, W.M.; Sosnovik, D.E.; Jaffer, F.A.; Tung, C.H.; Weissleder, R. Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: Implications for fxiii therapy and antithrombin use in myocardial infarction. Eur. Heart J. 2008, 29, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and Th1-Th17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, D.; Tamura, T.; Yanai, H.; Taniguchi, T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 2010, 59, 489–510. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.E.; Whyte, C.S.; Gordon, P.; Barker, R.N.; Rees, A.J.; Wilson, H.M. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology 2014, 141, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Stewart, K.N.; Bishop, E.; Marek, C.J.; Kluth, D.C.; Rees, A.J.; Wilson, H.M. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J. Immunol. 2008, 180, 6270–6278. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Holdbrooks, A.T.; Liu, Y.; Reynolds, S.L.; Yanagisawa, L.L.; Benveniste, E.N. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 2012, 189, 3439–3448. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.S.; Bishop, E.T.; Ruckerl, D.; Gaspar-Pereira, S.; Barker, R.N.; Allen, J.E.; Rees, A.J.; Wilson, H.M. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J. Leukoc. Biol. 2011, 90, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Sachithanandan, N.; Graham, K.L.; Galic, S.; Honeyman, J.E.; Fynch, S.L.; Hewitt, K.A.; Steinberg, G.R.; Kay, T.W. Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes 2011, 60, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Yasukawa, H.; Hoshijima, M.; Sasaki, K.; Futamata, N.; Fukui, D.; Mawatari, K.; Nagata, T.; Kyogoku, S.; Ohshima, H.; et al. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J. Am. Coll. Cardiol. 2012, 59, 838–852. [Google Scholar] [CrossRef] [PubMed]
- Tsujioka, H.; Imanishi, T.; Ikejima, H.; Kuroi, A.; Takarada, S.; Tanimoto, T.; Kitabata, H.; Okochi, K.; Arita, Y.; Ishibashi, K.; et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Shantsila, E.; Wrigley, B.; Tapp, L.; Apostolakis, S.; Montoro-Garcia, S.; Drayson, M.T.; Lip, G.Y. Immunophenotypic characterization of human monocyte subsets: Possible implications for cardiovascular disease pathophysiology. J. Thromb. Haemost. 2011, 9, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, M.D.; Keliher, E.J.; Heidt, T.; Leuschner, F.; Truelove, J.; Sena, B.F.; Gorbatov, R.; Iwamoto, Y.; Dutta, P.; Wojtkiewicz, G.; et al. Monocyte-directed rnai targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 2013, 127, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Xie, Z.; Tang, Y.; Bai, L.; Yao, Y.; Fu, C.; Ma, G. Photoluminescent mesoporous silicon nanoparticles with SICCR2 improve the effects of mesenchymal stromal cell transplantation after acute myocardial infarction. Theranostics 2015, 5, 1068–1082. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ter Horst, E.N.; Hakimzadeh, N.; Van der Laan, A.M.; Krijnen, P.A.J.; Niessen, H.W.M.; Piek, J.J. Modulators of Macrophage Polarization Influence Healing of the Infarcted Myocardium. Int. J. Mol. Sci. 2015, 16, 29583-29591. https://doi.org/10.3390/ijms161226187
Ter Horst EN, Hakimzadeh N, Van der Laan AM, Krijnen PAJ, Niessen HWM, Piek JJ. Modulators of Macrophage Polarization Influence Healing of the Infarcted Myocardium. International Journal of Molecular Sciences. 2015; 16(12):29583-29591. https://doi.org/10.3390/ijms161226187
Chicago/Turabian StyleTer Horst, Ellis N., Nazanin Hakimzadeh, Anja M. Van der Laan, Paul A. J. Krijnen, Hans W. M. Niessen, and Jan J. Piek. 2015. "Modulators of Macrophage Polarization Influence Healing of the Infarcted Myocardium" International Journal of Molecular Sciences 16, no. 12: 29583-29591. https://doi.org/10.3390/ijms161226187