
Active Site Mutations as a Suitable Tool Contributing to Explain a Mechanism of Aristolochic Acid I Nitroreduction by Cytochromes P450 1A1, 1A2 and 1B1

 , and
, and
Abstract
:
1. Introduction
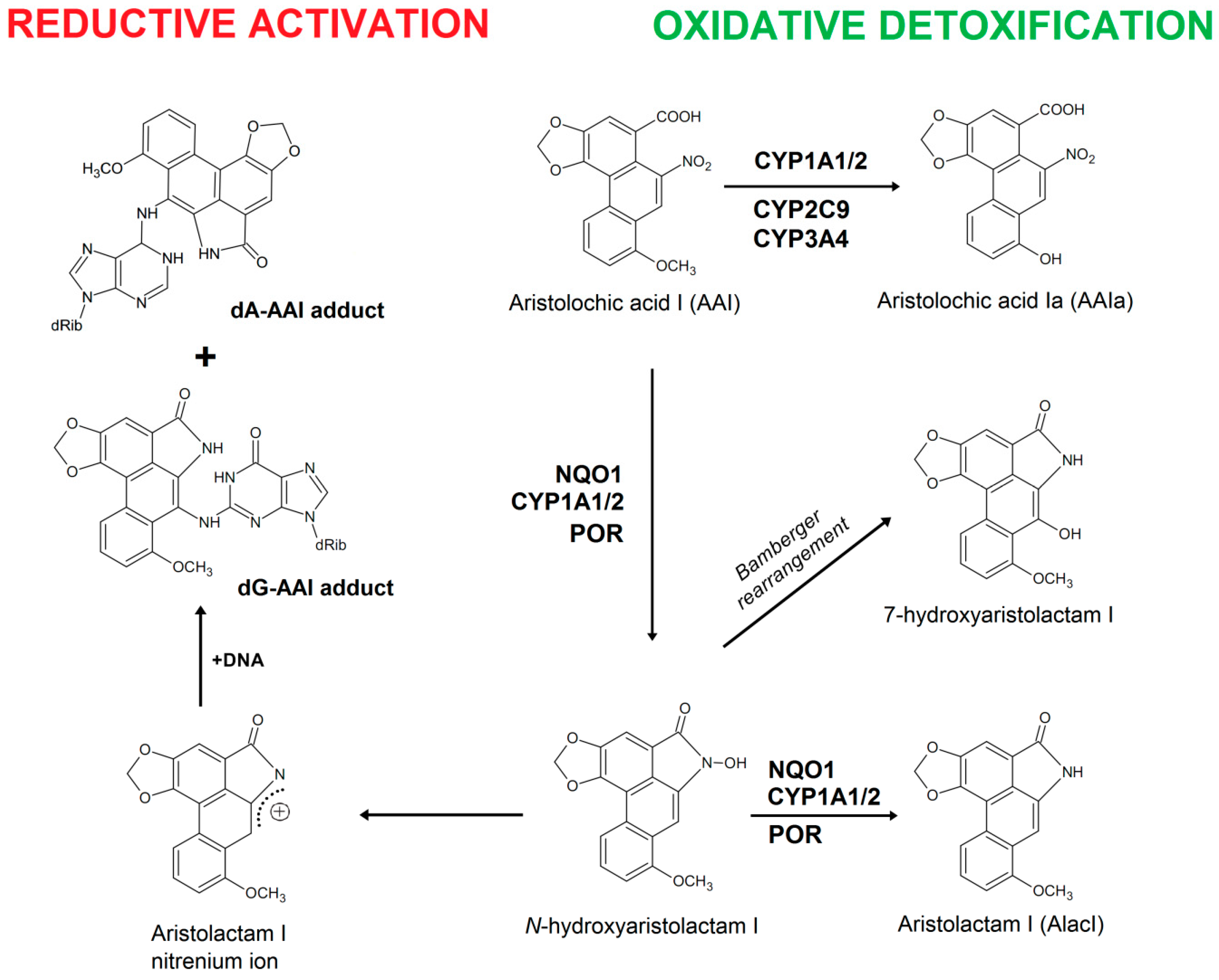
2. Results and Discussion
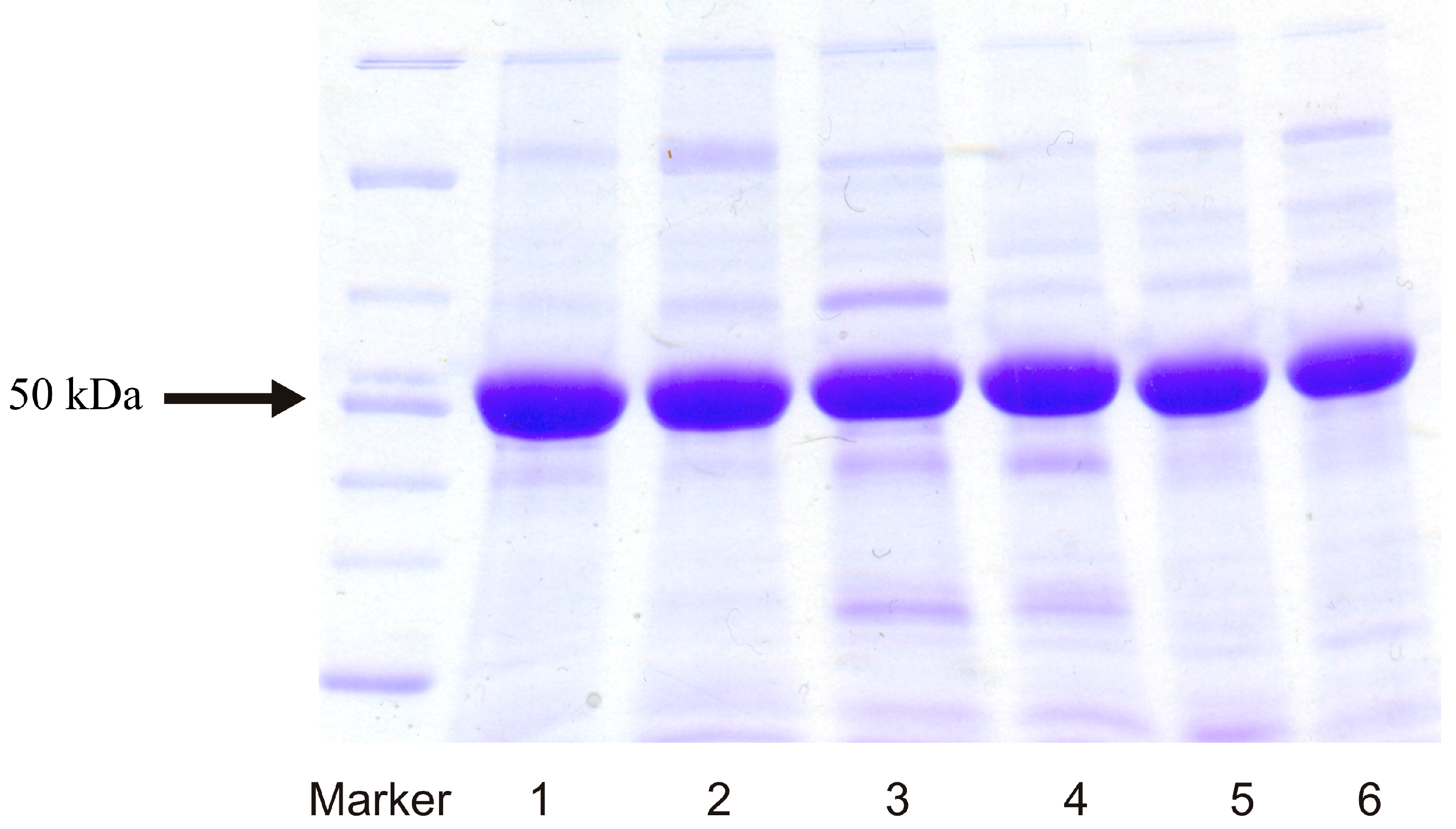
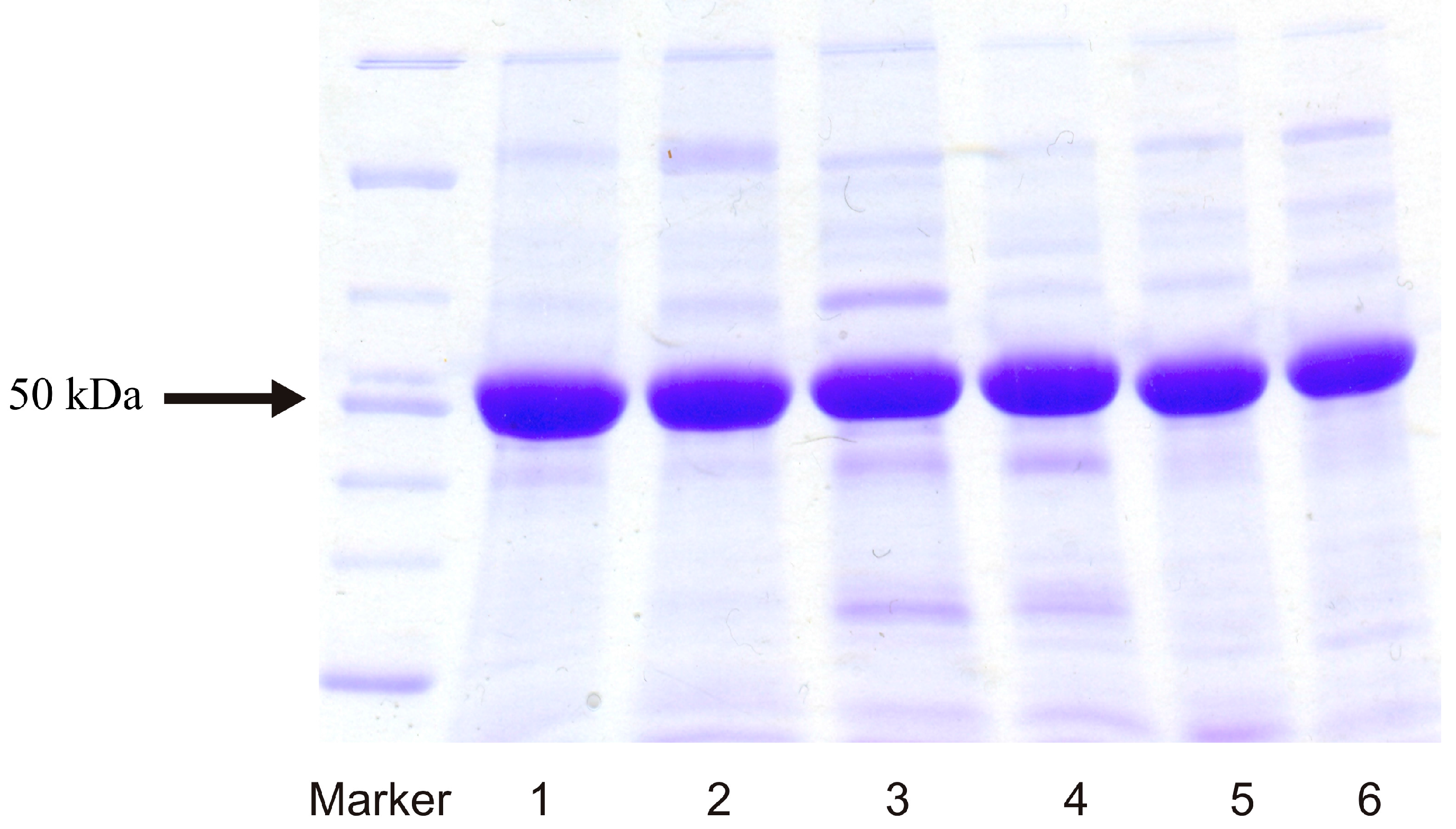
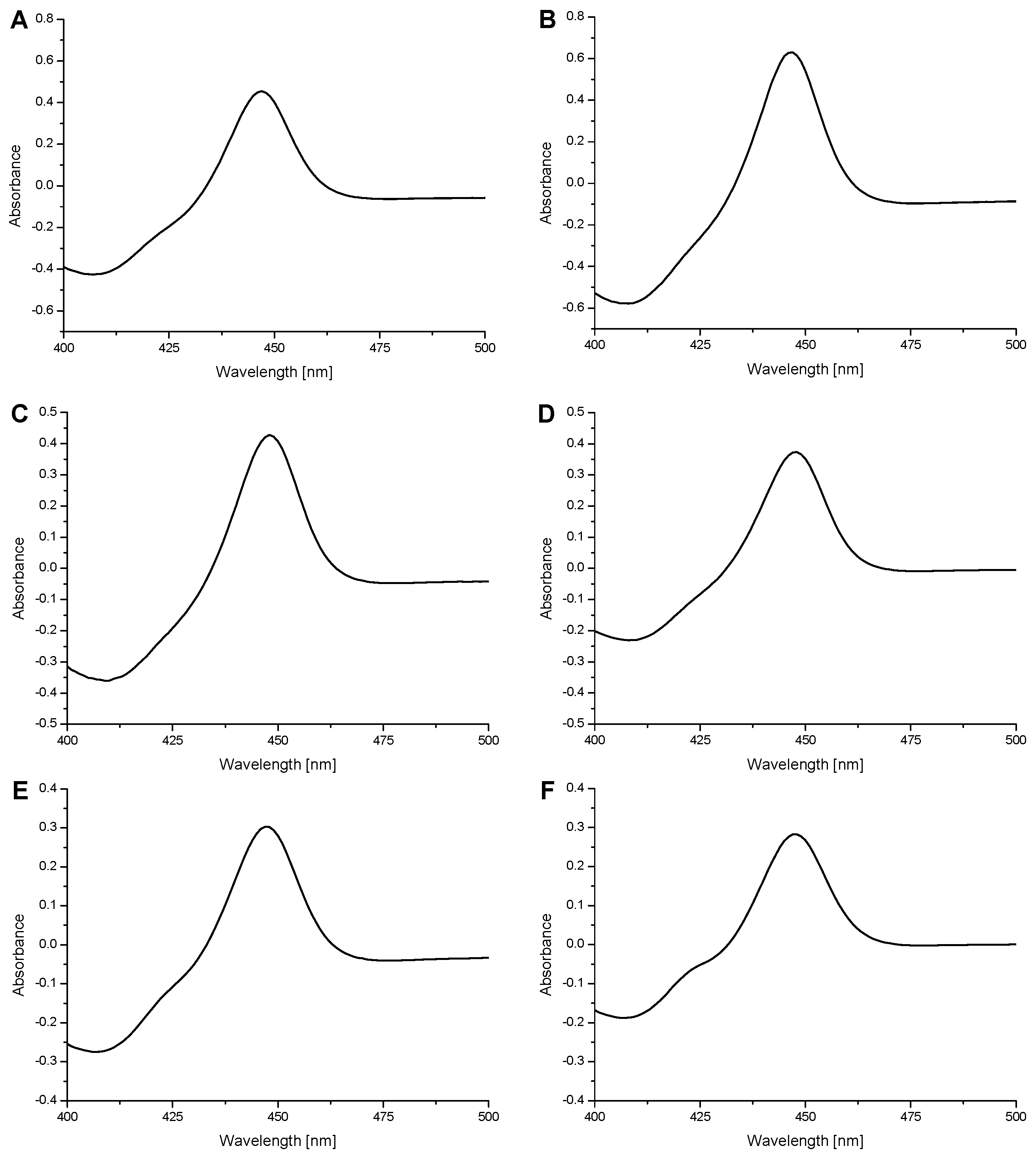
2.1. Expression of Human Wild-Type and Mutant CYP1A1, 1A2, and 1B1 Enzymes in E. coli and Their Purification

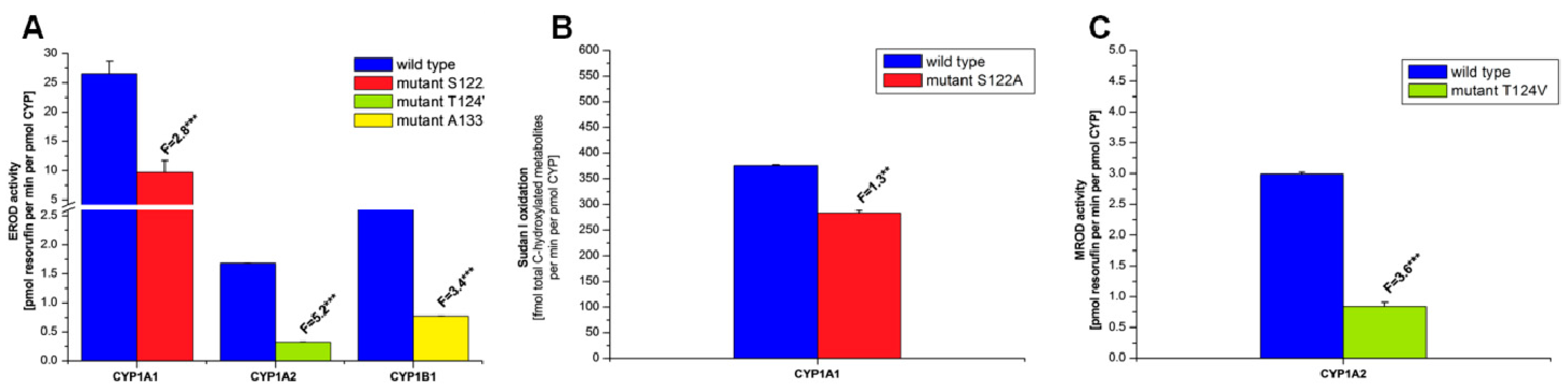
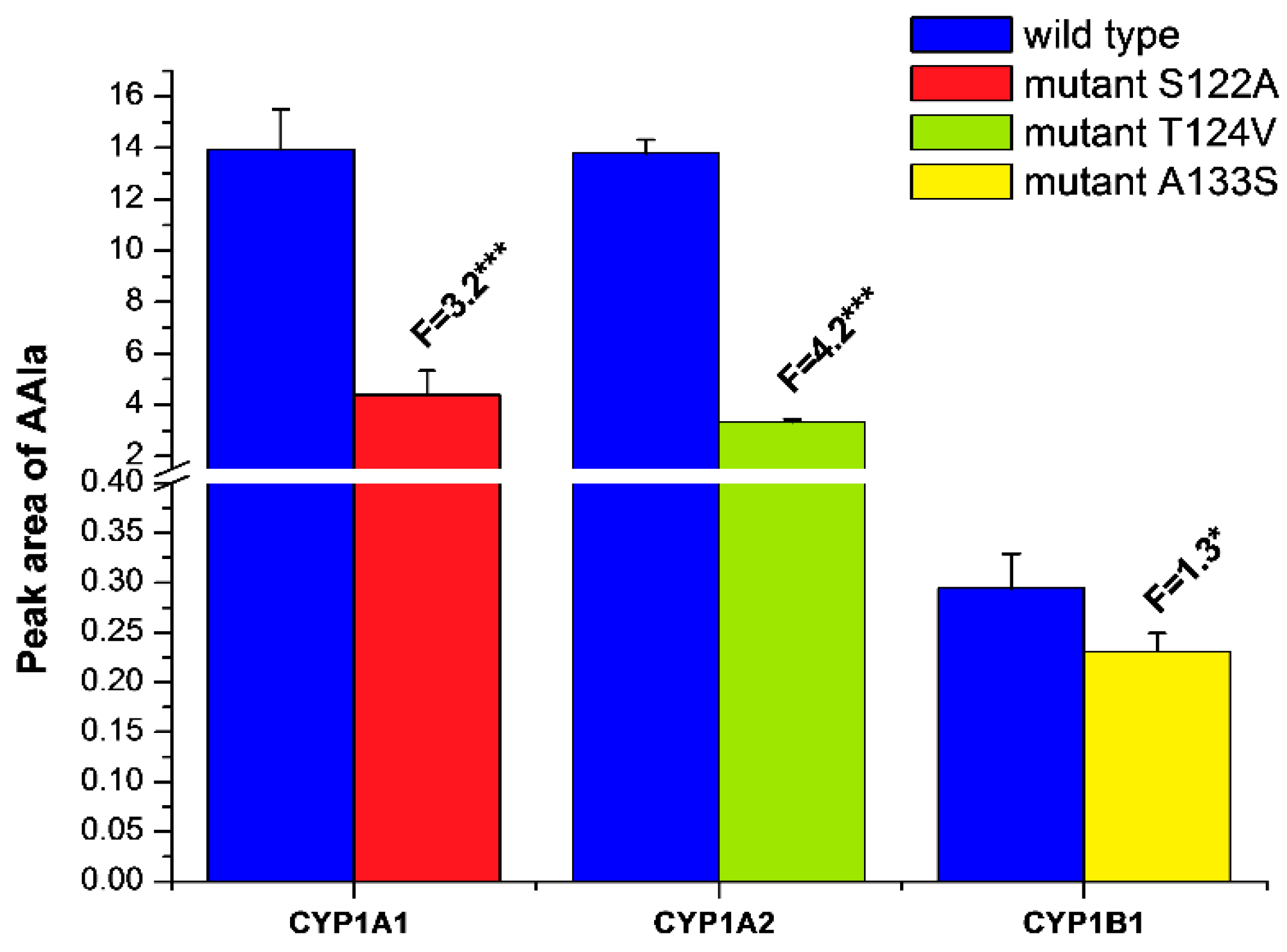
2.2. Examination of the Catalytic Activity of Human CYP1A1, 1A2 and 1B1 and Their Mutants to Oxidize Marker Substrates and AAI
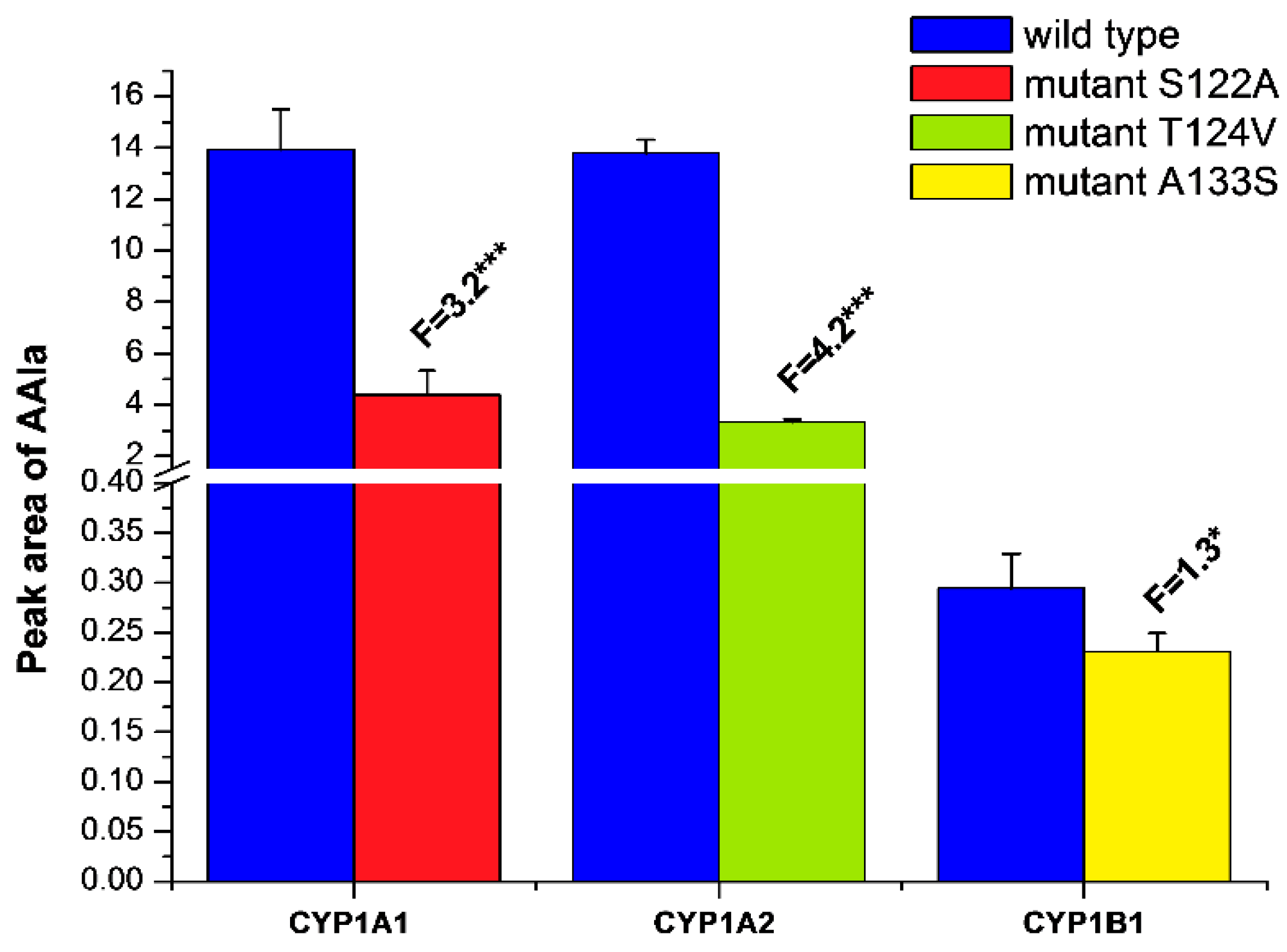
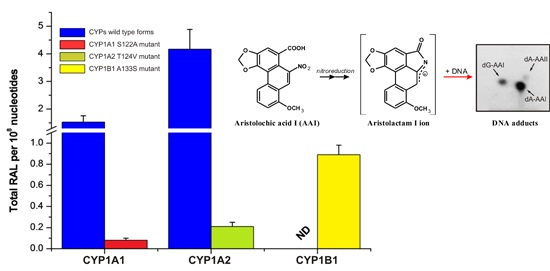
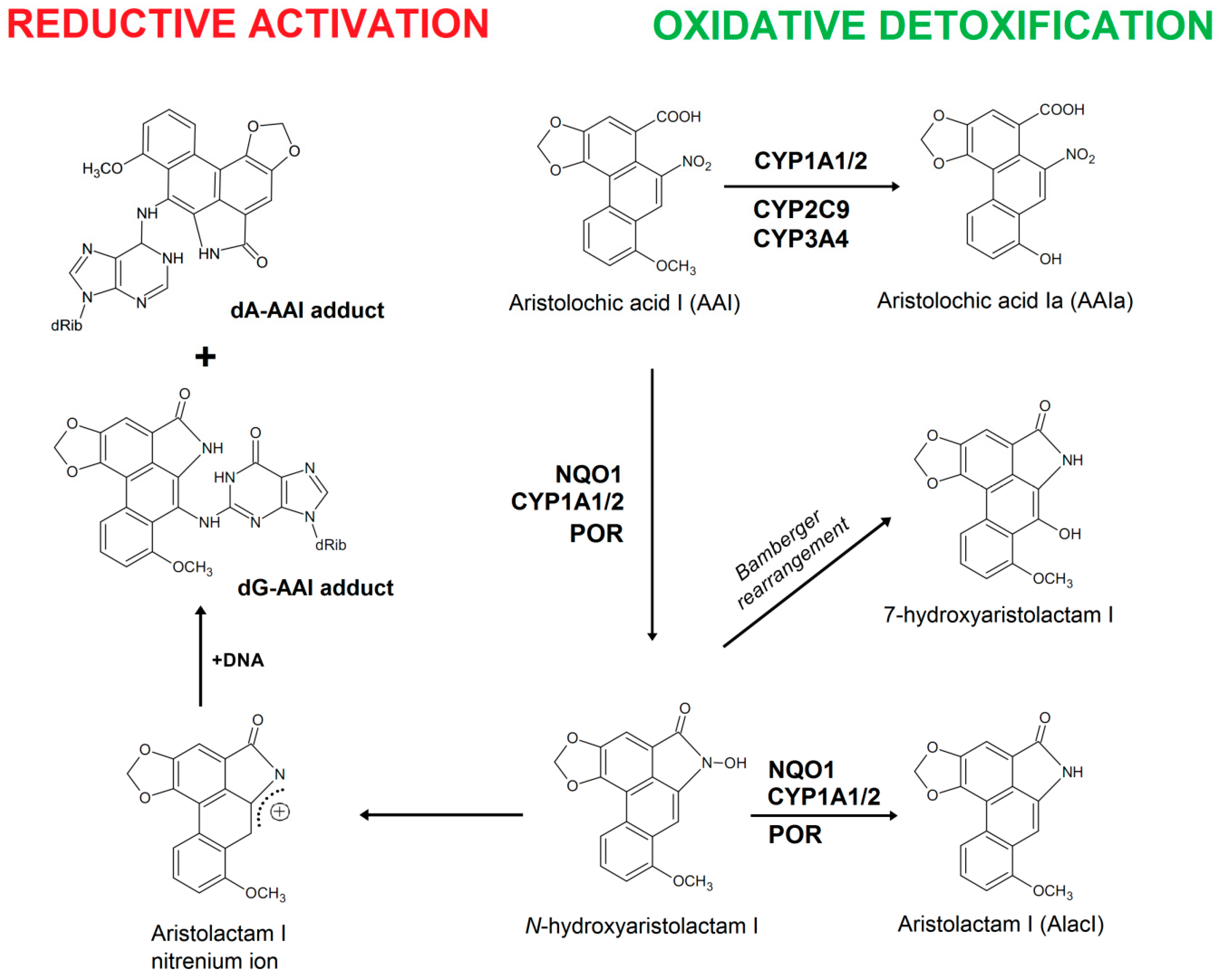
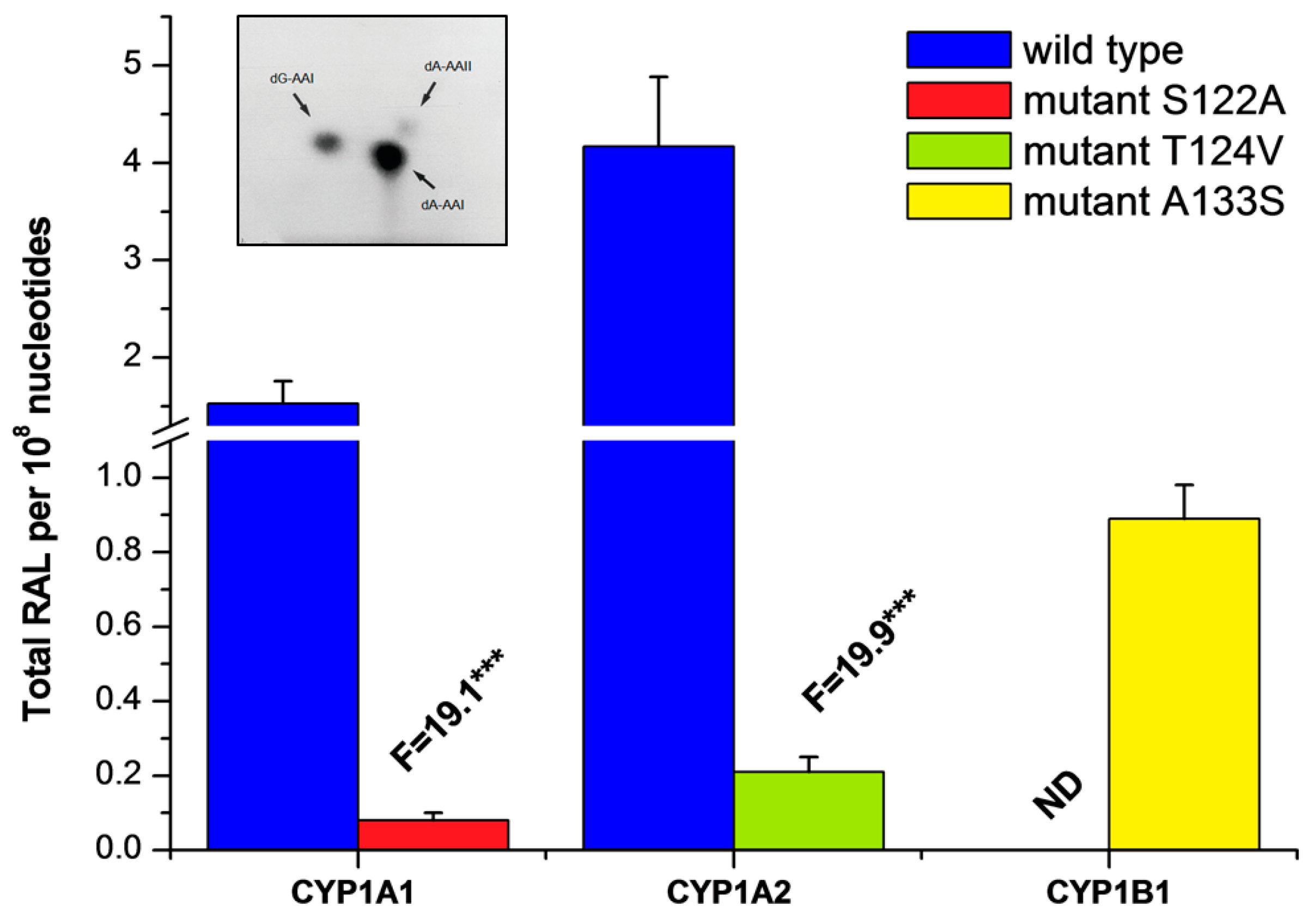
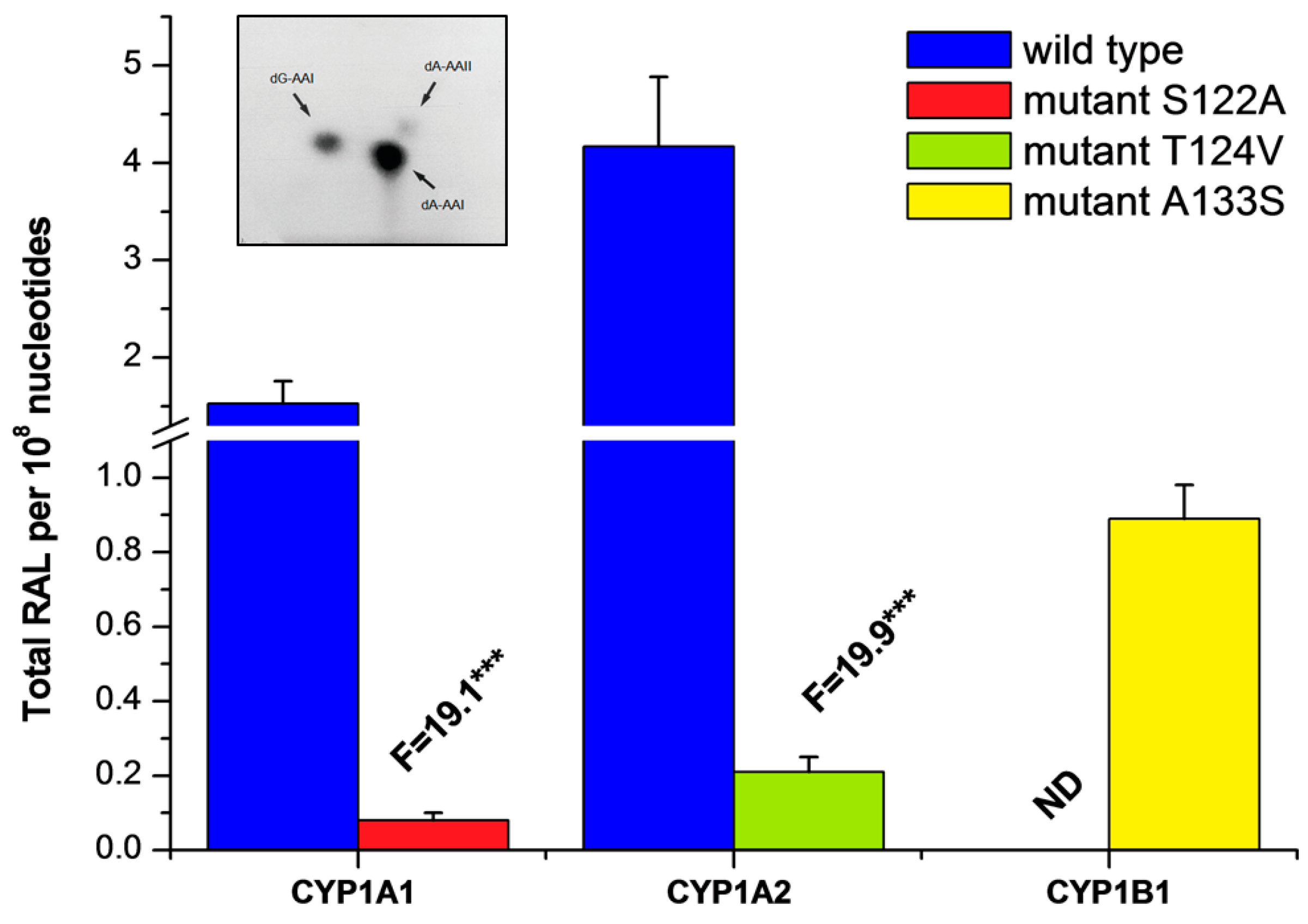
2.3. Examination of the Catalytic Activity of Human CYP1A1, 1A2 and 1B1 and Their Mutants to Reduce AAI to Species Forming AAI-DNA Adducts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymatic System | Levels of DNA Adducts in RAL a (Mean ± SD/108 Nucleotides) | |||
|---|---|---|---|---|
| dG-AAI | dA-AAI | dA-AAII | Total | |
| POR | 0.22 ± 0.05 | 0.61 ± 0.08 | 0.12 ± 0.03 | 0.95 ± 0.14 |
| CYP1A1 + POR | 0.43 ± 0.08 | 1.89 ± 0.34 | 0.17 ± 0.04 | 2.49 ± 0.38 *** |
| CYP1A1-S122A mutant + POR | 0.22 ± 0.06 | 0.71 ± 0.10 | 0.11 ± 0.05 | 1.04 ± 0.21 |
| CYP1A2 + POR | 0.89 ± 0.10 | 3.85 ± 0.48 | 0.38 ± 0.05 | 5.12 ± 0.87 *** |
| CYP1A2-T124V mutant + POR | 0.24 ± 0.05 | 0.82 ± 0.10 | 0.10 ± 0.03 | 1.16 ± 0.23 |
| CYP1B1 + POR | 0.22 ± 0.05 | 0.61 ± 0.10 | 0.10 ± 0.05 | 0.93 ± 0.18 |
| CYP1B1-A133S mutant + POR | 0.40 ± 0.08 | 1.22 ± 0.14 | 0.21 ± 0.04 | 1.83 ± 0.19 *** |
3. Experimental Section
3.1. Vectors
| CYP1A1 | WT | MLFPISMSATEFLLASVIFCLVFWVM |
| Modified | MAFPISMSATEFLLASVIFCLVFWVM | |
| CYP1A2 | WT | MALSQSVPFSATELLLASAIFCLVFW |
| Modified | MA------------LLAVFLFCLVFW | |
| CYP1B1 | WT | MGTSLSPNDPWPLN |
| Modified | M---LSPNDPWPLN |
3.2. Construction of CYP Mutants
| 1A1 | Correction of I171L |
| Forward GAGGCTGAGGTCCTGATAAGCACGTTGCAGG | |
| Reverse CCTGCAACGTGCTTATCAGGACCTCAGCCTC | |
| 1a1 | Histidine tag modification |
| Forward GAATTCATATGGCTTTTCCAATTTCAATG | |
| Reverse TATCTAAGCTTCATTAATGATGATGATGATGATGAGAGCGCAGCTGCATTTG | |
| 1A1 | S122A mutation |
| Forward GTAATGGTCAGAGCATGGCCTTCAGCCCAGACTC | |
| Reverse GAGTCTGGGCTGAAGGCCATGCTCTGACCATTAC | |
| 1A2 | Histidine tag modification |
| Forward GAATTCCATATGGCTCTGTTATTAGCAG | |
| Reverse TATCTAAGCTTCATTAATGATGATGATGATGATGATTGATGGAGAAGCGCAG | |
| 1A2 | T124V mutation |
| Forward CTGGCCAGAGCTTGGTCTTCAGCACAGACTCTG | |
| Reverse CAGAGTCTGTGCTGAAGACCAAGCTCTGGCCAG | |
| 1B1 | N-terminal and histidine tag modification |
| Forward GACGAATTCATATGCTTTCTCCAAATGATCCATGGCCGCTAAACCCG | |
| Reverse GCCAAGGAAACTTGCCAACATCATCATCATCATCATTAATGAAGCTTAGATA | |
| 1B1 | Correction of L432V |
| Forward CTGTGAATCATGACCCACTGAAGTGGCCTAACCCG | |
| Reverse CGGGTTAGGCCACTTCAGTGGGTCATGATTCACAG | |
| 1B1 | A133S mutation |
| Forward CGGCGGCCGCAGCATGTCTTTCGGCCACTACTC | |
| Reverse GAGTAGTGGCCGAAAGACATGCTGCGGCCGCCG |
3.3. Expression of Human CYPs and Their Purification
3.4. Rat POR Expression and Purification
3.5. Determination of CYP and Protein Contents
3.6. Measurement of CYP1A1-, 1A2- and 1B1-Mediated EROD Activities
3.7. Measurement of CYP1A1-Mediated Oxidation of Sudan I
3.8. Measurement of CYP1A2-Mediated MROD Activity
3.9. Incubations to Study AAI Oxidation to AAIa by Human Recombinant CYPs
3.10. Determination of AAI-DNA Adduct Formation by 32P-Postlabelling
3.11. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arlt, V.M.; Stiborova, M.; Schmeiser, H.H. Aristolochic acid as a probable human cancer hazard in herbal remedies: A review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Stiborová, M.; Arlt, V.M. Chemical and molecular basis of the carcinogenicity of Aristolochia plants. Curr. Opin. Drug Discov. Dev. 2009, 12, 141–148. [Google Scholar]
- Gökmen, M.R.; Cosyns, J.P.; Arlt, V.M.; Stiborová, M.; Phillips, D.H.; Schmeiser, H.H.; Simmonds, M.S.J.; Look, H.T.; Vanherweghem, J.L.; Nortier, J.L.; et al. The epidemiology, diagnosis and management of Aristolochic Acid Nephropathy: A narrative review. Ann. Intern. Med. 2013, 158, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Vanherweghem, J.L.; Tielemans, C.; Abramowicz, D.; Depierreux, M.; Vanhaelen-Fastre, R.; Vanhaelen, M.; Dratwa, M.; Richard, C.; Vandervelde, D.; Verbeelen, D.; et al. Rapidly progressive interstitial renal fibrosis in young women: Association with slimming regimen including Chinese herbs. Lancet 1993, 341, 387–391. [Google Scholar] [CrossRef]
- Nortier, J.L.; Martinez, M.C.; Schmeiser, H.H.; Arlt, V.M.; Bieler, C.A.; Petein, M.; Depierreux, M.F.; de Pauw, L.; Abramowicz, D.; Vereerstraeten, P.; et al. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). N. Engl. J. Med. 2000, 342, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Rosenquist, T.A.; Sidorenko, V.; Iden, C.R.; Chen, C.H.; Pu, Y.S.; Bonala, R.; Johnson, F.; Dickman, K.G.; Grollman, A.P.; et al. Biomonitoring of aristolactam-DNA adducts in human tissues using ultra-performance liquid chromatography/ion-trap mass spectrometry. Chem. Res. Toxicol. 2012, 25, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). A review of human CARCINOGENS: Pharmaceuticals. In Environ. Health Criteria Monographs; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Arlt, V.M.; Stiborova, M.; vom Brocke, J.; Simoes, M.L.; Lord, G.M.; Nortier, J.L.; Hollstein, M.; Phillips, D.H.; Schmeiser, H.H. Aristolochic acid mutagenesis: Molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis 2007, 28, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Shibutani, S.; Moriya, M.; Miller, F.; Wu, L.; Moll, U.; Suzuki, N.; Fernandes, A.; Rosenquist, T.; Medverec, Z.; et al. Aristolochic acid and the etiology of endemic Balkan nephropathy. Proc. Natl. Acad. Sci. USA 2007, 104, 12129–12134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmeiser, H.H.; Kucab, J.E.; Arlt, V.M.; Phillips, D.H.; Hollstein, M.; Gluhovschi, G.; Gluhovschi, C.; Modilca, M.; Daminescu, L.; Petrica, L.; et al. Evidence of exposure to aristolochic acid in patients with urothelial cancer from a Balkan endemic nephropathy region of Romania. Environ. Mol. Mutagen. 2012, 53, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenko, D.V.; Wu, L.; Turesky, R.J.; Wu, X.R.; et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Metabolic activation of carcinogenic aristolochic acid, a risk factor for Balkan endemic nephropathy. Mutat. Res. 2008, 658, 55–67. [Google Scholar]
- Stiborová, M.; Frei, E.; Schmeiser, H.H. Biotransformation enzymes in development of renal injury and urothelial cancer caused by aristolochic acid. Kidney Int. 2008, 73, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Martínek, V.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Enzymes metabolizing aristolochic acid and their contribution to the development of Aristolochic acid nephropathy and urothelial cancer. Curr. Drug Metab. 2013, 14, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Knock-out and humanized mice as suitable tools to identify enzymes metabolizing the human carcinogen aristolochic acid. Xenobiotica 2014, 44, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Bieler, C.A.; Wiessler, M.; van Ypersele de Strihou, C.; Cosyns, J.P. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res. 1996, 56, 2025–2028. [Google Scholar] [PubMed]
- Stiborová, M.; Frei, E.; Breuer, A.; Bieler, C.A.; Schmeiser, H.H. Aristolactam I a metabolite of aristolochic acid I upon activation forms an adduct found in DNA of patients with Chinese herbs nephropathy. Exp. Toxic. Pathol. 1999, 51, 421–427. [Google Scholar] [CrossRef]
- Arlt, V.M.; Ferluga, D.; Stiborova, M.; Pfohl-Leszkowicz, A.; Vukelic, M.; Ceovic, S.; Schmeiser, H.H.; Cosyns, J.P. Is aristolochic acid a risk factor for Balkan endemic nephropathy-associated urothelial cancer? Int. J. Cancer 2002, 101, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Nortier, J.L.; Singh, R.; Gamboa da Costa, G.; Sennesael, J.; Cassuto-Viguier, E.; Ambrosetti, D.; Rorive, S.; Pozdzik, A.; Phillips, D.H.; et al. Exceptionally long-term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int. J. Cancer 2014, 135, 562–567. [Google Scholar] [CrossRef]
- Lord, G.M.; Hollstein, M.; Arlt, V.M.; Roufosse, C.; Pusey, C.D.; Cook, T.; Schmeiser, H.H. DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J. Kidney Dis. 2014, 43, e18.1–e18.7. [Google Scholar] [CrossRef]
- Nedelko, T.; Arlt, V.M.; Phillips, D.H.; Hollstein, M. TP53 mutation signature supports involvement of aristolochic acid in the aetiology of endemic nephropathy-associated tumours. Int. J. Cancer 2009, 124, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J.E.; Phillips, D.H.; Arlt, V.M. Linking environmental carcinogen exposure to TP53 mutations in human tumours using the human TP53 knock-in (Hupki) mouse model. FEBS J. 2010, 277, 2567–2583. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.L.; Pang, S.T.; McPherson, J.R.; Yu, W.; Huang, K.K.; Guan, P.; Weng, W.H.; Siew, E.Y.; Liu, Y.; Heng, H.L.; et al. Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci. Transl. Med. 2013, 5, 197ra101. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Chen, C.H.; Sidorenko, V.S.; He, J.; Dickman, K.G.; Yun, B.H.; Moriya, M.; Niknafs, N.; Douville, C.; Karchin, R.; et al. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med. 2013, 5, 197ra102. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Schmeiser, H.H.; Straif, K.; Wild, C.P. Upper urinary tract urothelial cancer: Where it is A:T. Nat. Rev. 2012, 12, 503–504. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Kucab, J.E.; Morganella, S.; Glodzik, D.; Alexandrov, L.B.; Arlt, V.M.; Weninger, A.; Hollstein, M.; Stratton, M.R.; Phillips, D.H. The genome as a record of environmental exposure. Mutagenesis 2015, 30, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Sopko, B.; Wiessler, M.; Schmeiser, H.H. Carcinogenic aristolochic acids upon activation by DT-diaphorase form adducts found in DNA of patients with Chinese herbs nephropathy. Carcinogenesis 2002, 23, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Sopko, B.; Sopková, K.; Marková, V.; Laňková, M.; Kumstýřová, T.; Wiessler, M.; Schmeiser, H.H. Human cytosolic enzymes involved in the metabolic activation of carcinogenic aristolochic acid: Evidence for reductive activation by human NAD(P)H:quinone oxidoreductase. Carcinogenesis 2003, 24, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gong, L.; Qi, X.; Xing, G.; Luan, Y.; Wu, Y.; Xiao, Y.; Yao, J.; Li, Y.; Xue, X.; et al. Inhibition of renal NQO1 activity by dicoumarol suppresses nitroreduction of aristolochic acid I and attenuates its nephrotoxicity. Toxicol. Sci. 2011, 122, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Levová, K.; Bárta, F.; Šulc, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The influence of dicoumarol on the bioactivation of the carcinogen aristolochic acid I in rats. Mutagenesis 2014, 29, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Mareš, J.; Frei, E.; Arlt, V.M.; Martínek, V.; Schmeiser, H.H. The human carcinogen aristolochic acid I is activated to form DNA adducts by human NAD(P)H:quinone oxidoreductase without the contribution of acetyltransferases or sulfotransferases. Environ. Mol. Mutagen. 2011, 52, 448–459. [Google Scholar]
- Martínek, V.; Kubickova, B.; Arlt, V.M.; Frei, E.; Schmeiser, H.H.; Hudeček, J.; Stiborova, M. Comparison of activation of aristolochic acid I and II with NADPH:quinone oxidoreductase, sulphotransferases and N-acetyltransferases. Neuro. Endocrinol. Lett. 2011, 32 (Suppl. 1), S57–S70. [Google Scholar]
- Sidorenko, V.S.; Attaluri, S.; Zaitseva, I.; Iden, C.R.; Dickman, K.G.; Johnson, F.; Grollman, A.P. Bioactivation of the human carcinogen aristolochic acid. Carcinogenesis 2014, 35, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Wiessler, M.; Schmeiser, H.H. Human enzymes involved in the metabolic activation of carcinogenic aristolochic acids: Evidence for reductive activation by cytochromes P450 1A1 and 1A2. Chem. Res. Toxicol. 2001, 14, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Hájek, M.; Frei, E.; Schmeiser, H.H. Carcinogenic and nephrotoxic alkaloids aristolochic acids upon activation by NADPH:cytochrome P450 reductase form adducts found in DNA of patients with Chinese herbs nephropathy. Gen. Physiol. Biophys. 2001, 20, 375–392. [Google Scholar] [PubMed]
- Stiborová, M.; Frei, E.; Hodek, P.; Wiessler, M.; Schmeiser, H.H. Human hepatic and renal microsomes, cytochromes P450 1A1/2, NADPH:CYP reductase and prostaglandin H synthase mediate the formation of aristolochic acid DNA-adducts found in patients with urothelial cancer. Int. J. Cancer 2005, 113, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Jerabek, P.; Martinek, V.; Stiborova, M. Theoretical investigation of differences in nitroreduction of aristolochic acid I by cytochromes P450 1A1, 1A2 and 1B1. Neuro Endocrinol. Lett. 2012, 33 (Suppl. 3), S25–S32. [Google Scholar]
- Stiborová, M.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. Mechanisms of enzyme-catalyzed reduction of two carcinogenic nitro-aromatics, 3-nitrobenzanthrone and aristolochic acid I: Experimental and theoretical approaches. Int. J. Mol. Sci. 2014, 15, 10271–10295. [Google Scholar] [CrossRef] [PubMed]
- Sistkova, J.; Hudecek, J.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Stiborova, M. Human cytochromes P450 1A1 and 1A2 participate in detoxication of carcinogenic aristolochic acid. Neuro Endocrinol. Lett. 2008, 29, 733–737. [Google Scholar] [PubMed]
- Rosenquist, T.A.; Einolf, H.J.; Dickman, K.G.; Wang, L.; Smith, A.; Grollman, A.P. Cytochrome P450 1A2 detoxicates aristolochic acid in the mouse. Drug Metab. Dispos. 2010, 38, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Bonala, R.R.; Rosenquist, T.; Rieger, R.; Suzuki, N.; Johnson, F.; Miller, F.; Grollman, A.P. Detoxification of aristolochic acid I by O-demethylation: Less nephrotoxicity and genotoxicity of aristolochic acid Ia in rodents. Int. J. Cancer 2010, 127, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Levova, K.; Barta, F.; Shi, Z.; Evans, J.D.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Stiborova, M. Role of P450 1A1 and P450 1A2 in bioactivation versus detoxication of the renal carcinogen aristolochic acid I: Studies in Cyp1a1(−/−), Cyp1a2(−/−), and Cyp1a1/1a2(−/−) mice. Chem. Res. Toxicol. 2011, 24, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Levová, K.; Bárta, F.; Shi, Z.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Arlt, V.M. Bioactivation versus detoxication of the urothelial carcinogen aristolochic acid I by human cytochrome P450 1A1 and 1A2. Toxicol. Sci. 2012, 125, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Bárta, F.; Levová, K.; Hodek, P.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. Contributions of cytochromes P450 to detoxification of a human carcinogen aristolochic acid I in human and rat livers: Experimental and theoretical approaches. Int. J. Mol. Sci. 2015, 16, 27561–27575. [Google Scholar] [CrossRef] [PubMed]
- Schyman, P.; Lai, W.; Chen, H.; Wang, Y.; Shaik, S. The directive of the protein: How does cytochrome P450 select the mechanism of dopamine formation? J. Am. Chem. Soc. 2011, 133, 7977–7984. [Google Scholar] [CrossRef] [PubMed]
- Bornhorst, J.A.; Falke, J.J. Purification of proteins using polyhistidine affinity tags. Methods Enzymol. 2000, 326, 245–254. [Google Scholar] [PubMed]
- Yun, C.H.; Miller, G.P.; Guengerich, F.P. Rate-determining steps in phenacetin oxidations by human cytochrome P450 1A2 and selected mutants. Biochemistry 2000, 39, 11319–11329. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.L.; Porter, T.D.; Wilson, T.E.; Kasper, C.B. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J. Biol. Chem. 1989, 264, 7584–7589. [Google Scholar] [PubMed]
- Burke, M.D.; Mayer, R.T. Ethoxyresorufin: Direct fluorimetric assay of a microsomal O-dealkylation which is preferentially inducible by 3-methylcholanthrene. Drug Metab. Dispos. 1974, 2, 583–588. [Google Scholar] [PubMed]
- Stiborová, M.; Martínek, V.; Rýdlová, H.; Hodek, P.; Frei, E. Sudan I is a potential carcinogen for humans: Evidence for its metabolic activation and detoxication by human recombinant cytochrome P450 1A1 and liver microsomes. Cancer Res. 2002, 62, 5678–5684. [Google Scholar] [PubMed]
- Levová, K.; Mizerovská, M.; Kotrbová, V.; Šulc, M.; Henderson, C.J.; Wolf, C.R.; Philips, D.H.; Frei, E.; Schmeiser, H.H.; Mareš, J.; et al. Role of cytochromes P450 1A1/2 in detoxication and activation of carcinogenic aristolochic acid I: Studies with the hepatic NADPH:cytochrome P450 reductase null (HRN) mouse model. Toxicol. Sci. 2011, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Sopko, B.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Hudeček, J. The binding of aristolochic acid I to the active site of human cytochromes P450 1A1 and 1A2 explains their potential to reductively activate this human carcinogen. Cancer Lett. 2005, 229, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bieler, C.A.; Stiborová, M.; Wiessler, M.; Cosyns, J.-P.; van Ypersele de Strihou, C.; Schmeiser, H.H. 32P-postlabelling analysis of DNA adducts formed by aristolochic acid in tissues from patients with Chinese herbs nephropathy. Carcinogenesis 1997, 18, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.W.; Caudle, D.L.; Martin-Wixtrom, C.; Quattrochi, L.C.; Tukey, R.H.; Waterman, M.R.; Estabrook, R.W. High-level expression of functional human cytochrome P450 1A2 in Escherichia coli. FASEB J. 1992, 6, 759–764. [Google Scholar] [PubMed]
- Guo, Z.; Gillam, E.M.; Ohmori, S.; Tukey, R.H.; Guengerich, F.P. Expression of modified human cytochrome P450 1A1 in Escherichia coli: Effects of 5’ substitution, stabilization, purification, spectral characterization, and catalytic properties. Arch. Biochem. Biophys. 1994, 312, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, P.; Guo, Z.; Baba, T.; Martin, M.V.; Tukey, R.H.; Guengerich, F.P. Expression of modified human cytochrome P450 1A2 in Escherichia coli: Stabilization, purification, spectral characterization, and catalytic activities of the enzyme. Arch. Biochem. Biophys. 1994, 309, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Wunsch, R.M.; Hanna, I.H.; Sutter, T.R.; Guengerich, F.P.; Gillam, E.M.J. Recombinant human cytochrome P450 1B1 expression in Escherichia coli. Arch. Biochem. Biophys. 1998, 357, 111–120. [Google Scholar] [CrossRef] [PubMed]
- The Human Cytochrome P450 (CYP): Allele Nomenclature Database. Available online: http://www.cypalleles.ki.se/ (accessed on 29 January 2016).
- Harlow, G.R.; Halpert, J.R. Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P450 3A4: Role of residues 210 and 211 in flavonoid activation and substrate specifity. J. Biol. Chem. 1997, 272, 5396–5402. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Sato, R. The carbon monoxide-binding pigment of liver microsomes: I Evidence for its hemoprotein nature. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [PubMed]
- Wiechelman, K.J.; Braun, R.D.; Fitzpatrick, J.D. Investigation of the bicinchoninic acid protein assay: Identification of the groups responsible for color formation. Anal. Biochem. 1988, 175, 231–237. [Google Scholar] [CrossRef]
- Hodek, P.; Koblihova, J.; Kizek, R.; Frei, E.; Arlt, V.M.; Stiborova, M. The relationship between DNA adduct formation by benzo[a]pyrene and expression of its activation enzyme cytochrome P450 1A1 in rats. Environ. Toxicol. Pharmacol. 2013, 36, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Bieler, C.A.; Wiessler, M.; Frei, E. The anticancer agent ellipticine on activation by cytochrome P450 forms covalent DNA adducts. Biochem. Pharmacol. 2001, 62, 1675–1684. [Google Scholar] [CrossRef]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milichovský, J.; Bárta, F.; Schmeiser, H.H.; Arlt, V.M.; Frei, E.; Stiborová, M.; Martínek, V. Active Site Mutations as a Suitable Tool Contributing to Explain a Mechanism of Aristolochic Acid I Nitroreduction by Cytochromes P450 1A1, 1A2 and 1B1. Int. J. Mol. Sci. 2016, 17, 213. https://doi.org/10.3390/ijms17020213
Milichovský J, Bárta F, Schmeiser HH, Arlt VM, Frei E, Stiborová M, Martínek V. Active Site Mutations as a Suitable Tool Contributing to Explain a Mechanism of Aristolochic Acid I Nitroreduction by Cytochromes P450 1A1, 1A2 and 1B1. International Journal of Molecular Sciences. 2016; 17(2):213. https://doi.org/10.3390/ijms17020213
Chicago/Turabian StyleMilichovský, Jan, František Bárta, Heinz H. Schmeiser, Volker M. Arlt, Eva Frei, Marie Stiborová, and Václav Martínek. 2016. "Active Site Mutations as a Suitable Tool Contributing to Explain a Mechanism of Aristolochic Acid I Nitroreduction by Cytochromes P450 1A1, 1A2 and 1B1" International Journal of Molecular Sciences 17, no. 2: 213. https://doi.org/10.3390/ijms17020213