
α-Tocopherol and Hippocampal Neural Plasticity in Physiological and Pathological Conditions

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Vitamin E Structure and Mechanisms of Actions
Methodological Issues Concerning Experimental Manipulations of Vitamin E Intake
3. α-Tocopherol and Brain Development
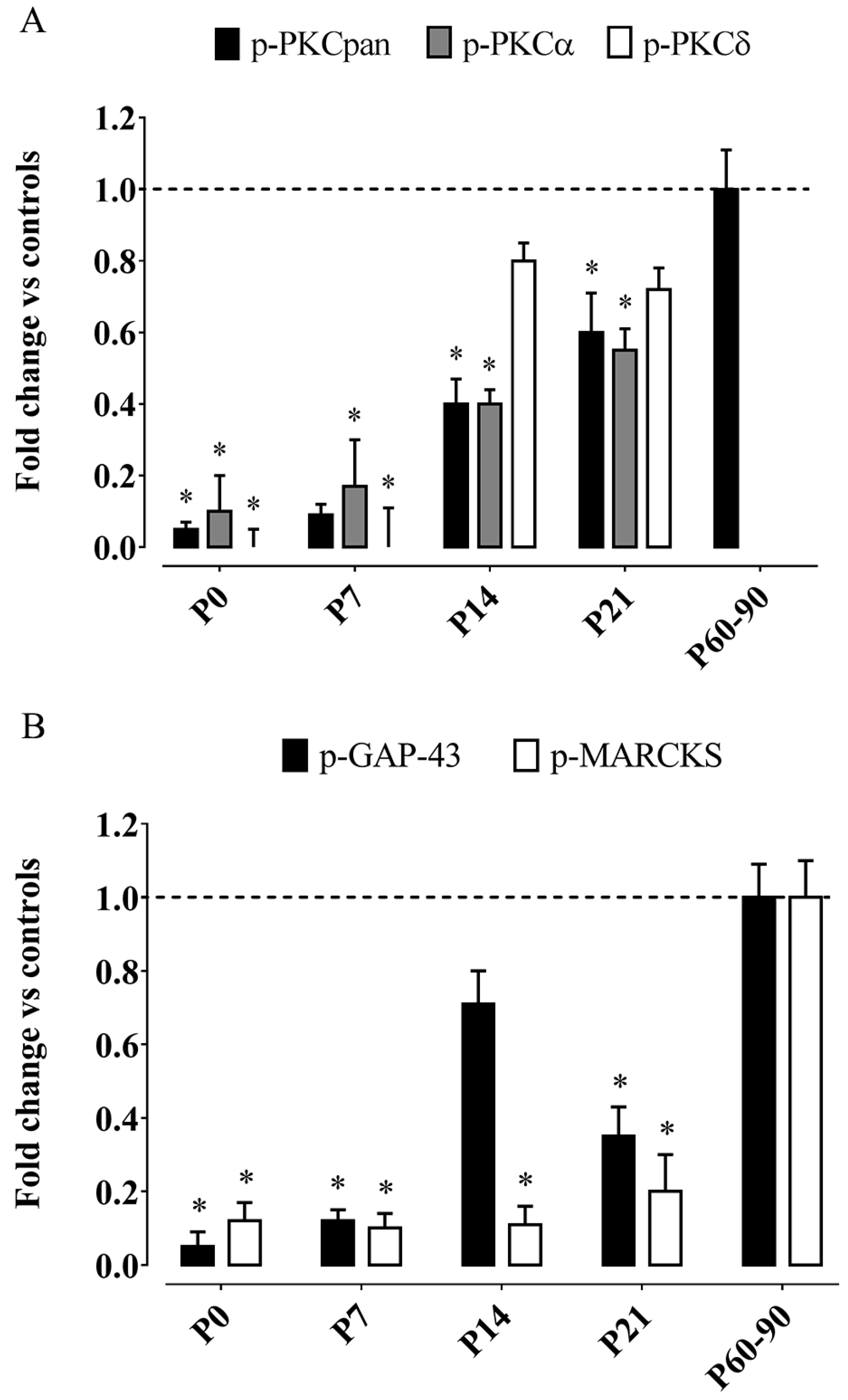
3.1. Effects on Postnatal Development
3.2. Long-Lasting Effects in Adulthood
4. α-Tocopherol, Synaptic Plasticity and Cognitive Functions
4.1. Evidence in Animal Models
4.2. Possible Mechanisms
4.3. Human Studies
5. α-Tocopherol and Adult Hippocampal Neurogenesis
Possible Mechanisms
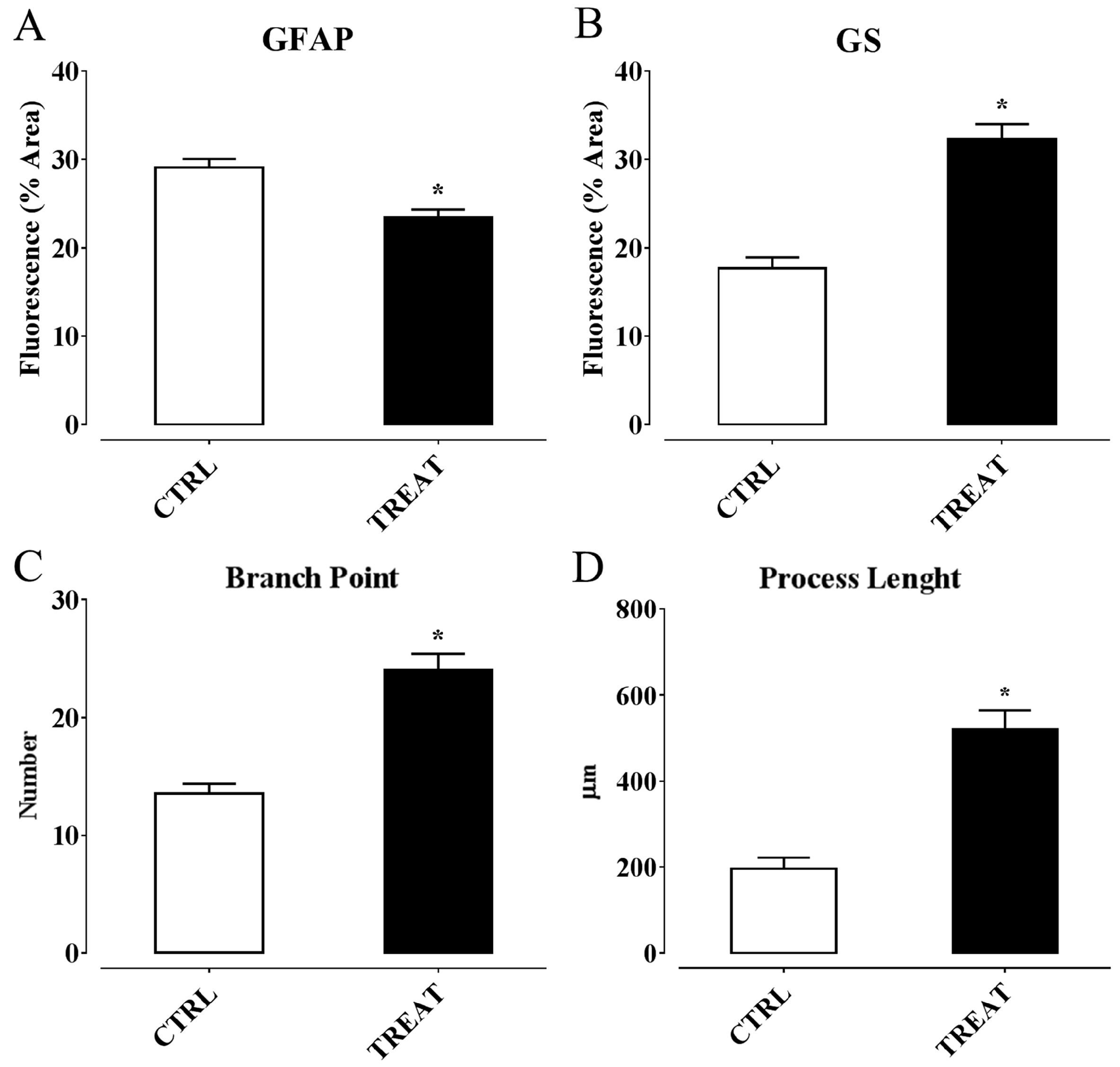
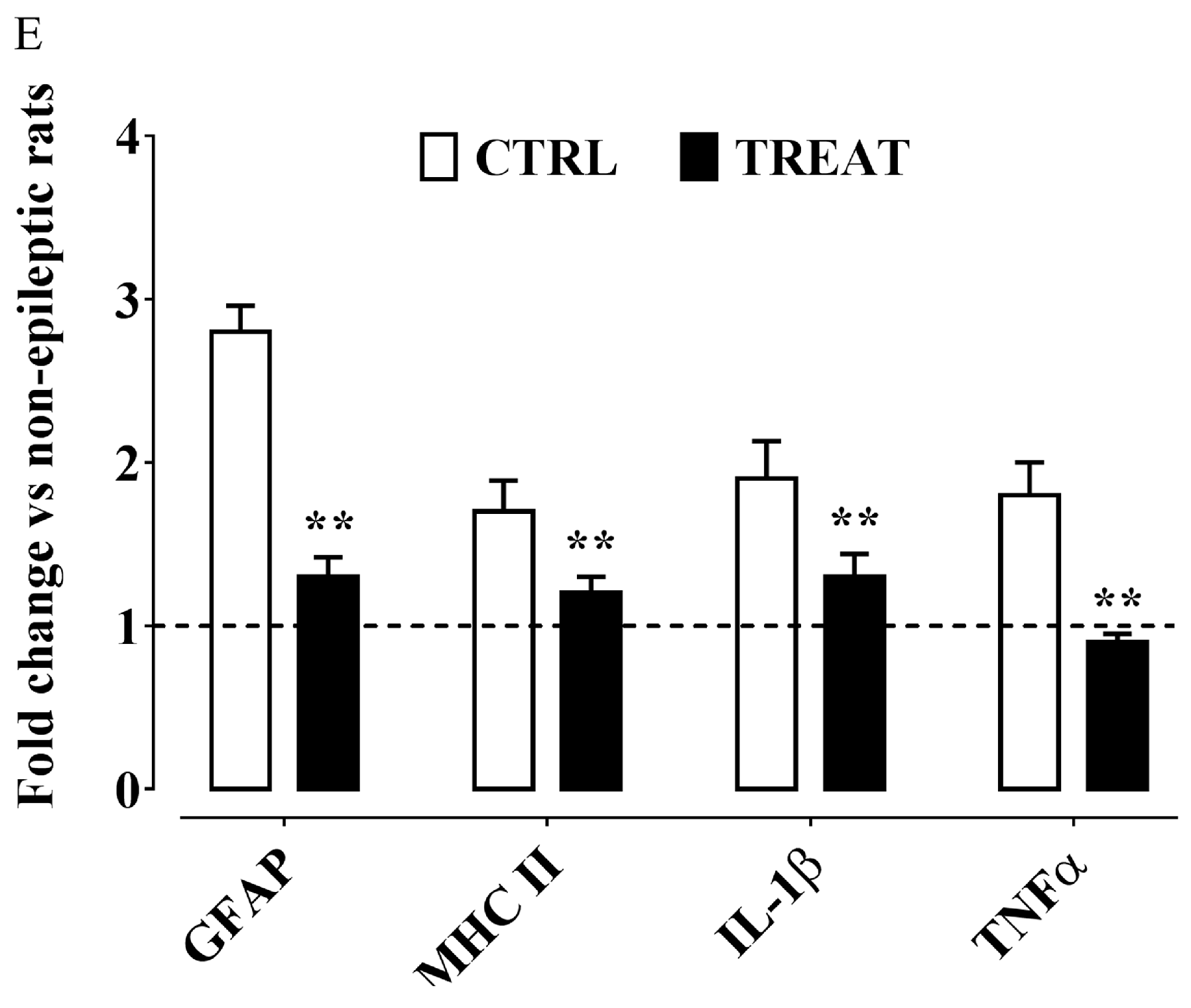
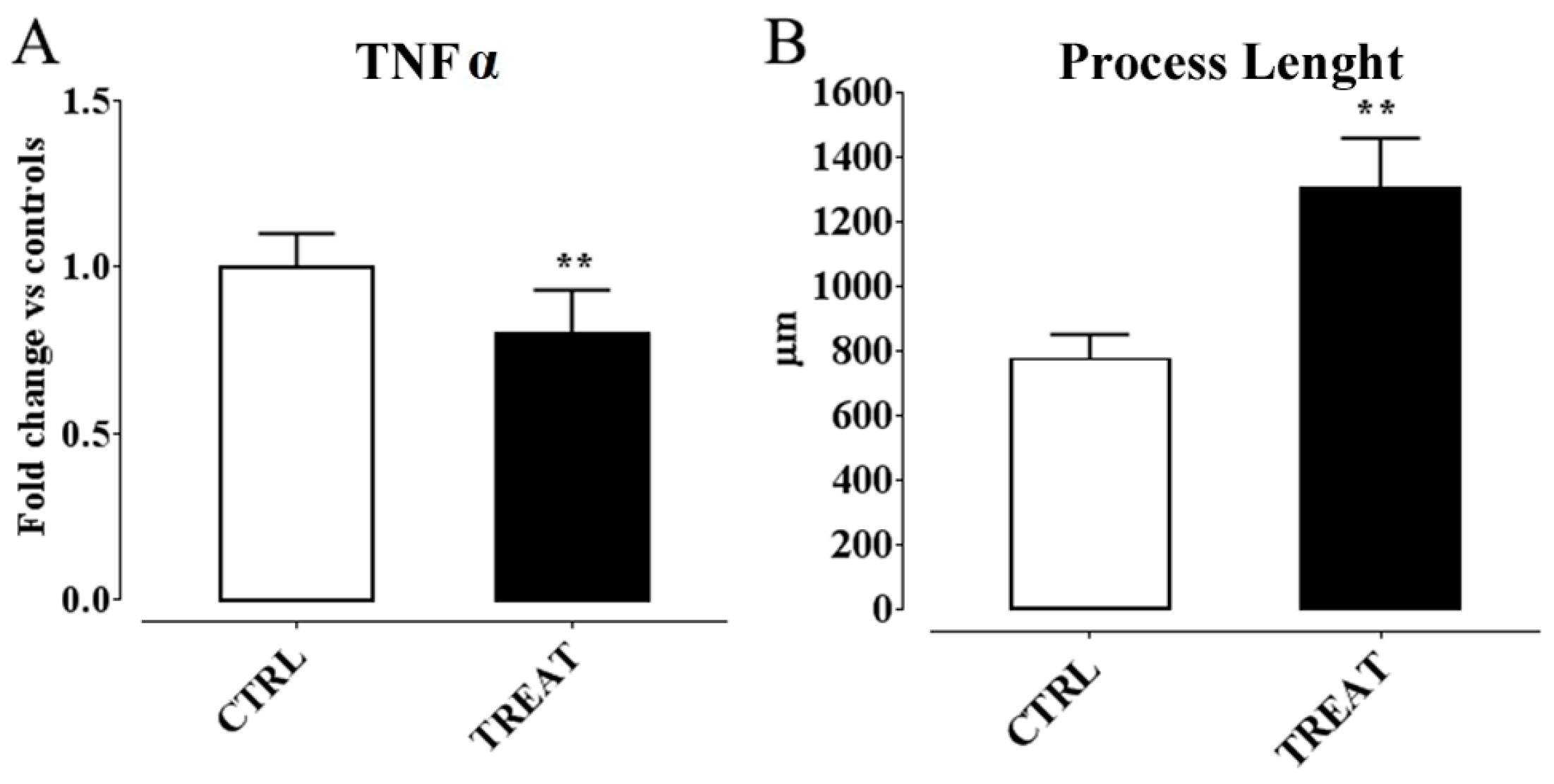
6. α-Tocopherol and Epilepsy
Possible Mechanisms
7. Conclusions
Conflicts of Interest
References
- Pascual-Leone, A.; Amedi, A.; Fregni, F.; Merabet, L.B. The plastic human brain cortex. Annu. Rev. Neurosci. 2005, 28, 377–401. [Google Scholar] [CrossRef] [PubMed]
- Wefelmeyer, W.; Puhl, C.J.; Burrone, J. Homeostatic plasticity of subcellular neuronal structures: From inputs to outputs. Trends Neurosci. 2016, 39, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Aimone, J.B.; Li, Y.; Lee, S.W.; Clemenson, G.D.; Deng, W.; Gage, F.H. Regulation and function of adult neurogenesis: From genes to cognition. Physiol. Rev. 2014, 94, 991–1026. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Lomo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef] [PubMed]
- Maren, S.; Baudry, M. Properties and mechanisms of long-term synaptic plasticity in the mammalian brain: Relationships to learning and memory. Neurobiol. Learn. Mem. 1995, 63, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.A. Long-term potentiation and memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Stress and the aging hippocampus. Front. Neuroendocrinol. 1999, 20, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Doidge, N. The Brain that Changes Itself Stories of Personal Triumph from the Frontiers of Brain Science; Viking Press: New York, NY, USA, 2007. [Google Scholar]
- Galli, F.; Azzi, A.; Birringer, M.; Cook-Mills, J.M.; Eggersdorfer, M.; Frank, J.; Cruciani, G.; Lorkowski, S.; Kartal Özer, N. Vitamin E: Emerging aspects and new directions. Free Radic. Biol. Med. 2017, 102, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Gysin, R.; Kempna, P.; Munteanu, A.; Negis, Y.; Villacorta, L.; Visarius, T.; Zingg, J.M. Vitamin E mediates cell signaling and regulation of gene expression. Ann. N. Y. Acad. Sci. 2004, 1031, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Aratri, E.; Boscoboinik, D.; Clement, S.; Ozer, N.K.; Ricciarelli, R.; Spycher, S. Molecular basis of α-tocopherol control of smooth muscle cell proliferation. BioFactors 1998, 7, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, R.; Nowicka, B.; Kruk, J. Vitamin E—Occurrence, Biosynthesis by Plants and Functions in Human Nutrition. Mini. Rev. Med. Chem. 2016. Available online: https://www.ncbi.nlm.nih.gov/pubmed/27457214 (accessed on 12 December 2016). [Google Scholar] [CrossRef]
- Evans, H.M.; Bishop, K.S. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 1922, 56, 650–651. [Google Scholar] [CrossRef] [PubMed]
- Olcott, H.S.; Emerson, O.H. Antioxidants and the autoxidation of fats. IX. The antioxidant properties of the tocopherols. J. Am. Chem. Soc. 1937, 59, 1008–1009. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Vitamin E as an in vitro and in vivo antioxidant. Ann. N. Y. Acad. Sci. 1989, 570, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, P.; Kluth, D.; Landes, N.; Bumke-Vogt, C.; Brigelius-Flohe, R. Vitamin E: Underestimated as an antioxidant. Redox Rep. 2004, 9, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R. Bioactivity of vitamin E. Nutr. Res. Rev. 2006, 19, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Niki, E.; Noguchi, N. Comparative study on the action of tocopherols and tocotrienols as antioxidant: Chemical and physical effects. Chem. Phys. Lipids 2003, 123, 63–75. [Google Scholar] [CrossRef]
- Muller, L.; Theile, K.; Bohm, V. In vitro antioxidant activity of tocopherols and tocotrienols and comparison of vitamin E concentration and lipophilic antioxidant capacity in human plasma. Mol. Nutr. Food Res. 2010, 54, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Podda, M.; Weber, C.; Traber, M.G.; Packer, L. Simultaneous determination of tissue tocopherols, tocotrienols, ubiquinols, and ubiquinones. J. Lipid Res. 1996, 37, 893–901. [Google Scholar] [PubMed]
- Betti, M.; Minelli, A.; Canonico, B.; Castaldo, P.; Magi, S.; Aisa, M.C.; Piroddi, M.; di Tomaso, V.; Galli, F. Antiproliferative effects of tocopherols (vitamin E) on murine glioma C6 cells: Homologue-specific control of PKC/ERK and cyclin signaling. Free Radic. Biol. Med. 2006, 41, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Chuang, H.C.; Chou, C.C.; Wang, H.; Lee, S.L.; Yang, H.C.; Chiu, H.C.; Kapuriya, N.; Wang, D.; Kulp, S.K.; et al. Vitamin E facilitates the inactivation of the kinase Akt by the phosphatase PHLPP1. Sci. Signal. 2013, 6, ra19. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Fukuhara, A.; Nishio, K.; Hayakawa, M.; Ogawa, Y.; Sakamoto, H.; Fujii, K.; Yoshida, Y.; Niki, E. Characterization of cellular uptake and distribution of coenzyme Q10 and vitamin E in PC12 cells. J. Nutr. Biochem. 2009, 20, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Barve, A.; Khor, T.O.; Nair, S.; Reuhl, K.; Suh, N.; Reddy, B.; Newmark, H.; Kong, A.N. γ-tocopherol-enriched mixed tocopherol diet inhibits prostate carcinogenesis in tramp mice. Int. J. Cancer 2009, 124, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.K.; Omaye, S.T. α-tocopherol modulates human umbilical vein endothelial cell expression of Cu/Zn superoxide dismutase and catalase and lipid peroxidation. Nutr. Res. 2008, 28, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Jiang, Q.; Christen, S. Anti-inflammatory properties of α- and γ-tocopherol. Mol. Asp. Med. 2007, 28, 668–691. [Google Scholar] [CrossRef] [PubMed]
- Jack Yang, N.Y.; Desai, I.D. Effect of high levels of dietary vitamin E on liver and plasma lipids and fat soluble vitamins in rats. J. Nutr. 1977, 107, 1418–1426. [Google Scholar] [PubMed]
- Machlin, L.J.; Gabriel, E. Kinetics of tissue α-tocopherol uptake and depletion following administration of high levels of vitamin E. Ann. N. Y. Acad. Sci. 1982, 393, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Mardones, P.; Rigotti, A. Cellular mechanisms of vitamin E uptake: Relevance in α-tocopherol metabolism and potential implications for disease. J. Nutr. Biochem. 2004, 15, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Gohil, K.; Oommen, S.; Quach, H.T.; Vasu, V.T.; Aung, H.H.; Schock, B.; Cross, C.E.; Vatassery, G.T. Mice lacking α-tocopherol transfer protein gene have severe α-tocopherol deficiency in multiple regions of the central nervous system. Brain Res. 2008, 1201, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Igarashi, K.; Uchihara, T.; Jishage, K.; Tomita, H.; Inaba, A.; Li, Y.; Arita, M.; Suzuki, H.; Mizusawa, H.; et al. Delayed-onset ataxia in mice lacking α-tocopherol transfer protein: Model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. USA 2001, 98, 15185–15190. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R. Vitamin E: The shrew waiting to be tamed. Free Radic. Biol. Med. 2009, 46, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Traber, M.G.; Acuff, R.V.; Walters, D.N.; Kayden, H.; Hughes, L.; Ingold, K.U. Human plasma and tissue α-tocopherol concentrations in response to supplementation with deuterated natural and synthetic vitamin E. Am. J. Clin. Nutr. 1998, 67, 669–684. [Google Scholar] [PubMed]
- Nishio, K.; Ishida, N.; Saito, Y.; Ogawa-Akazawa, Y.; Shichiri, M.; Yoshida, Y.; Hagihara, Y.; Noguchi, N.; Chirico, J.; Atkinson, J.; et al. α-tocopheryl phosphate: Uptake, hydrolysis, and antioxidant action in cultured cells and mouse. Free Radic. Biol. Med. 2011, 50, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.; Harroun, T.; Wassall, S.R.; Stillwell, W.; Katsaras, J. The location and behavior of α-tocopherol in membranes. Mol. Nutr. Food Res. 2010, 54, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.A.; Lynch, M.A. Dietary supplementation with vitamin E reverses the age-related deficit in long term potentiation in dentate gyrus. J. Biol. Chem. 1998, 273, 12161–12168. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.C.; de Avila, D.S.; Schneider, C.Y.; Hermes, F.S.; Furian, A.F.; Oliveira, M.S.; Rubin, M.A.; Lehmann, M.; Krieglstein, J.; Mello, C.F. α-tocopherol protects against pentylenetetrazol- and methylmalonate-induced convulsions. Epilepsy Res. 2005, 66, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Tome, A.R.; Feng, D.; Freitas, R.M. The effects of α-tocopherol on hippocampal oxidative stress prior to in pilocarpine-induced seizures. Neurochem. Res. 2010, 35, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Lodge, J.K.; Hall, W.L.; Jeanes, Y.M.; Proteggente, A.R. Physiological factors influencing vitamin E biokinetics. Ann. N. Y. Acad. Sci. 2004, 1031, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Sivan, E.; Reece, E.A.; Wu, Y.K.; Homko, C.J.; Polansky, M.; Borenstein, M. Dietary vitamin E prophylaxis and diabetic embryopathy: Morphologic and biochemical analysis. Am. J. Obstet. Gynecol. 1996, 175, 793–799. [Google Scholar] [CrossRef]
- Cederberg, J.; Eriksson, U.J. Antioxidative treatment of pregnant diabetic rats diminishes embryonic dysmorphogenesis. Birth Defects Res. A Clin. Mol. Teratol. 2005, 73, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Cederberg, J.; Siman, C.M.; Eriksson, U.J. Combined treatment with vitamin E and vitamin C decreases oxidative stress and improves fetal outcome in experimental diabetic pregnancy. Pediatr. Res. 2001, 49, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.; Castro, M.; Barbas, C.; Herrera, E.; Bonet, B. Effect of different doses of vitamin E on the incidence of malformations in pregnant diabetic rats. Ann. Nutr. Metab. 2003, 47, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Reece, E.A.; Wu, Y.K.; Zhao, Z.; Dhanasekaran, D. Dietary vitamin and lipid therapy rescues aberrant signaling and apoptosis and prevents hyperglycemia-induced diabetic embryopathy in rats. Am. J. Obstet. Gynecol. 2006, 194, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Shirpoor, A.; Salami, S.; Khadem-Ansari, M.H.; Minassian, S.; Yegiazarian, M. Protective effect of vitamin E against ethanol-induced hyperhomocysteinemia, DNA damage, and atrophy in the developing male rat brain. Alcohol. Clin. Exp. Res. 2009, 33, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Erdemli, M.E.; Turkoz, Y.; Altinoz, E.; Elibol, E.; Dogan, Z. Investigation of the effects of acrylamide applied during pregnancy on fetal brain development in rats and protective role of the vitamin E. Hum. Exp. Toxicol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rumbold, A.; Crowther, C.A. Vitamin C supplementation in pregnancy. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef]
- Rumbold, A.; Duley, L.; Crowther, C.A.; Haslam, R.R. Antioxidants for preventing pre-eclampsia. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Rumbold, A.; Ota, E.; Hori, H.; Miyazaki, C.; Crowther, C.A. Vitamin E supplementation in pregnancy. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Boskovic, R.; Gargaun, L.; Oren, D.; Djulus, J.; Koren, G. Pregnancy outcome following high doses of vitamin E supplementation. Reprod. Toxicol. 2005, 20, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Bastani, P.; Hamdi, K.; Abasalizadeh, F.; Navali, N. Effects of vitamin E supplementation on some pregnancy health indices: A randomized clinical trial. Int. J. Gen. Med. 2011, 4, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.E.; Hong, Y.C.; Lee, K.H.; Kim, Y.J.; Kim, W.K.; Chang, N.S.; Park, E.A.; Park, H.S.; Hann, H.J. Influence of maternal serum levels of vitamins C and E during the second trimester on birth weight and length. Eur. J. Clin. Nutr. 2004, 58, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Scholl, T.O.; Chen, X.; Sims, M.; Stein, T.P. Vitamin E: Maternal concentrations are associated with fetal growth. Am. J. Clin. Nutr. 2006, 84, 1442–1448. [Google Scholar] [PubMed]
- Poston, L.; Briley, A.L.; Seed, P.T.; Kelly, F.J.; Shennan, A.H. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): Randomised placebo-controlled trial. Lancet 2006, 367, 1145–1154. [Google Scholar] [CrossRef]
- Martin, M.M.; Hurley, L.S. Effect of large amounts of vitamin E during pregnancy and lactation. Am. J. Clin. Nutr. 1977, 30, 1629–1637. [Google Scholar] [PubMed]
- Smedts, H.P.; de Vries, J.H.; Rakhshandehroo, M.; Wildhagen, M.F.; Verkleij-Hagoort, A.C.; Steegers, E.A.; Steegers-Theunissen, R.P. High maternal vitamin E intake by diet or supplements is associated with congenital heart defects in the offspring. BJOG 2009, 116, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Debier, C.; Larondelle, Y. Vitamins a and e: Metabolism, roles and transfer to offspring. Br. J. Nutr. 2005, 93, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Zarban, A.; Toroghi, M.M.; Asli, M.; Jafari, M.; Vejdan, M.; Sharifzadeh, G. Effect of vitamin C and e supplementation on total antioxidant content of human breastmilk and infant urine. Breastfeed. Med. 2015, 10, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Melo, L.R.; Clemente, H.A.; Bezerra, D.F.; Dantas, R.C.; Ramalho, H.M.; Dimenstein, R. Effect of maternal supplementation with vitamin E on the concentration of α-tocopherol in colostrum. J. Pediatr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pires Medeiros, J.F.; Ribeiro, K.D.; Lima, M.S.; das Neves, R.A.; Lima, A.C.; Dantas, R.C.; da Silva, A.B.; Dimenstein, R. α-tocopherol in breast milk of women with preterm delivery after a single postpartum oral dose of vitamin E. Br. J. Nutr. 2016, 115, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Cuppini, R.; Ciaroni, S.; Cecchini, T.; Ambrogini, P.; Ferri, P.; Cuppini, C.; Ninfali, P.; del Grande, P. Tocopherols enhance neurogenesis in dentate gyrus of adult rats. Int. J. Vitam. Nutr. Res. 2002, 72, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Ferri, P.; Cecchini, T.; Ambrogini, P.; Betti, M.; Cuppini, R.; del Grande, P.; Ciaroni, S. α-tocopherol affects neuronal plasticity in adult rat dentate gyrus: The possible role of pkcdelta. J. Neurobiol. 2006, 66, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, T.; Ciaroni, S.; Ferri, P.; Ambrogini, P.; Cuppini, R.; Santi, S.; del Grande, P. α-tocopherol, an exogenous factor of adult hippocampal neurogenesis regulation. J. Neurosci. Res. 2003, 73, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Ferri, P.; Cecchini, T.; Ciaroni, S.; Ambrogini, P.; Cuppini, R.; Santi, S.; Benedetti, S.; Pagliarani, S.; del Grande, P.; Papa, S. Vitamin E affects cell death in adult rat dentate gyrus. J. Neurocytol. 2003, 32, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. The protein kinase C family and lipid mediators for transmembrane signaling and cell regulation. Alcohol. Clin. Exp. Res. 2001, 25, 3S–7S. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Hashimoto, K.; Chen, C.; Abeliovich, A.; Aiba, A.; Kurihara, H.; Watanabe, M.; Inoue, Y.; Tonegawa, S. Impaired synapse elimination during cerebellar development in PKCγmutant mice. Cell 1995, 83, 1223–1231. [Google Scholar] [CrossRef]
- Hama, H.; Hara, C.; Yamaguchi, K.; Miyawaki, A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron 2004, 41, 405–415. [Google Scholar] [CrossRef]
- Kapfhammer, J.P. Cellular and molecular control of dendritic growth and development of cerebellar purkinje cells. Prog. Histochem. Cytochem. 2004, 39, 131–182. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Feng, L. Neuregulin induces proliferation of neural progenitor cells via PLC/PKC pathway. Biochem. Biophys. Res. Commun. 2004, 319, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.T.; Fleming, M.R.; Leu, B. Presynaptic protein kinase C controls maturation and branch dynamics of developing retinotectal arbors: Possible role in activity-driven sharpening. J. Neurobiol. 2004, 58, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Kolkova, K.; Stensman, H.; Berezin, V.; Bock, E.; Larsson, C. Distinct roles of PKC isoforms in NCAM-mediated neurite outgrowth. J. Neurochem. 2005, 92, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Brandt, N.; Franke, K.; Rasin, M.R.; Baumgart, J.; Vogt, J.; Khrulev, S.; Hassel, B.; Pohl, E.E.; Sestan, N.; Nitsch, R.; et al. The neural EGF family member CALEB/NGC mediates dendritic tree and spine complexity. EMBO J. 2007, 26, 2371–2386. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Derr-Yellin, E.C.; Kodavanti, P.R. Alterations in brain protein kinase C isoforms following developmental exposure to a polychlorinated biphenyl mixture. Brain Res. Mol. Brain Res. 2003, 111, 123–135. [Google Scholar] [CrossRef]
- Xu, S.Z.; Bullock, L.; Shan, C.J.; Cornelius, K.; Rajanna, B. PKC isoforms were reduced by lead in the developing rat brain. Int. J. Dev. Neurosci. 2005, 23, 53–64. [Google Scholar] [CrossRef] [PubMed]
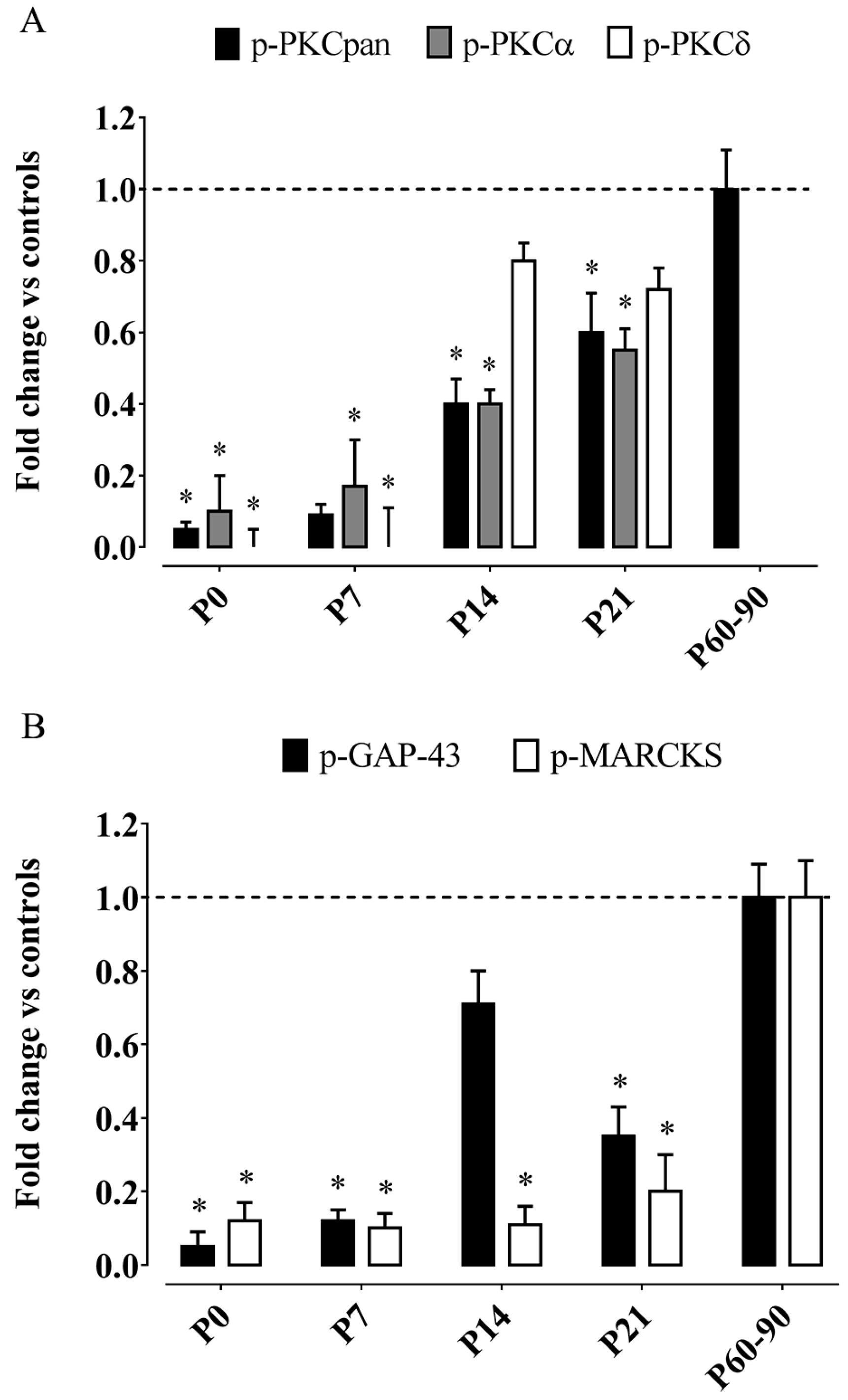
- Betti, M.; Ambrogini, P.; Minelli, A.; Floridi, A.; Lattanzi, D.; Ciuffoli, S.; Bucherelli, C.; Prospero, E.; Frontini, A.; Santarelli, L.; et al. Maternal dietary loads of α-tocopherol depress protein kinase C signaling and synaptic plasticity in rat postnatal developing hippocampus and promote permanent deficits in adult offspring. J. Nutr. Biochem. 2011, 22, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Ambrogini, P.; Ciuffoli, S.; Lattanzi, D.; Minelli, A.; Bucherelli, C.; Baldi, E.; Betti, M.; Cuppini, R. Maternal dietary loads of α-tocopherol differentially influence fear conditioning and spatial learning in adult offspring. Physiol. Behav. 2011, 104, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Ambrogini, P.; Lattanzi, D.; Betti, M.; Gobbi, P.; Galati, C.; Galli, F.; Cuppini, R.; Minelli, A. Maternal dietary loads of α-tocopherol increase synapse density and glial synaptic coverage in the hippocampus of adult offspring. Eur. J. Histochem. 2014, 58, 2355. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, L.I.; Routtenberg, A. GAP-43: An intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997, 20, 84–91. [Google Scholar] [CrossRef]
- Arbuzova, A.; Schmitz, A.A.; Vergeres, G. Cross-talk unfolded: Marcks proteins. Biochem. J. 2002, 362, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.; Akyeli, J.; Kirischuk, S.; Grantyn, R. GABAA receptor activity and PKC control inhibitory synaptogenesis in CNS tissue slices. Mol. Cell. Neurosci. 2003, 23, 600–613. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Alkon, D.L. A structural basis for enhancement of long-term associative memory in single dendritic spines regulated by PKC. Proc. Natl. Acad. Sci. USA 2007, 104, 19571–19576. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 2008, 585, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Meiri, K.F.; Saffell, J.L.; Walsh, F.S.; Doherty, P. Neurite outgrowth stimulated by neural cell adhesion molecules requires growth-associated protein-43 (GAP-43) function and is associated with GAP-43 phosphorylation in growth cones. J. Neurosci. 1998, 18, 10429–10437. [Google Scholar] [PubMed]
- Maier, D.L.; Mani, S.; Donovan, S.L.; Soppet, D.; Tessarollo, L.; McCasland, J.S.; Meiri, K.F. Disrupted cortical map and absence of cortical barrels in growth-associated protein (GAP)-43 knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 9397–9402. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.K.; Lenox, R.H. Distribution of the protein kinase C substrates MARCKS and MRP in the postnatal developing rat brain. J. Comp. Neurol. 1998, 397, 337–356. [Google Scholar] [CrossRef]
- McNamara, R.K.; Stumpo, D.J.; Morel, L.M.; Lewis, M.H.; Wakeland, E.K.; Blackshear, P.J.; Lenox, R.H. Effect of reduced myristoylated alanine-rich C kinase substrate expression on hippocampal mossy fiber development and spatial learning in mutant mice: Transgenic rescue and interactions with gene background. Proc. Natl. Acad. Sci. USA 1998, 95, 14517–14522. [Google Scholar] [CrossRef] [PubMed]
- Dumas, T.C. Late postnatal maturation of excitatory synaptic transmission permits adult-like expression of hippocampal-dependent behaviors. Hippocampus 2005, 15, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.S.; Raynor, R.L.; Mazzei, G.J.; Girard, P.R.; Kuo, J.F. Developmental studies of phospholipid-sensitive Ca2+-dependent protein kinase and its substrates and of phosphoprotein phosphatases in rat brain. Proc. Natl. Acad. Sci. USA 1984, 81, 3143–3147. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, S.M.; Igarashi, M.; Fishman, M.C. GAP-43 amino terminal peptides modulate growth cone morphology and neurite outgrowth. J. Neurosci. 1994, 14, 5503–5513. [Google Scholar] [PubMed]
- Spencer, S.A.; Schuh, S.M.; Liu, W.S.; Willard, M.B. GAP-43, a protein associated with axon growth, is phosphorylated at three sites in cultured neurons and rat brain. J. Biol. Chem. 1992, 267, 9059–9064. [Google Scholar] [PubMed]
- Wikstrom, M.A.; Matthews, P.; Roberts, D.; Collingridge, G.L.; Bortolotto, Z.A. Parallel kinase cascades are involved in the induction of LTP at hippocampal CA1 synapses. Neuropharmacology 2003, 45, 828–836. [Google Scholar] [CrossRef]
- Hussain, R.J.; Carpenter, D.O. A comparison of the roles of protein kinase C in long-term potentiation in rat hippocampal areas CA1 and CA3. Cell. Mol. Neurobiol. 2005, 25, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, Y.; Saitoh, Y.; Ozawa, F.; Ohta, Y.; Mizuno, K.; Inokuchi, K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 2003, 38, 447–460. [Google Scholar] [CrossRef]
- Leahy, J.C.; Luo, Y.; Kent, C.S.; Meiri, K.F.; Vallano, M.L. Demonstration of presynaptic protein kinase C activation following long-term potentiation in rat hippocampal slices. Neuroscience 1993, 52, 563–574. [Google Scholar] [CrossRef]
- Calabrese, B.; Halpain, S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron 2005, 48, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.; Jones, P.B.; Murray, R.M. Obstetric complications and schizophrenia: Historical and meta-analytic review. Am. J. Psychiatry 2002, 159, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, K.A.; Brunson, K.L.; Baram, T.Z. Hippocampal neuroplasticity induced by early-life stress: Functional and molecular aspects. Front. Neuroendocrinol. 2006, 27, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Morgane, P.J.; Mokler, D.J.; Galler, J.R. Effects of prenatal protein malnutrition on the hippocampal formation. Neurosci. Biobehav. Rev. 2002, 26, 471–483. [Google Scholar] [CrossRef]
- Nagapan, G.; Meng Goh, Y.; Shameha Abdul Razak, I.; Nesaretnam, K.; Ebrahimi, M. The effects of prenatal and early postnatal tocotrienol-rich fraction supplementation on cognitive function development in male offspring rats. BMC Neurosci. 2013, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Railey, A.M.; Micheli, T.L.; Wanschura, P.B.; Flinn, J.M. Alterations in fear response and spatial memory in pre- and post-natal zinc supplemented rats: Remediation by copper. Physiol. Behav. 2010, 100, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.X.; Han, H.L.; Tian, M.; Cao, J.; Xiu, J.B.; Song, N.N.; Huang, Y.; Xu, T.L.; Ding, Y.Q.; Xu, L. Enhanced contextual fear memory in central serotonin-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11981–11986. [Google Scholar] [CrossRef] [PubMed]
- Jarrard, L.E. On the role of the hippocampus in learning and memory in the rat. Behav. Neural Biol. 1993, 60, 9–26. [Google Scholar] [CrossRef]
- Mizuno, K.; Giese, K.P. Hippocampus-dependent memory formation: Do memory type-specific mechanisms exist? J. Pharmacol. Sci. 2005, 98, 191–197. [Google Scholar] [CrossRef] [PubMed]
- DeFelipe, J.; Conti, F.; Van Eyck, S.L.; Manzoni, T. Demonstration of glutamate-positive axon terminals forming asymmetric synapses in cat neocortex. Brain Res. 1988, 455, 162–165. [Google Scholar] [CrossRef]
- Chen, C.; Kano, M.; Abeliovich, A.; Chen, L.; Bao, S.; Kim, J.J.; Hashimoto, K.; Thompson, R.F.; Tonegawa, S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell 1995, 83, 1233–1242. [Google Scholar] [CrossRef]
- Garner, C.C.; Waites, C.L.; Ziv, N.E. Synapse development: Still looking for the forest, still lost in the trees. Cell Tissue Res. 2006, 326, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Paus, T.; Keshavan, M.; Giedd, J.N. Why do many psychiatric disorders emerge during adolescence? Nat. Rev. Neurosci. 2008, 9, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Araque, A. Glia modulates synaptic transmission. Brain Res. Rev. 2010, 63, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Orkand, R.K.; Kettenmann, H. Glial calcium: Homeostasis and signaling function. Physiol. Rev. 1998, 78, 99–141. [Google Scholar] [PubMed]
- Filosa, A.; Paixao, S.; Honsek, S.D.; Carmona, M.A.; Becker, L.; Feddersen, B.; Gaitanos, L.; Rudhard, Y.; Schoepfer, R.; Klopstock, T.; et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat. Neurosci. 2009, 12, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.R.; Baimoukhametova, D.V.; Hewitt, S.A.; Rajapaksha, W.R.; Fisher, T.E.; Bains, J.S. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat. Neurosci. 2005, 8, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.; Rusakov, D.A. Long-term potentiation depends on release of d-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, H.; Tanaka, K.; Manabe, T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur. J. Neurosci. 2001, 14, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Theodosis, D.T.; Mothet, J.P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H. Glia-derived d-serine controls NMDA receptor activity and synaptic memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.; Melone, M.; Bellesi, M.; Safiulina, V.; Aida, T.; Tanaka, K.; Cherubini, E.; Conti, F. Up-regulation of GLT-1 severely impairs LTD at mossy fibre—CA3 synapses. J. Physiol. 2009, 587, 4575–4588. [Google Scholar] [CrossRef] [PubMed]
- Hruska, M.; Dalva, M.B. Ephrin regulation of synapse formation, function and plasticity. Mol. Cell. Neurosci. 2012, 50, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Carmona, M.A.; Murai, K.K.; Wang, L.; Roberts, A.J.; Pasquale, E.B. Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc. Natl. Acad. Sci. USA 2009, 106, 12524–12529. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Nicoll, R.A. Long-term potentiation—A decade of progress? Science 1999, 285, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Benzi, G.; Moretti, A. Age- and peroxidative stress-related modifications of the cerebral enzymatic activities linked to mitochondria and the glutathione system. Free Radic. Biol. Med. 1995, 19, 77–101. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, I.Y.; Bi, X.; Thompson, R.F.; Doctrow, S.R.; Malfroy, B.; Baudry, M. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc. Natl. Acad. Sci. USA 2003, 100, 8526–8531. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.J.; Dubey, A.; Dawson, K.M.; Stutts, W.A.; Lal, H.; Sohal, R.S. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc. Natl. Acad. Sci. USA 1996, 93, 4765–4769. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Mockett, R.J.; Orr, W.C. Mechanisms of aging: An appraisal of the oxidative stress hypothesis. Free Radic. Biol. Med. 2002, 33, 575–586. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [PubMed]
- Takatsu, H.; Owada, K.; Abe, K.; Nakano, M.; Urano, S. Effect of vitamin E on learning and memory deficit in aged rats. J. Nutr. Sci. Vitaminol. 2009, 55, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Thakurta, I.G.; Banerjee, P.; Bagh, M.B.; Ghosh, A.; Sahoo, A.; Chattopadhyay, S.; Chakrabarti, S. Combination of n-acetylcysteine, α-lipoic acid and α-tocopherol substantially prevents the brain synaptosomal alterations and memory and learning deficits of aged rats. Exp. Gerontol. 2014, 50, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Taridi, N.M.; Yahaya, M.F.; Teoh, S.L.; Latiff, A.A.; Ngah, W.Z.; Das, S.; Mazlan, M. Tocotrienol rich fraction (TRF) supplementation protects against oxidative DNA damage and improves cognitive functions in wistar rats. Clin. Ther. 2011, 162, 93–98. [Google Scholar]
- Taridi, N.M.; Abd Rani, N.; Abd Latiff, A.; Ngah, W.Z.; Mazlan, M. Tocotrienol rich fraction reverses age-related deficits in spatial learning and memory in aged rats. Lipids 2014, 49, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Salehi, I.; Karamian, R.; Komaki, A.; Tahmasebi, L.; Taheri, M.; Nazari, M.; Shahidi, S.; Sarihi, A. Effects of vitamin E on lead-induced impairments in hippocampal synaptic plasticity. Brain Res. 2015, 1629, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Khodamoradi, N.; Komaki, A.; Salehi, I.; Shahidi, S.; Sarihi, A. Effect of vitamin E on lead exposure-induced learning and memory impairment in rats. Physiol. Behav. 2015, 144, 90–94. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Zhang, T. Vitamins c and e reverse melamine-induced deficits in spatial cognition and hippocampal synaptic plasticity in rats. Neurotoxicology 2014, 44, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.H.; Khabour, O.F.; Salah, H.A.; Hasan, Z. Vitamin E prevents high-fat high-carbohydrates diet-induced memory impairment: The role of oxidative stress. Physiol. Behav. 2013, 119, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.H.; Khabour, O.F.; Rashid, B.A.; Damaj, I.M.; Salah, H.A. The neuroprotective effect of vitamin E on chronic sleep deprivation-induced memory impairment: The role of oxidative stress. Behav. Brain Res. 2012, 226, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Tagliari, B.; Scherer, E.B.; Machado, F.R.; Ferreira, A.G.; Dalmaz, C.; Wyse, A.T. Antioxidants prevent memory deficits provoked by chronic variable stress in rats. Neurochem. Res. 2011, 36, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Aiguo, W.; Zhe, Y.; Gomez-Pinilla, F. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil. Neural Repair 2010, 24, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Hasanein, P.; Shahidi, S. Effects of combined treatment with vitamins C and E on passive avoidance learning and memory in diabetic rats. Neurobiol. Learn. Mem. 2010, 93, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Tuzcu, M.; Baydas, G. Effect of melatonin and vitamin E on diabetes-induced learning and memory impairment in rats. Eur. J. Pharmacol. 2006, 537, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Yoshida, Y.; Ishida, N.; Hagihara, Y.; Iwahashi, H.; Tamai, H.; Niki, E. α-tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 2011, 50, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Sastry, B.R. Impairment of long-term potentiation in rats fed with vitamin E-deficient diet. Brain Res. 1995, 681, 193–196. [Google Scholar] [CrossRef]
- Fukui, K.; Omoi, N.O.; Hayasaka, T.; Shinnkai, T.; Suzuki, S.; Abe, K.; Urano, S. Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann. N. Y. Acad. Sci. 2002, 959, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.H.; Rigotti, A.; Huerta, P.T. Age-related influence of the HDL receptor SR-BI on synaptic plasticity and cognition. Neurobiol. Aging 2009, 30, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Onodera, K.; Shinkai, T.; Suzuki, S.; Urano, S. Impairment of learning and memory in rats caused by oxidative stress and aging, and changes in antioxidative defense systems. Ann. N. Y. Acad. Sci. 2001, 928, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Serrano, F.; Klann, E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 2004, 3, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Bodhinathan, K.; Kumar, A.; Foster, T.C. Intracellular redox state alters NMDA receptor response during aging through Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 2010, 30, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Hota, S.K.; Hota, K.B.; Prasad, D.; Ilavazhagan, G.; Singh, S.B. Oxidative-stress-induced alterations in Sp factors mediate transcriptional regulation of the NR1 subunit in hippocampus during hypoxia. Free Radic. Biol. Med. 2010, 49, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.; Galeffi, F.; Ahrens, R.; Jordan, G.; Schwartz-Bloom, R.D. Modulation of the GABAA-gated chloride channel by reactive oxygen species. J. Neurochem. 2002, 80, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Sesti, F. Oxidation of ion channels in the aging nervous system. Brain Res. 2016, 1639, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Saito, M.; Takatsu, H.; Fukui, K.; Urano, S. Dysfunction of the fusion of pre-synaptic plasma membranes and synaptic vesicles caused by oxidative stress, and its prevention by vitamin E. J. Alzheimers Dis. 2011, 24, 759–766. [Google Scholar] [PubMed]
- Lu, Q.; Harris, V.A.; Sun, X.; Hou, Y.; Black, S.M. Ca2+/calmodulin-dependent protein kinase II contributes to hypoxic ischemic cell death in neonatal hippocampal slice cultures. PLoS ONE 2013, 8, e70750. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Pao, M.; Holland, S.M.; Klann, E. Nadph oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J. Neurochem. 2005, 94, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kemmerling, U.; Munoz, P.; Muller, M.; Sanchez, G.; Aylwin, M.L.; Klann, E.; Carrasco, M.A.; Hidalgo, C. Calcium release by ryanodine receptors mediates hydrogen peroxide-induced activation of ERK and CREB phosphorylation in N2a cells and hippocampal neurons. Cell Calcium 2007, 41, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Calcium-dependent mitochondrial superoxide modulates nuclear CREB phosphorylation in hippocampal neurons. Mol. Cell. Neurosci. 2003, 24, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Kaneai, N.; Arai, M.; Takatsu, H.; Fukui, K.; Urano, S. Vitamin E inhibits oxidative stress-induced denaturation of nerve terminal proteins involved in neurotransmission. J. Alzheimers Dis. 2012, 28, 183–189. [Google Scholar] [PubMed]
- Kaneai, N.; Fukui, K.; Koike, T.; Urano, S. Vitamin E prevents hyperoxia-induced loss of soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor proteins in the rat neuronal cytoplasm. Biol. Pharm. Bull. 2013, 36, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Vereker, E.; O'Donnell, E.; Lynch, M.A. The inhibitory effect of interleukin-1β on long-term potentiation is coupled with increased activity of stress-activated protein kinases. J. Neurosci. 2000, 20, 6811–6819. [Google Scholar] [PubMed]
- Dolu, N.; Khan, A.; Dokutan, S. Effect of vitamin E administration on learning of the young male rats. J. Exp. Neurosci. 2015, 9, 81–85. [Google Scholar] [PubMed]
- Xie, Z.; Sastry, B.R. Induction of hippocampal long-term potentiation by α-tocopherol. Brain Res. 1993, 604, 173–179. [Google Scholar] [CrossRef]
- Yang, T.T.; Wang, S.J. Facilitatory effect of glutamate exocytosis from rat cerebrocortical nerve terminals by α-tocopherol, a major vitamin E component. Neurochem. Int. 2008, 52, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Eidi, A.; Eidi, M.; Mahmoodi, G.; Oryan, S. Effect of vitamin E on memory retention in rats: Possible involvement of cholinergic system. Eur. Neuropsychopharmacol. 2006, 16, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Villayandre, B.; Paniagua, M.A.; Fernandez-Lopez, A.; Calvo, P. Effect of δ-aminolevulinic acid and vitamin E treatments on the N-methyl-d-aspartate receptor at different ages in the striatum of rat brain. Brain Res. 2006, 1114, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Martinez Villayandre, B.; Paniagua, M.A.; Fernandez-Lopez, A.; Chinchetru, M.A.; Calvo, P. Effect of vitamin E treatment on N-methyl-d-aspartate receptor at different ages in the rat brain. Brain Res. 2004, 1028, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Rafnsson, S.B.; Dilis, V.; Trichopoulou, A. Antioxidant nutrients and age-related cognitive decline: A systematic review of population-based cohort studies. Eur. J. Nutr. 2013, 52, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Shen, L.; Ji, H.F. Dietary intakes of vitamin E, vitamin C, and β-carotene and risk of Alzheimer's disease: A meta-analysis. J. Alzheimers Dis. 2012, 31, 253–258. [Google Scholar] [PubMed]
- Crichton, G.E.; Bryan, J.; Murphy, K.J. Dietary antioxidants, cognitive function and dementia—A systematic review. Plant Foods Hum. Nutr. 2013, 68, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Nooyens, A.C.; Milder, I.E.; van Gelder, B.M.; Bueno-de-Mesquita, H.B.; van Boxtel, M.P.; Verschuren, W.M. Diet and cognitive decline at middle age: The role of antioxidants. Br. J. Nutr. 2015, 113, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Kareholt, I.; Hooshmand, B.; Cecchetti, R.; Fratiglioni, L.; Soininen, H.; Laatikainen, T.; Mecocci, P.; Kivipelto, M. Serum levels of vitamin E forms and risk of cognitive impairment in a finnish cohort of older adults. Exp. Gerontol. 2013, 48, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Xu, W.; Kivipelto, M.; Costanzi, E.; Ercolani, S.; Pigliautile, M.; Cecchetti, R.; Baglioni, M.; Simmons, A.; Soininen, H.; et al. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol. Aging 2012, 33, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Farina, N.; Isaac, M.G.; Clark, A.R.; Rusted, J.; Tabet, N. Vitamin E for Alzheimer's dementia and mild cognitive impairment. Cochrane Database Syst. Rev. 2012, 11, CD002854. [Google Scholar] [PubMed]
- Morris, M.C.; Schneider, J.A.; Li, H.; Tangney, C.C.; Nag, S.; Bennett, D.A.; Honer, W.G.; Barnes, L.L. Brain tocopherols related to Alzheimer's disease neuropathology in humans. Alzheimers Dement. 2015, 11, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.; Roupas, P. Effect of vitamin intake on cognitive decline in older adults: Evaluation of the evidence. J. Nutr. Health Aging 2015, 19, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.C.; Holroyd-Leduc, J.M.; Poulin, M.J.; Hogan, D.B. Effect of nutrients, dietary supplements and vitamins on cognition: A systematic review and meta-analysis of randomized controlled trials. Can. Geriatr. J. 2015, 18, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Aisen, P.S.; Andrews, H.F.; Tsai, W.Y.; Lai, F.; Dalton, A.J. Vitamin E in aging persons with Down syndrome: A randomized, placebo-controlled clinical trial. Neurology 2016, 86, 2071–2076. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Arozamena, C.; Marti-Mari, O.; Estrada, M.; de la Fuente Revenga, M.; Rodriguez-Franco, M.I. Recent advances in neurogenic small molecules as innovative treatments for neurodegenerative diseases. Molecules 2016, 21, 1165. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, S. Neural repair in the adult brain. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Rusznak, Z.; Henskens, W.; Schofield, E.; Kim, W.S.; Fu, Y. Adult neurogenesis and gliogenesis: Possible mechanisms for neurorestoration. Exp. Neurobiol. 2016, 25, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.; Das, G.D. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 1965, 124, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Ming, G.L.; Song, H. Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Wiskott, L.; Gage, F.H. Functional significance of adult neurogenesis. Curr. Opin. Neurobiol. 2004, 14, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Ambrogini, P.; Lattanzi, D.; Ciuffoli, S.; Agostini, D.; Bertini, L.; Stocchi, V.; Santi, S.; Cuppini, R. Morpho-functional characterization of neuronal cells at different stages of maturation in granule cell layer of adult rat dentate gyrus. Brain Res. 2004, 1017, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Ambrogini, P.; Minelli, A.; Lattanzi, D.; Ciuffoli, S.; Fanelli, M.; Cuppini, R. Synaptically-silent immature neurons show GABA and glutamate receptor-mediated currents in adult rat dentate gyrus. Arch. Ital. Biol. 2006, 144, 115–126. [Google Scholar] [PubMed]
- Toni, N.; Schinder, A.F. Maturation and functional integration of new granule cells into the adult hippocampus. Cold Spring Harb. Perspect. Biol. 2016, 8, a018903. [Google Scholar] [CrossRef] [PubMed]
- Ambrogini, P.; Cuppini, R.; Cuppini, C.; Ciaroni, S.; Cecchini, T.; Ferri, P.; Sartini, S.; del Grande, P. Spatial learning affects immature granule cell survival in adult rat dentate gyrus. Neurosci. Lett. 2000, 286, 21–24. [Google Scholar] [CrossRef]
- Ambrogini, P.; Orsini, L.; Mancini, C.; Ferri, P.; Ciaroni, S.; Cuppini, R. Learning may reduce neurogenesis in adult rat dentate gyrus. Neurosci. Lett. 2004, 359, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Gage, F.H. Experience-dependent regulation of adult hippocampal neurogenesis: Effects of long-term stimulation and stimulus withdrawal. Hippocampus 1999, 9, 321–332. [Google Scholar] [CrossRef]
- Kitamura, T.; Saitoh, Y.; Takashima, N.; Murayama, A.; Niibori, Y.; Ageta, H.; Sekiguchi, M.; Sugiyama, H.; Inokuchi, K. Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 2009, 139, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Lledo, P.M.; Alonso, M.; Grubb, M.S. Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 2006, 7, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Temple, S.; Alvarez-Buylla, A. Stem cells in the adult mammalian central nervous system. Curr. Opin. Neurobiol. 1999, 9, 135–141. [Google Scholar] [CrossRef]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Jinno, S. Aging affects new cell production in the adult hippocampus: A quantitative anatomic review. J. Chem. Neuroanat. 2016, 76, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011, 476, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Femenia, T.; Gomez-Galan, M.; Lindskog, M.; Magara, S. Dysfunctional hippocampal activity affects emotion and cognition in mood disorders. Brain Res. 2012, 1476, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhong, C.; Bonaguidi, M.A.; Sun, G.J.; Hsu, D.; Gu, Y.; Meletis, K.; Huang, Z.J.; Ge, S.; Enikolopov, G.; et al. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 2012, 489, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Grote, H.E.; Hannan, A.J. Regulators of adult neurogenesis in the healthy and diseased brain. Clin. Exp. Pharmacol. Physiol. 2007, 34, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, V.A.; Vadodaria, K.C.; Jha, S. Neurotransmitter regulation of adult neurogenesis: Putative therapeutic targets. CNS Neurol. Disord. Drug Targets 2007, 6, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Guterman, P.; Yagi, S.; Chow, C.; Galea, L.A. Hippocampal learning, memory, and neurogenesis: Effects of sex and estrogens across the lifespan in adults. Horm. Behav. 2015, 74, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Ciaroni, S.; Cecchini, T.; Ferri, P.; Cuppini, R.; Ambrogini, P.; Santi, S.; Benedetti, S.; del Grande, P.; Papa, S. Neural precursor proliferation and newborn cell survival in the adult rat dentate gyrus are affected by vitamin E deficiency. Neurosci. Res. 2002, 44, 369–377. [Google Scholar] [CrossRef]
- Ciaroni, S.; Cuppini, R.; Cecchini, T.; Ferri, P.; Ambrogini, P.; Cuppini, C.; del Grande, P. Neurogenesis in the adult rat dentate gyrus is enhanced by vitamin E deficiency. J. Comp. Neurol. 1999, 411, 495–502. [Google Scholar] [CrossRef]
- Cuppini, R.; Ciaroni, S.; Cecchini, T.; Ambrogini, P.; Ferri, P.; del Grande, P.; Papa, S. α-tocopherol controls cell proliferation in the adult rat dentate gyrus. Neurosci. Lett. 2001, 303, 198–200. [Google Scholar] [CrossRef]
- Cecchini, T.; Cuppini, R.; Ciaroni, S.; Barili, P.; de Matteis, R.; del Grande, P. Changes in the number of primary sensory neurons in normal and vitamin-E-deficient rats during aging. Somatosens. Mot. Res. 1995, 12, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, T.; Cuppini, R.; Ciaroni, S.; de Matteis, R.; del Grande, P. Increased number of sciatic sensory neurons in vitamin-E-deficient rats. Somatosens. Mot. Res. 1994, 11, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, T.; Cuppini, R.; Ciaroni, S.; del Grande, P. Increased number of dorsal root ganglion neurons in vitamin-E-deficient rats. Somatosens. Mot. Res. 1993, 10, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Devor, M.; Govrin-Lippmann, R.; Frank, I.; Raber, P. Proliferation of primary sensory neurons in adult rat dorsal root ganglion and the kinetics of retrograde cell loss after sciatic nerve section. Somatosens. Res. 1985, 3, 139–167. [Google Scholar] [CrossRef] [PubMed]
- Goldring, C.E.; Rice-Evans, C.A.; Burdon, R.H.; Rao, R.; Haq, I.; Diplock, A.T. α-tocopherol uptake and its influence on cell proliferation and lipid peroxidation in transformed and nontransformed baby hamster kidney cells. Arch. Biochem. Biophys. 1993, 303, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Ricciarelli, R.; Zingg, J.M. Non-antioxidant molecular functions of α-tocopherol (vitamin E). FEBS Lett. 2002, 519, 8–10. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E 80th anniversary: A double life, not only fighting radicals. IUBMB Life 2001, 52, 71–76. [Google Scholar] [PubMed]
- Zingg, J.M.; Azzi, A. Non-antioxidant activities of vitamin E. Curr. Med. Chem. 2004, 11, 1113–1133. [Google Scholar] [CrossRef] [PubMed]
- Rimbach, G.; Minihane, A.M.; Majewicz, J.; Fischer, A.; Pallauf, J.; Virgli, F.; Weinberg, P.D. Regulation of cell signalling by vitamin E. Proc. Nutr. Soc. 2002, 61, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Kelly, F.J.; Salonen, J.T.; Neuzil, J.; Zingg, J.M.; Azzi, A. The european perspective on vitamin E: Current knowledge and future research. Am. J. Clin. Nutr. 2002, 76, 703–716. [Google Scholar] [PubMed]
- Boscoboinik, D.; Szewczyk, A.; Azzi, A. α-tocopherol (vitamin E) regulates vascular smooth muscle cell proliferation and protein kinase C activity. Arch. Biochem. Biophys. 1991, 286, 264–269. [Google Scholar] [CrossRef]
- Mahoney, C.W.; Azzi, A. Vitamin E inhibits protein kinase C activity. Biochem. Biophys. Res. Commun. 1988, 154, 694–697. [Google Scholar] [CrossRef]
- Martin-Nizard, F.; Boullier, A.; Fruchart, J.C.; Duriez, P. α-tocopherol but not β-tocopherol inhibits thrombin-induced PKC activation and endothelin secretion in endothelial cells. J. Cardiovasc. Risk 1998, 5, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Azzi, A. Regulation of recombinant PKC α activity by protein phosphatase 1 and protein phosphatase 2A. Arch. Biochem. Biophys. 1998, 355, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Tasinato, A.; Clement, S.; Ozer, N.K.; Boscoboinik, D.; Azzi, A. α-tocopherol specifically inactivates cellular protein kinase C α by changing its phosphorylation state. Biochem. J. 1998, 334, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Adams-Huet, B.; Fuller, C.J.; Jialal, I. Dose-response comparison of RRR-α-tocopherol and all-racemic α-tocopherol on LDL oxidation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Li, D.; Jialal, I. The effects of α tocopherol supplementation on monocyte function. Decreased lipid oxidation, interleukin 1β secretion, and monocyte adhesion to endothelium. J. Clin. Investig. 1996, 98, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Farhat, J.H.; Loscalzo, J.; Keaney, J.F., Jr. α-tocopherol inhibits aggregation of human platelets by a protein kinase C-dependent mechanism. Circulation 1996, 94, 2434–2440. [Google Scholar] [CrossRef] [PubMed]
- Hehenberger, K.; Hansson, A. High glucose-induced growth factor resistance in human fibroblasts can be reversed by antioxidants and protein kinase C-inhibitors. Cell Biochem. Funct. 1997, 15, 197–201. [Google Scholar] [CrossRef]
- Kanno, T.; Utsumi, T.; Kobuchi, H.; Takehara, Y.; Akiyama, J.; Yoshioka, T.; Horton, A.A.; Utsumi, K. Inhibition of stimulus-specific neutrophil superoxide generation by α-tocopherol. Free Radic. Res. 1995, 22, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Weber, T.; Schroder, A.; Lu, M.; Ostermann, G.; Gellert, N.; Mayne, G.C.; Olejnicka, B.; Negre-Salvayre, A.; Sticha, M.; et al. Induction of cancer cell apoptosis by α-tocopheryl succinate: Molecular pathways and structural requirements. FASEB J. 2001, 15, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, P.W.; McIntyre, B.S.; Gapor, A.; Briski, K.P. Vitamin E inhibition of normal mammary epithelial cell growth is associated with a reduction in protein kinase Cα activation. Cell Prolif. 2001, 34, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Ishii, H.; Isogai, S. Protective effect of d-α-tocopherol on the function of human mesangial cells exposed to high glucose concentrations. Metabolism 1997, 46, 779–784. [Google Scholar] [CrossRef]
- Tasinato, A.; Boscoboinik, D.; Bartoli, G.M.; Maroni, P.; Azzi, A. d-α-tocopherol inhibition of vascular smooth muscle cell proliferation occurs at physiological concentrations, correlates with protein kinase C inhibition, and is independent of its antioxidant properties. Proc. Natl. Acad. Sci. USA 1995, 92, 12190–12194. [Google Scholar] [CrossRef] [PubMed]
- Yasunari, K.; Kohno, M.; Kano, H.; Yokokawa, K.; Minami, M.; Yoshikawa, J. Antioxidants improve impaired insulin-mediated glucose uptake and prevent migration and proliferation of cultured rabbit coronary smooth muscle cells induced by high glucose. Circulation 1999, 99, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Boscoboinik, D.; Clement, S.; Ozer, N.; Ricciarelli, R.; Stocker, A. Vitamin E mediated response of smooth muscle cell to oxidant stress. Diabetes Res. Clin. Pract. 1999, 45, 191–198. [Google Scholar] [CrossRef]
- Kasparek, S. Chemistry of tocopherols and tocotrienols. In Vitamin E: A Comprehensive Treatise; Machlin, L.J., Ed.; Marcel Dekker: New York, NY, USA, 1980; pp. 7–65. [Google Scholar]
- Ceballos-Picot, I. The Role of Oxidative Stress in Neuronal Death; Springer-Verlag Berlin Heidelberg: Berlin, Germany, 1997. [Google Scholar]
- De Nigris, F.; Franconi, F.; Maida, I.; Palumbo, G.; Anania, V.; Napoli, C. Modulation by α- and γ-tocopherol and oxidized low-density lipoprotein of apoptotic signaling in human coronary smooth muscle cells. Biochem. Pharmacol. 2000, 59, 1477–1487. [Google Scholar] [CrossRef]
- Numakawa, Y.; Numakawa, T.; Matsumoto, T.; Yagasaki, Y.; Kumamaru, E.; Kunugi, H.; Taguchi, T.; Niki, E. Vitamin E protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J. Neurochem. 2006, 97, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Zeiher, A.M.; Dimmeler, S. Vitamin C and E prevent lipopolysaccharide-induced apoptosis in human endothelial cells by modulation of Bcl-2 and Bax. Eur. J. Pharmacol. 1996, 317, 407–411. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Hidaka, R.; Fujimaki, S.; Asashima, M.; Kuwabara, T. Micrornas and epigenetics in adult neurogenesis. Adv. Genet. 2014, 86, 27–44. [Google Scholar] [PubMed]
- Rimbach, G.; Moehring, J.; Huebbe, P.; Lodge, J.K. Gene-regulatory activity of α-tocopherol. Molecules 2010, 15, 1746–1761. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Cui, J.G.; Li, Y.Y.; Zhao, Y.; Culicchia, F.; Lukiw, W.J. Micro RNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci. Lett. 2010, 476, 18–22. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Epilepsy: Epidemiology, Aetiology and Prognosis; WHO Fact sheets N°165; World Health Organization: Geneva, Switzerland, 2001. [Google Scholar]
- Aguiar, C.C.; Almeida, A.B.; Araujo, P.V.; de Abreu, R.N.; Chaves, E.M.; do Vale, O.C.; Macedo, D.S.; Woods, D.J.; Fonteles, M.M.; Vasconcelos, S.M. Oxidative stress and epilepsy: Literature review. Oxid. Med. Cell. Longev. 2012, 2012, 795259. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J. Epileptogenesis: More than just the latent period. Epilepsy Curr. 2016, 16, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Engel, J., Jr. Mesial temporal lobe epilepsy: What have we learned? Neuroscientist 2001, 7, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Sutula, T.P. Seizure-induced plasticity and adverse long-term effects of early-life seizures. Ann. Neurol. 2004, 56, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Iori, V.; Frigerio, F.; Vezzani, A. Modulation of neuronal excitability by immune mediators in epilepsy. Curr. Opin. Pharmacol. 2016, 26, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Pernot, F.; Heinrich, C.; Barbier, L.; Peinnequin, A.; Carpentier, P.; Dhote, F.; Baille, V.; Beaup, C.; Depaulis, A.; Dorandeu, F. Inflammatory changes during epileptogenesis and spontaneous seizures in a mouse model of mesiotemporal lobe epilepsy. Epilepsia 2011, 52, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Roseti, C.; Fucile, S.; Lauro, C.; Martinello, K.; Bertollini, C.; Esposito, V.; Mascia, A.; Catalano, M.; Aronica, E.; Limatola, C.; et al. Fractalkine/CX3CL1 modulates GABAA currents in human temporal lobe epilepsy. Epilepsia 2013, 54, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Aycicek, A.; Iscan, A. The effects of carbamazepine, valproic acid and phenobarbital on the oxidative and antioxidative balance in epileptic children. Eur. Neurol. 2007, 57, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Yis, U.; Seckin, E.; Kurul, S.H.; Kuralay, F.; Dirik, E. Effects of epilepsy and valproic acid on oxidant status in children with idiopathic epilepsy. Epilepsy Res. 2009, 84, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Ezz, H.S.; Khadrawy, Y.A.; Noor, N.A. The neuroprotective effect of curcumin and nigella sativa oil against oxidative stress in the pilocarpine model of epilepsy: A comparison with valproate. Neurochem. Res. 2011, 36, 2195–2204. [Google Scholar] [PubMed]
- Junior, J.S.; de Almeida, A.A.; Tome Ada, R.; Cito, A.M.; Saffi, J.; de Freitas, R.M. Evaluation of possible antioxidant and anticonvulsant effects of the ethyl acetate fraction from Platonia insignis Mart. (Bacuri) on epilepsy models. Epilepsy Behav. 2011, 22, 678–684. [Google Scholar] [PubMed]
- Gupta, Y.K.; Briyal, S.; Chaudhary, G. Protective effect of trans-resveratrol against kainic acid-induced seizures and oxidative stress in rats. Pharmacol. Biochem. Behav. 2002, 71, 245–249. [Google Scholar] [CrossRef]
- Shin, S.M.; Cho, I.J.; Kim, S.G. Resveratrol protects mitochondria against oxidative stress through AMP-activated protein kinase-mediated glycogen synthase kinase-3β inhibition downstream of poly(ADP-ribose)polymerase-LKB1 pathway. Mol. Pharmacol. 2009, 76, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Zaja-Milatovic, S.; Gupta, R.C.; Aschner, M.; Montine, T.J.; Milatovic, D. Pharmacologic suppression of oxidative damage and dendritic degeneration following kainic acid-induced excitotoxicity in mouse cerebrum. Neurotoxicology 2008, 29, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Simeone, K.A.; Matthews, S.A.; Samson, K.K.; Simeone, T.A. Targeting deficiencies in mitochondrial respiratory complex I and functional uncoupling exerts anti-seizure effects in a genetic model of temporal lobe epilepsy and in a model of acute temporal lobe seizures. Exp. Neurol. 2014, 251, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, V.M.; Kryzhanovskii, G.N.; Kovalenko, V.S.; Pronina, I.G.; Nikushkin, E.V. α-tocopherol in the complex treatment of several forms of epilepsy. Zh. Nevropatol. Psikhiatr. Im. SS Korsakova 1984, 84, 892–897. [Google Scholar]
- Mehvari, J.; Motlagh, F.G.; Najafi, M.; Ghazvini, M.R.; Naeini, A.A.; Zare, M. Effects of vitamin E on seizure frequency, electroencephalogram findings, and oxidative stress status of refractory epileptic patients. Adv. Biomed. Res. 2016, 5, 36. [Google Scholar] [PubMed]
- Galic, M.A.; Riazi, K.; Heida, J.G.; Mouihate, A.; Fournier, N.M.; Spencer, S.J.; Kalynchuk, L.E.; Teskey, G.C.; Pittman, Q.J. Postnatal inflammation increases seizure susceptibility in adult rats. J. Neurosci. 2008, 28, 6904–6913. [Google Scholar] [CrossRef] [PubMed]
- Somera-Molina, K.C.; Nair, S.; Van Eldik, L.J.; Watterson, D.M.; Wainwright, M.S. Enhanced microglial activation and proinflammatory cytokine upregulation are linked to increased susceptibility to seizures and neurologic injury in a "two-hit" seizure model. Brain Res. 2009, 1282, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Noe, F.; Zardoni, D.; Vaghi, V.; Sifringer, M.; Vezzani, A. Interleukin converting enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1β production. Neurobiol. Dis. 2008, 31, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.A.; Wang, L.; Ribak, C.E. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 2008, 49, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Turrin, N.P.; Rivest, S. Unraveling the molecular details involved in the intimate link between the immune and neuroendocrine systems. Exp. Biol. Med. 2004, 229, 996–1006. [Google Scholar]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Schafers, M.; Sorkin, L. Effect of cytokines on neuronal excitability. Neurosci. Lett. 2008, 437, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav. Immun. 2008, 22, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Annahazi, A.; Mracsko, E.; Sule, Z.; Karg, E.; Penke, B.; Bari, F.; Farkas, E. Pre-treatment and post-treatment with α-tocopherol attenuates hippocampal neuronal damage in experimental cerebral hypoperfusion. Eur. J. Pharmacol. 2007, 571, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, O.; Gonzalez-Castaneda, R.E.; Huerta, M.; Luquin, S.; Gomez-Pinedo, U.; Sanchez-Almaraz, E.; Navarro-Ruiz, A.; Garcia-Estrada, J. Beneficial effects of α-lipoic acid plus vitamin E on neurological deficit, reactive gliosis and neuronal remodeling in the penumbra of the ischemic rat brain. Neurosci. Lett. 2002, 321, 100–104. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Mrak, R.E.; Griffin, W.S. Vitamin E suppression of microglial activation is neuroprotective. J. Neurosci. Res. 2001, 66, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Stolzing, A.; Widmer, R.; Jung, T.; Voss, P.; Grune, T. Tocopherol-mediated modulation of age-related changes in microglial cells: Turnover of extracellular oxidized protein material. Free Radic. Biol. Med. 2006, 40, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Pitkanen, A.; Sutula, T.P. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002, 1, 173–181. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
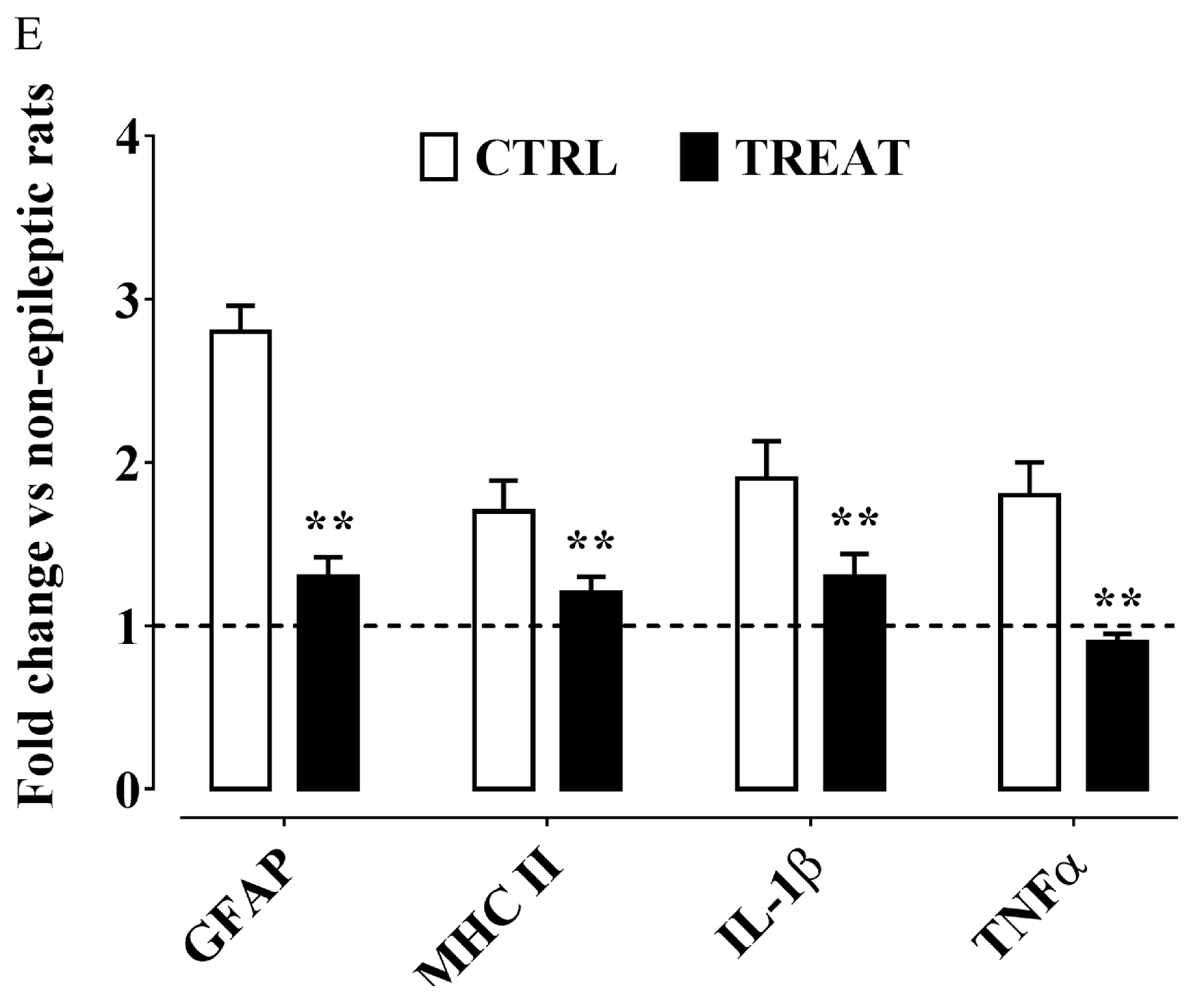
- Ambrogini, P.; Minelli, A.; Galati, C.; Betti, M.; Lattanzi, D.; Ciffolilli, S.; Piroddi, M.; Galli, F.; Cuppini, R. Post-seizure α-tocopherol treatment decreases neuroinflammation and neuronal degeneration induced by status epilepticus in rat hippocampus. Mol. Neurobiol. 2014, 50, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Eid, T.; Tu, N.; Lee, T.S.; Lai, J.C. Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem. Int. 2013, 63, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Azzi, A. Present trends in vitamin E research. BioFactors 2010, 36, 33–42. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrogini, P.; Betti, M.; Galati, C.; Di Palma, M.; Lattanzi, D.; Savelli, D.; Galli, F.; Cuppini, R.; Minelli, A. α-Tocopherol and Hippocampal Neural Plasticity in Physiological and Pathological Conditions. Int. J. Mol. Sci. 2016, 17, 2107. https://doi.org/10.3390/ijms17122107
Ambrogini P, Betti M, Galati C, Di Palma M, Lattanzi D, Savelli D, Galli F, Cuppini R, Minelli A. α-Tocopherol and Hippocampal Neural Plasticity in Physiological and Pathological Conditions. International Journal of Molecular Sciences. 2016; 17(12):2107. https://doi.org/10.3390/ijms17122107
Chicago/Turabian StyleAmbrogini, Patrizia, Michele Betti, Claudia Galati, Michael Di Palma, Davide Lattanzi, David Savelli, Francesco Galli, Riccardo Cuppini, and Andrea Minelli. 2016. "α-Tocopherol and Hippocampal Neural Plasticity in Physiological and Pathological Conditions" International Journal of Molecular Sciences 17, no. 12: 2107. https://doi.org/10.3390/ijms17122107
APA StyleAmbrogini, P., Betti, M., Galati, C., Di Palma, M., Lattanzi, D., Savelli, D., Galli, F., Cuppini, R., & Minelli, A. (2016). α-Tocopherol and Hippocampal Neural Plasticity in Physiological and Pathological Conditions. International Journal of Molecular Sciences, 17(12), 2107. https://doi.org/10.3390/ijms17122107