
Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma

 ,
, 
Abstract
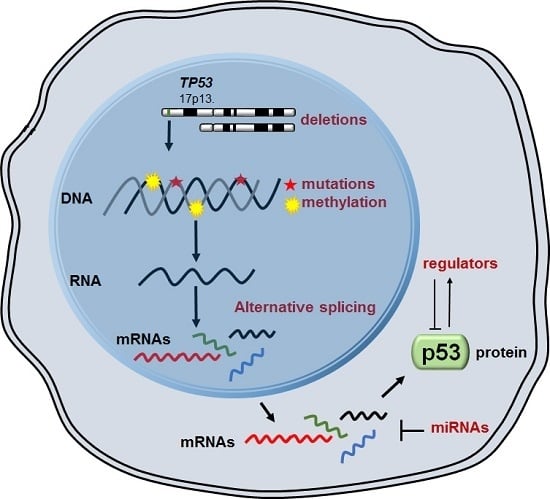
:
{kind=link}
{kind=link}
1. Introduction
2. Alterations of TP53 Gene in Human Cancers and Particularly in Multiple Myeloma (MM)
3. Alterations of p53 Regulators
4. Deregulation of p53 through Epigenetic Modifications
4.1. TP53 DNA Methylation
4.2. miRNAs that Regulate the Activity of p53
5. Altered Pattern of Human p53 Isoforms
6. p53-Based Antitumor Therapy
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ashcroft, M.; Kubbutat, M.H.; Vousden, K.H. Regulation of p53 function and stability by phosphorylation. Mol. Cell. Biol. 1999, 19, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.M.; Gu, W. Understanding the non-canonical pathways involved in p53-mediated tumor suppression. Carcinogenesis 2014, 35, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Wasylishen, A.R.; Lozano, G. Attenuating the p53 Pathway in Human Cancers: Many Means to the Same End. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Lens, D.; de Schouwer, P.J.; Hamoudi, R.A.; Abdul-Rauf, M.; Farahat, N.; Matutes, E.; Crook, T.; Dyer, M.J.; Catovsky, D. p53 abnormalities in B-cell prolymphocytic leukemia. Blood 1997, 89, 2015–2023. [Google Scholar] [PubMed]
- Stirewalt, D.L.; Kopecky, K.J.; Meshinchi, S.; Appelbaum, F.R.; Slovak, M.L.; Willman, C.L.; Radich, J.P. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood 2001, 97, 3589–3595. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, E.W. p53 gene mutations in lymphoid diseases and their possible relevance to drug resistance. Leuk. Lymphoma 1995, 17, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Wattel, E.; Preudhomme, C.; Hecquet, B.; Vanrumbeke, M.; Quesnel, B.; Dervite, I.; Morel, P.; Fenaux, P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood 1994, 84, 3148–3157. [Google Scholar] [PubMed]
- Lionetti, M.; Barbieri, M.; Manzoni, M.; Fabris, S.; Bandini, C.; Todoerti, K.; Nozza, F.; Rossi, D.; Musto, P.; Baldini, L.; et al. Molecular spectrum of TP53 mutations in plasma cell dyscrasias by next generation sequencing: An Italian cohort study and overview of the literature. Oncotarget 2016, 7, 21353–21361. [Google Scholar] [PubMed]
- Dimopoulos, M.; Kyle, R.; Fermand, J.-P.; Rajkumar, S.V.; San Miguel, J.; Chanan-Khan, A.; Ludwig, H.; Joshua, D.; Mehta, J.; Gertz, M.; et al. International Myeloma Workshop Consensus Panel 3 Consensus recommendations for standard investigative workup: Report of the International Myeloma Workshop Consensus Panel 3. Blood 2011, 117, 4701–4705. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Mateos, M.-V.; Gutierrez, N.C.; Rajkumar, S.V.; San Miguel, J.F. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood 2013, 121, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar] [PubMed]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Eldar, A.; Rozenberg, H.; Diskin-Posner, Y.; Rohs, R.; Shakked, Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013, 41, 8748–8759. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 2007, 97, 1–23. [Google Scholar] [PubMed]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci. 2011, 32, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.D.; Ross, F.M.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gonzalez, D.; Walker, B.A.; Hockley, S.L.; Wardell, C.P.; et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes. Chromosomes Cancer 2011, 50, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Glebov, O.; Bergsagel, P.L.; Kuehl, W.M. Genetic events in the pathogenesis of multiple myeloma. Best Pract. Res. Clin. Haematol. 2007, 20, 571–596. [Google Scholar] [CrossRef] [PubMed]
- Xu-Monette, Z.Y.; Medeiros, L.J.; Li, Y.; Orlowski, R.Z.; Andreeff, M.; Bueso-Ramos, C.E.; Greiner, T.C.; McDonnell, T.J.; Young, K.H. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood 2012, 119, 3668–3683. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Price-Troska, T.; Gonzalez-Paz, N.; van Wier, S.; Jacobus, S.; Blood, E.; Henderson, K.; Oken, M.; van Ness, B.; Greipp, P.; et al. Clinical significance of TP53 mutation in myeloma. Leukemia 2007, 21, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Drach, J.; Ackermann, J.; Fritz, E.; Krömer, E.; Schuster, R.; Gisslinger, H.; DeSantis, M.; Zojer, N.; Fiegl, M.; Roka, S.; et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood 1998, 92, 802–809. [Google Scholar] [PubMed]
- Gutiérrez, N.C.; Castellanos, M.V.; Martín, M.L.; Mateos, M.V.; Hernández, J.M.; Fernández, M.; Carrera, D.; Rosiñol, L.; Ribera, J.M.; Ojanguren, J.M.; et al. Prognostic and biological implications of genetic abnormalities in multiple myeloma undergoing autologous stem cell transplantation: t(4;14) is the most relevant adverse prognostic factor, whereas RB deletion as a unique abnormality is not associated with adverse prognosis. Leukemia 2007, 21, 143–150. [Google Scholar] [PubMed]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; van Ness, B.; van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-H.; Qi, C.X.Y.; Saha, M.N.; Chang, H. p53 nuclear expression correlates with hemizygous TP53 deletion and predicts an adverse outcome for patients with relapsed/refractory multiple myeloma treated with lenalidomide. Am. J. Clin. Pathol. 2012, 137, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Greipp, P.R.; Litzow, M.R.; Henderson, K.J.; van Wier, S.A.; Ahmann, G.J.; Fonseca, R. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and -17p13 in myeloma patients treated with high-dose therapy. Blood 2005, 106, 2837–2840. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Zepeda, V.H.; Neme-Yunes, Y.; Braggio, E. Chromosome abnormalities defined by conventional cytogenetics in plasma cell leukemia: What have we learned about its biology? Eur. J. Haematol. 2011, 87, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Elnenaei, M.O.; Gruszka-Westwood, A.M.; A’Hernt, R.; Matutes, E.; Sirohi, B.; Powles, R.; Catovsky, D. Gene abnormalities in multiple myeloma; the relevance of TP53, MDM2, and CDKN2A. Haematologica 2003, 88, 529–537. [Google Scholar] [PubMed]
- Deng, S.; Xu, Y.; An, G.; Sui, W.; Zou, D.; Zhao, Y.; Qi, J.; Li, F.; Hao, M.; Qiu, L. Features of extramedullary disease of multiple myeloma: High frequency of p53 deletion and poor survival: A retrospective single-center study of 834 cases. Clin. Lymphoma Myeloma Leuk. 2015, 15, 286–291. [Google Scholar] [CrossRef] [PubMed]
- López-Anglada, L.; Gutiérrez, N.C.; García, J.L.; Mateos, M.V.; Flores, T.; San Miguel, J.F. P53 deletion may drive the clinical evolution and treatment response in multiple myeloma. Eur. J. Haematol. 2010, 84, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.; Hielscher, T.; Seckinger, A.; Hose, D.; Mai, E.K.; Raab, M.S.; Goldschmidt, H.; Jauch, A.; Hillengass, J. Baseline characteristics, chromosomal alterations, and treatment affecting prognosis of deletion 17p in newly diagnosed myeloma. Am. J. Hematol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mangiacavalli, S.; Pochintesta, L.; Cocito, F.; Pompa, A.; Bernasconi, P.; Cazzola, M.; Corso, A. Correlation between burden of 17P13.1 alteration and rapid escape to plasma cell leukaemia in multiple myeloma. Br. J. Haematol. 2013, 162, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Qi, C.; Yi, Q.-L.; Reece, D.; Stewart, A.K. p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood 2005, 105, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Rosiñol, L.; Oriol, A.; Teruel, A.I.; Hernández, D.; López-Jiménez, J.; de la Rubia, J.; Granell, M.; Besalduch, J.; Palomera, L.; González, Y.; et al. Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pretransplantation therapy in multiple myeloma: A randomized phase 3 PETHEMA/GEM study. Blood 2012, 120, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Li, Z.; Tai, Y.-T.; Acharya, C.; Li, Q.; Qin, X.; Yi, S.; Xu, Y.; Feng, X.; Li, C.; et al. The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2148–2156. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Fakharzadeh, S.S.; Trusko, S.P.; George, D.L. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991, 10, 1565–1569. [Google Scholar] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Poyurovsky, M.V.; Priest, C.; Kentsis, A.; Borden, K.; Pan, Z.; Pavletich, N.; Prives, C. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 2007, 26, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Bradley, A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 15608–15612. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Quesnel, B.; Preudhomme, C.; Oscier, D.; Lepelley, P.; Collyn-d’Hooghe, M.; Facon, T.; Zandecki, M.; Fenaux, P. Over-expression of the MDM2 gene is found in some cases of haematological malignancies. Br. J. Haematol. 1994, 88, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Teoh, G.; Urashima, M.; Ogata, A.; Chauhan, D.; DeCaprio, J.A.; Treon, S.P.; Schlossman, R.L.; Anderson, K.C. MDM2 protein overexpression promotes proliferation and survival of multiple myeloma cells. Blood 1997, 90, 1982–1992. [Google Scholar] [PubMed]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Wang, X.; Feng, Z. The regulation of the p53/MDM2 feedback loop by microRNAs. RNA Dis. Houst. Tex 2015, 2, e502. [Google Scholar]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [PubMed]
- Shvarts, A.; Bazuine, M.; Dekker, P.; Ramos, Y.F.; Steegenga, W.T.; Merckx, G.; van Ham, R.C.; van der Houven van Oordt, W.; van der Eb, A.J.; Jochemsen, A.G. Isolation and identification of the human homolog of a new p53-binding protein, Mdmx. Genomics 1997, 43, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.-G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S.; et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, S.; Gembarska, A.; Denecker, G.; Maetens, M.; Naessens, M.; Haigh, K.; Haigh, J.J.; Marine, J.-C. Widespread overexpression of epitope-tagged Mdm4 does not accelerate tumor formation in vivo. Mol. Cell. Biol. 2010, 30, 5394–5405. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vousden, K.H. The role of ubiquitin modification in the regulation of p53. Biochim. Biophys. Acta 2014, 1843, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Collavin, L.; Lunardi, A.; del Sal, G. p53-family proteins and their regulators: Hubs and spokes in tumor suppression. Cell Death Differ. 2010, 17, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.-S.; Liang, R.; Leung, M.-H.; Kwong, Y.-L. Aberrant gene methylation implicated in the progression of monoclonal gammopathy of undetermined significance to multiple myeloma. J. Clin. Pathol. 2007, 60, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.; Mass, M.J. CpG Methylation Inactivates the Transcriptional Activity of the Promoter of the Humanp53Tumor Suppressor Gene. Biochem. Biophys. Res. Commun. 1997, 235, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Pogribna, M.; Christman, J.K.; James, S.J. Single-site methylation within the p53 promoter region reduces gene expression in a reporter gene construct: Possible in vivo relevance during tumorigenesis. Cancer Res. 2000, 60, 588–594. [Google Scholar] [PubMed]
- Hossain, M.M.; Ray, S.K. EWS Knockdown and Taxifolin Treatment Induced Differentiation and Removed DNA Methylation from p53 Promoter to Promote Expression of Puma and Noxa for Apoptosis in Ewing’s Sarcoma. J. Cancer Ther. 2014, 5, 1092–1113. [Google Scholar] [CrossRef] [PubMed]
- Jesionek-Kupnicka, D.; Szybka, M.; Malachowska, B.; Fendler, W.; Potemski, P.; Piaskowski, S.; Jaskolski, D.; Papierz, W.; Skowronski, W.; Och, W.; et al. TP53 promoter methylation in primary glioblastoma: Relationship with TP53 mRNA and protein expression and mutation status. DNA Cell Biol. 2014, 33, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Zornoza, A.; Agirre, X.; Martín-Palanco, V.; Martín-Subero, J.I.; San José-Eneriz, E.; Garate, L.; Álvarez, S.; Miranda, E.; Rodríguez-Otero, P.; Rifón, J.; et al. Frequent and simultaneous epigenetic inactivation of TP53 pathway genes in acute lymphoblastic leukemia. PLoS ONE 2011, 6, e17012. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Novo, F.J.; Calasanz, M.J.; Larráyoz, M.J.; Lahortiga, I.; Valgañón, M.; García-Delgado, M.; Vizmanos, J.L. TP53 is frequently altered by methylation, mutation, and/or deletion in acute lymphoblastic leukaemia. Mol. Carcinog. 2003, 38, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; James, S.J. Reduction of p53 gene expression in human primary hepatocellular carcinoma is associated with promoter region methylation without coding region mutation. Cancer Lett. 2002, 176, 169–174. [Google Scholar] [CrossRef]
- Chmelarova, M.; Krepinska, E.; Spacek, J.; Laco, J.; Beranek, M.; Palicka, V. Methylation in the p53 promoter in epithelial ovarian cancer. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2013, 15, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kim, S.J.; Noh, D.Y.; Park, I.A.; Choe, K.J.; Yoo, O.J.; Kang, H.S. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: Correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab. Investig. J. Tech. Methods Pathol. 2001, 81, 573–579. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chung, T.H.; Sebastian, S.; Choo, S.N.; Yan, J.; Ng, S.B.; Fonseca, R.; Chng, W.J. p53 haploinsufficiency and functional abnormalities in multiple myeloma. Leukemia 2014, 28, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Reversal of p53 epigenetic silencing in multiple myeloma permits apoptosis by a p53 activator. Cancer Biol. Ther. 2006, 5, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Murao, K.; Kubo, Y.; Ohtani, N.; Hara, E.; Arase, S. Epigenetic abnormalities in cutaneous squamous cell carcinomas: Frequent inactivation of the RB1/p16 and p53 pathways. Br. J. Dermatol. 2006, 155, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, S.; Martin, E.; Gicquel, C.; Melki, J.; Clark, S.J.; Campbell, P.; Magarey, C.J.; Schulte, K.M.; Röher, H.D.; Delbridge, L.; et al. Mutation and methylation analysis of TP53 in adrenal carcinogenesis. Eur. J. Surg. Oncol. 2005, 31, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Papaggeli, P.C.; Kortsaris, A.C.; Matsouka, P.T. Aberrant methylation of c-myc and c-fos protooncogenes and p53 tumor suppressor gene in myelodysplastic syndromes and acute non-lymphocytic leukemia. J. BUON 2003, 8, 341–350. [Google Scholar] [PubMed]
- Radpour, R.; Barekati, Z.; Haghighi, M.M.; Kohler, C.; Asadollahi, R.; Torbati, P.M.; Holzgreve, W.; Zhong, X.Y. Correlation of telomere length shortening with promoter methylation profile of p16/Rb and p53/p21 pathways in breast cancer. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2010, 23, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Benard, A.; Zeestraten, E.C.M.; Goossens-Beumer, I.J.; Putter, H.; van de Velde, C.J.H.; Hoon, D.S.B.; Kuppen, P.J.K. DNA methylation of apoptosis genes in rectal cancer predicts patient survival and tumor recurrence. Apoptosis Int. J. Program. Cell Death 2014, 19, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Qing, Y.; Hu, H.; Liu, Y.; Feng, T.; Meng, W.; Jiang, L.; Sun, Y.; Yao, Y. Berberine induces apoptosis in human multiple myeloma cell line U266 through hypomethylation of p53 promoter. Cell Biol. Int. 2014, 38, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; An, J.; Huang, S.; Liao, H.; Weng, Y.; Cai, S.; Zhang, J. PLCE1 suppresses p53 expression in esophageal cancer cells. Cancer Investig. 2014, 32, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Peng, B.; Cherry, J.C.; Hurt, E.M.; Fox, S.D.; Kelley, J.A.; Munroe, D.J.; Farrar, W.L. Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res. 2005, 65, 4673–4682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Pan, X.; Xia, Y.; Xiao, Q.; Huang, X. Relationship between the methylation and mutation of p53 gene and endemic arsenism caused by coal-burning. Chin. J. Prev. Med. 2011, 45, 393–398. [Google Scholar]
- Najjar Sadeghi, R.; Vahedi, M.; Zojaji, H.; Zali, M.R. Correlation between global genome methylation and mutation at CpG codons of p53 gene. J. Dig. Dis. 2013, 14, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Yan, L.; Cheng, W.-H.; Uthus, E.O. Dietary selenomethionine increases exon-specific DNA methylation of the p53 gene in rat liver and colon mucosa. J. Nutr. 2011, 141, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Haider, S.; Jagannathan, S.; Anaissie, E.; Driscoll, J.J. MicroRNA theragnostics for the clinical management of multiple myeloma. Leukemia 2014, 28, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Di Martino, M.T.; Neri, A.; Tagliaferri, P.; Tassone, P. Non-coding RNA: A novel opportunity for the personalized treatment of multiple myeloma. Expert Opin. Biol. Ther. 2013, 13 (Suppl. 1), S125–S137. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, P.; Rossi, M.; di Martino, M.T.; Amodio, N.; Leone, E.; Gulla, A.; Neri, A.; Tassone, P. Promises and challenges of MicroRNA-based treatment of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; de Luca, A.; Morelli, E.; Giavaresi, G.; Tagliaferri, P.; Tassone, P.; Amodio, N. MicroRNAs: Novel Crossroads between Myeloma Cells and the Bone Marrow Microenvironment. BioMed Res. Int. 2016, 2016, 6504593. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, C.; Wu, R.; Hu, W. Tumor suppressor p53 meets microRNAs. J. Mol. Cell Biol. 2011, 3, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA control of p53. J. Cell. Biochem. 2016, 118, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.-S.; Rocci, A.; de Luca, L.; Taccioli, C.; Santhanam, R.; Zhou, W.; Benson, D.M.; Hofmainster, C.; Alder, H.; et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010, 18, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.-S.; Yoo, J.Y.; Nuovo, G.J.; Jeon, Y.-J.; Kim, S.; Lee, T.J.; Kim, T.; Bakàcs, A.; Alder, H.; Kaur, B.; et al. MicroRNAs/TP53 feedback circuitry in glioblastoma multiforme. Proc. Natl. Acad. Sci. USA 2012, 109, 5316–5321. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.-J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.-S.; et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Schwerk, J.; Savan, R. Translating the Untranslated Region. J. Immunol. Baltim. Md 1950 2015, 195, 2963–2971. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chan, C.S.; Wu, R.; Zhang, C.; Sun, Y.; Song, J.S.; Tang, L.H.; Levine, A.J.; Feng, Z. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol. Cell 2010, 38, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.N.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, J.-S.; Tang, X.; Tucker, L.D.; Quesenberry, P.; Rigoutsos, I.; Ramratnam, B. MicroRNA 125a and its regulation of the p53 tumor suppressor gene. FEBS Lett. 2009, 583, 3725–3730. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Huang, S.; Wu, S.; Guo, W.; Li, J.; He, X. MicroRNA-1285 inhibits the expression of p53 by directly targeting its 3’ untranslated region. Biochem. Biophys. Res. Commun. 2010, 396, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Lu, Z.; Takwi, A.A.L.; Chen, W.; Callander, N.S.; Ramos, K.S.; Young, K.H.; Li, Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene 2011, 30, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Leotta, M.; Biamonte, L.; Raimondi, L.; Ronchetti, D.; di Martino, M.T.; Botta, C.; Leone, E.; Pitari, M.R.; Neri, A.; Giordano, A.; et al. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J. Cell. Physiol. 2014, 229, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.Y.; Rushworth, S.A.; Zaitseva, L.; Bowles, K.M.; Macewan, D.J. Attenuation of dexamethasone-induced cell death in multiple myeloma is mediated by miR-125b expression. Cell Cycle Georget. Tex 2013, 12, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, N.C.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Delgado, M.; de Las Rivas, J.; Ticona, F.V.; Fermiñán, E.; Martín-Jiménez, P.; Chillón, C.; Risueño, A.; et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010, 24, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.-S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Pekarsky, Y.; Croce, C.M. Role of miR-15/16 in CLL. Cell Death Differ. 2015, 22, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yin, S.; Hao, Y.; Yang, J.; Zhang, H.; Sun, C.; Ma, M.; Chang, Q.; Xi, J.J. miR-19b promotes tumor growth and metastasis via targeting TP53. RNA (New York, NY) 2014, 20, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xing, R.; Zhang, X.; Dong, W.; Zhang, J.; Yan, Z.; Li, W.; Cui, J.; Lu, Y. miR-375 targets the p53 gene to regulate cellular response to ionizing radiation and etoposide in gastric cancer cells. DNA Repair 2013, 12, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, M.; Deva Magendhra Rao, A.K.; Arunkumar, G.; Rajkumar, K.S.; Rajaraman, R.; Munirajan, A.K. Down Regulation of miR-34a and miR-143 May Indirectly Inhibit p53 in Oral Squamous Cell Carcinoma: a Pilot Study. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 7619–7625. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Leone, E.; Amodio, N.; Foresta, U.; Lionetti, M.; Pitari, M.R.; Cantafio, M.E.G.; Gullà, A.; Conforti, F.; Morelli, E.; et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: In vitro and in vivo evidence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 6260–6270. [Google Scholar] [CrossRef] [PubMed]
- Misiewicz-Krzeminska, I.; Sarasquete, M.E.; Quwaider, D.; Krzeminski, P.; Ticona, F.V.; Paíno, T.; Delgado, M.; Aires, A.; Ocio, E.M.; García-Sanz, R.; et al. Restoration of microRNA-214 expression reduces growth of myeloma cells through positive regulation of p53 and inhibition of DNA replication. Haematologica 2013, 98, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Arai, N.; Nomura, D.; Yokota, K.; Wolf, D.; Brill, E.; Shohat, O.; Rotter, V. Immunologically distinct p53 molecules generated by alternative splicing. Mol. Cell. Biol. 1986, 6, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Khoury, M.P.; Bourdon, J.-C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets Ther. 2013, 7, 57–68. [Google Scholar] [PubMed]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.-C. Δ160p53 is a novel N-terminal p53 isoform encoded by Δ133p53 transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.-C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.-C.; et al. p53 mutant breast cancer patients expressing p53γ have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. BCR 2011, 13, R7. [Google Scholar] [CrossRef] [PubMed]
- Anensen, N.; Oyan, A.M.; Bourdon, J.-C.; Kalland, K.H.; Bruserud, O.; Gjertsen, B.T. A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 3985–3992. [Google Scholar] [CrossRef] [PubMed]
- Boldrup, L.; Bourdon, J.-C.; Coates, P.J.; Sjöström, B.; Nylander, K. Expression of p53 isoforms in squamous cell carcinoma of the head and neck. Eur. J. Cancer Oxf. Engl. 1990 2007, 43, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms δ133p53 and p53β are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Ånensen, N.; Hjelle, S.M.; van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.-C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lønning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012, 31, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Zhang, X.D.; Adams, L.J.; Scott, R.J.; Vojtesek, B.; Lane, D.P.; Hersey, P. Small molecular weight variants of p53 are expressed in human melanoma cells and are induced by the DNA-damaging agent cisplatin. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.K.; Martinez, J.D. The significance of p53 isoform expression in serous ovarian cancer. Future Oncol. Lond. Engl. 2012, 8, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Gahrton, G.; Bergsagel, P.L. Approach to the treatment of multiple myeloma: A clash of philosophies. Blood 2011, 118, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Lee, J.H.; Lahuerta, J.J.; Morgan, G.; Richardson, P.G.; Crowley, J.; Haessler, J.; Feather, J.; Hoering, A.; Moreau, P.; et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Leukemia 2012, 26, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G.; Wiman, K.G. Reactivation of mutant p53: Molecular mechanisms and therapeutic potential. Oncogene 2007, 26, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Jin, T.; Yang, Q.; Liu, W.; Liu, S.; Ji, M.; He, N.; Chen, C.; Shi, B.; Hou, P. PRIMA-1 selectively induces global DNA demethylation in p53 mutant-type thyroid cancer cells. J. Biomed. Nanotechnol. 2014, 10, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.G.C.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Kua, N.; Kaldis, P.; Lane, D.P. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell Death Differ. 2010, 17, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Qiu, L.; Chang, H. Targeting p53 by small molecules in hematological malignancies. J. Hematol. Oncol. 2013, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.T.; Vassilev, L. Small-molecule inhibitors of the p53-MDM2 interaction. Curr. Top. Microbiol. Immunol. 2011, 348, 151–172. [Google Scholar] [PubMed]
- Hao, Q.; Cho, W.C. Battle against cancer: An everlasting saga of p53. Int. J. Mol. Sci. 2014, 15, 22109–22127. [Google Scholar] [CrossRef] [PubMed]
- Stühmer, T.; Chatterjee, M.; Hildebrandt, M.; Herrmann, P.; Gollasch, H.; Gerecke, C.; Theurich, S.; Cigliano, L.; Manz, R.A.; Daniel, P.T.; et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood 2005, 106, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Chng, W.J. p53 abnormalities and potential therapeutic targeting in multiple myeloma. BioMed Res. Int. 2014, 2014, 717919. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Graves, B.; Packman, K.; Filipovic, Z.; Higgins, B.; Xia, M.; Tardell, C.; Garrido, R.; Lee, E.; Kolinsky, K.; et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013, 73, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.; Pecak, A.; Rys, B.; Wladyka, B.; Dömling, A.; Weber, L.; Holak, T.A.; Dubin, G. Mdm2 and MdmX inhibitors for the treatment of cancer: A patent review (2011-present). Expert Opin. Ther. Pat. 2013, 23, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, S.X.; Lu, H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell. Biochem. 2014, 85, 281–319. [Google Scholar] [PubMed]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y.; et al. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Chang, H. Molecular mechanisms of nutlin-induced apoptosis in multiple myeloma: Evidence for p53-transcription-dependent and -independent pathways. Cancer Biol. Ther. 2010, 10, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Jayakar, J.; Reece, D.; Branch, D.R.; Chang, H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol. Ther. 2010, 9, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Mukai, A.; Chang, H. RITA inhibits multiple myeloma cell growth through induction of p53-mediated caspase-dependent apoptosis and synergistically enhances nutlin-induced cytotoxic responses. Mol. Cancer Ther. 2010, 9, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, M.; Ozdowy, P.; D’Silva, L.; Rothweiler, U.; Holak, T.A. NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro. Nat. Med. 2005, 11, 1135–1137. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ahmed, A.; Poon, E.; Perusinghe, N.; de Haven Brandon, A.; Box, G.; Valenti, M.; Eccles, S.; Rouschop, K.; Wouters, B.; et al. Small-molecule activation of p53 blocks hypoxia-inducible factor 1alpha and vascular endothelial growth factor expression in vivo and leads to tumor cell apoptosis in normoxia and hypoxia. Mol. Cell. Biol. 2009, 29, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Nieves-Neira, W.; Rivera, M.I.; Kohlhagen, G.; Hursey, M.L.; Pourquier, P.; Sausville, E.A.; Pommier, Y. DNA protein cross-links produced by NSC 652287, a novel thiophene derivative active against human renal cancer cells. Mol. Pharmacol. 1999, 56, 478–484. [Google Scholar] [PubMed]
- Enge, M.; Bao, W.; Hedström, E.; Jackson, S.P.; Moumen, A.; Selivanova, G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell 2009, 15, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Bjorklund, C.C.; Baladandayuthapani, V.; Kuhn, D.J.; Orlowski, R.Z. Drug resistance to inhibitors of the human double minute-2 E3 ligase is mediated by point mutations of p53, but can be overcome with the p53 targeting agent RITA. Mol. Cancer Ther. 2012, 11, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Surget, S.; Descamps, G.; Brosseau, C.; Normant, V.; Maïga, S.; Gomez-Bougie, P.; Gouy-Colin, N.; odon, C.; Béné, M.; Moreau, P.; et al. RITA (Reactivating p53 and Inducing Tumor Apoptosis) is efficient against TP53 abnormal myeloma cells independently of the p53 pathway. BMC Cancer 2014, 14, 437. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Rökaeus, N.; Shen, J.; Eckhardt, I.; Bykov, V.J.N.; Wiman, K.G.; Wilhelm, M.T. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene 2010, 29, 6442–6451. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, M.-Q.-Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Besch-Williford, C.; Hyder, S.M. PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int. J. Oncol. 2009, 35, 1015–1023. [Google Scholar] [PubMed]
- Messina, R.L.; Sanfilippo, M.; Vella, V.; Pandini, G.; Vigneri, P.; Nicolosi, M.L.; Gianì, F.; Vigneri, R.; Frasca, F. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int. J. Cancer 2012, 130, 2259–2270. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, E.; Safa, M.; Sharifi, A.M.; Bashash, D. PRIMA-1 induces caspase-mediated apoptosis in acute promyelocytic leukemia NB4 cells by inhibition of nuclear factor-κB and downregulation of Bcl-2, XIAP, and c-Myc. Anti-Cancer. Drugs 2016. [Google Scholar] [CrossRef] [PubMed]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.; Mohammad, D.K.; Mujahed, H.; Jonson-Videsäter, K.; Nore, B.; Paul, C.; Lehmann, S. Anti-leukaemic effects induced by APR-246 are dependent on induction of oxidative stress and the NFE2L2/HMOX1 axis that can be targeted by PI3K and mTOR inhibitors in acute myeloid leukaemia cells. Br. J. Haematol. 2016, 174, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Grellety, T.; Laroche-Clary, A.; Chaire, V.; Lagarde, P.; Chibon, F.; Neuville, A.; Italiano, A. PRIMA-1(MET) induces death in soft-tissue sarcomas cell independent of p53. BMC Cancer 2015, 15, 684. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Bi, C.; Sintosebastian, C.; Tay, L.S.; Fonseca, R.; Chng, W.J. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget 2016. [Google Scholar] [CrossRef]
- Saha, M.N.; Jiang, H.; Yang, Y.; Reece, D.; Chang, H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol. Cancer Ther. 2013, 12, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J. PRIMA-1 as a cancer therapy restoring mutant p53: A review. Biosci. Horiz. 2015. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [PubMed]
- Bou-Hanna, C.; Jarry, A.; Lode, L.; Schmitz, I.; Schulze-Osthoff, K.; Kury, S.; Bezieau, S.; Mosnier, J.-F.; Laboisse, C.L. Acute cytotoxicity of MIRA-1/NSC19630, a mutant p53-reactivating small molecule, against human normal and cancer cells via a caspase-9-dependent apoptosis. Cancer Lett. 2015, 359, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Chen, Y.; Chen, M.-H.; Chen, G.; Chang, H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br. J. Cancer 2014, 110, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Shen, H.; Maki, C.G. Acquisition of p53 mutations in response to the non-genotoxic p53 activator Nutlin-3. Oncogene 2011, 30, 4678–4686. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Y.; El-Naggar, A.K.; Xiong, S.; Yang, P.; Jackson, J.G.; Chau, G.; Lozano, G. Therapeutic efficacy of p53 restoration in Mdm2-overexpressing tumors. Mol. Cancer Res. 2014, 12, 901–911. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutiérrez, N.C. Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma. Int. J. Mol. Sci. 2016, 17, 2003. https://doi.org/10.3390/ijms17122003
Herrero AB, Rojas EA, Misiewicz-Krzeminska I, Krzeminski P, Gutiérrez NC. Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma. International Journal of Molecular Sciences. 2016; 17(12):2003. https://doi.org/10.3390/ijms17122003
Chicago/Turabian StyleHerrero, Ana B., Elizabeta A. Rojas, Irena Misiewicz-Krzeminska, Patryk Krzeminski, and Norma C. Gutiérrez. 2016. "Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma" International Journal of Molecular Sciences 17, no. 12: 2003. https://doi.org/10.3390/ijms17122003
APA StyleHerrero, A. B., Rojas, E. A., Misiewicz-Krzeminska, I., Krzeminski, P., & Gutiérrez, N. C. (2016). Molecular Mechanisms of p53 Deregulation in Cancer: An Overview in Multiple Myeloma. International Journal of Molecular Sciences, 17(12), 2003. https://doi.org/10.3390/ijms17122003