
The Role of CC-Chemokines in the Regulation of Angiogenesis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
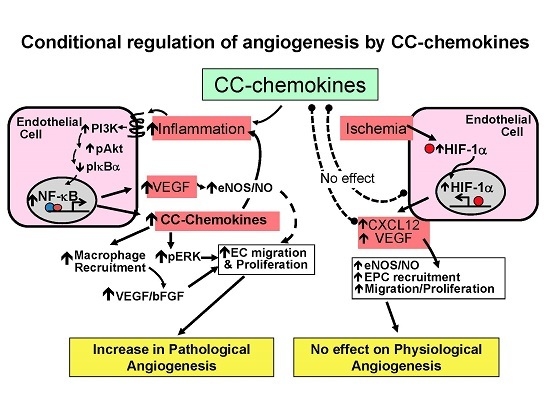
2. Conditional Regulation of Angiogenesis
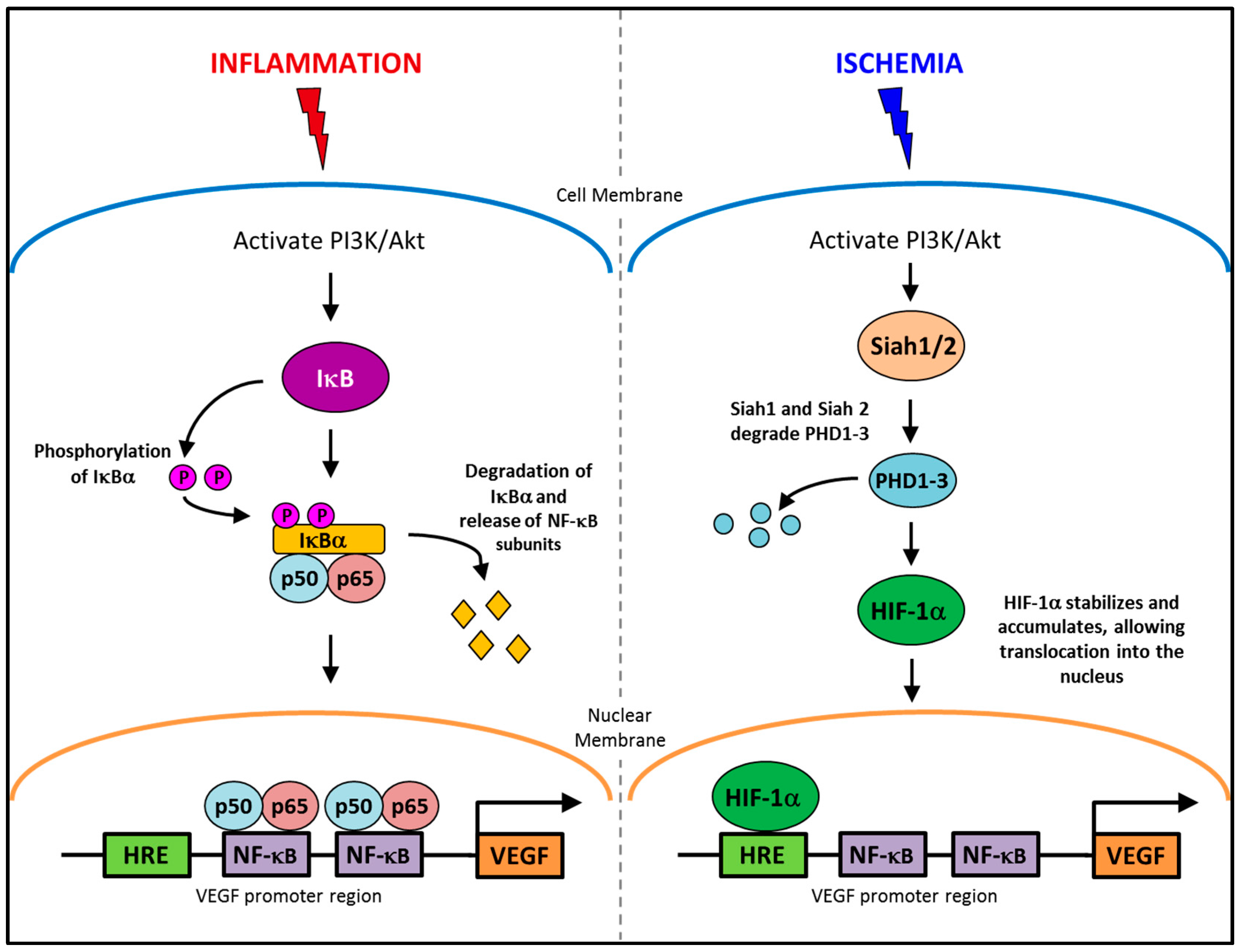
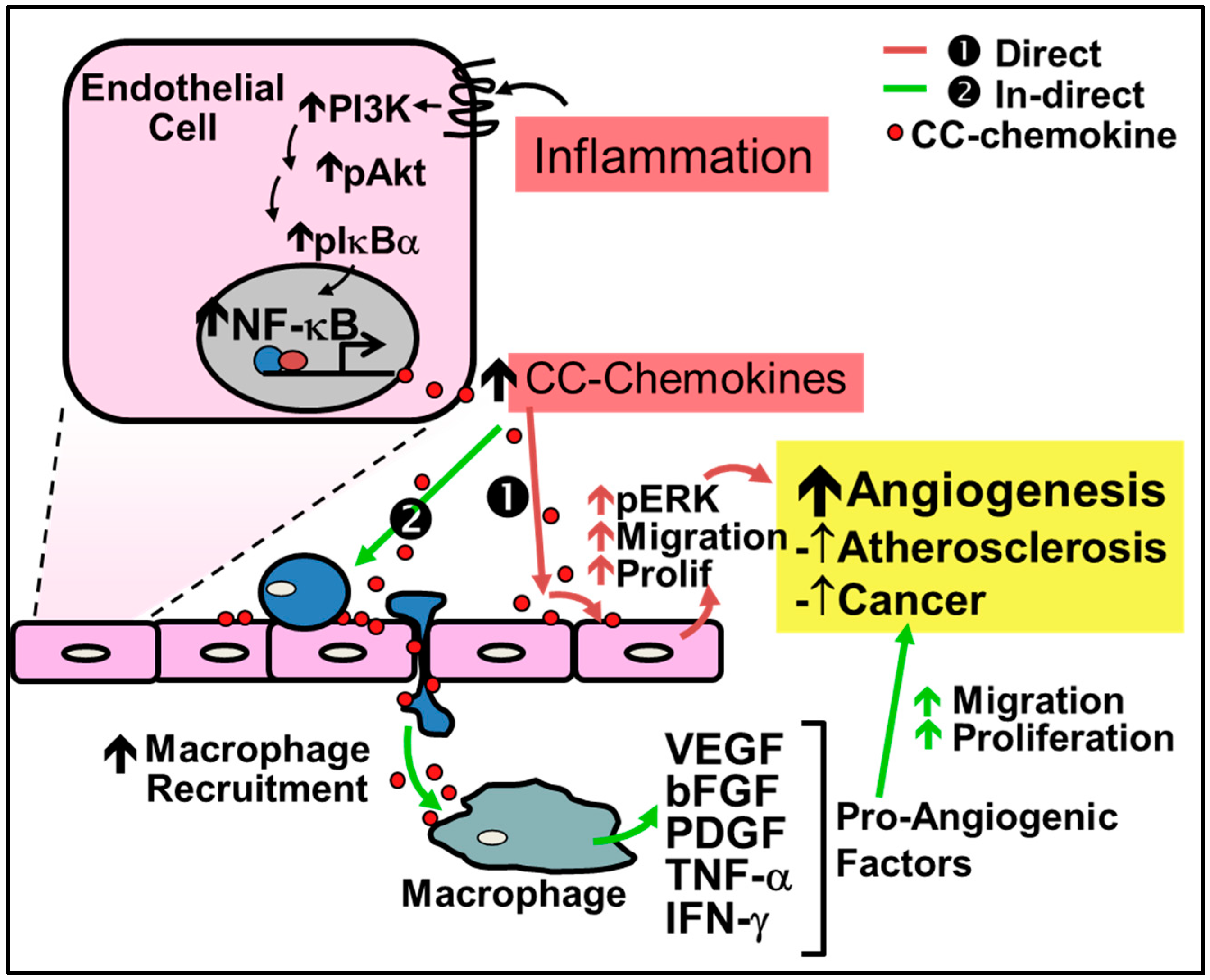
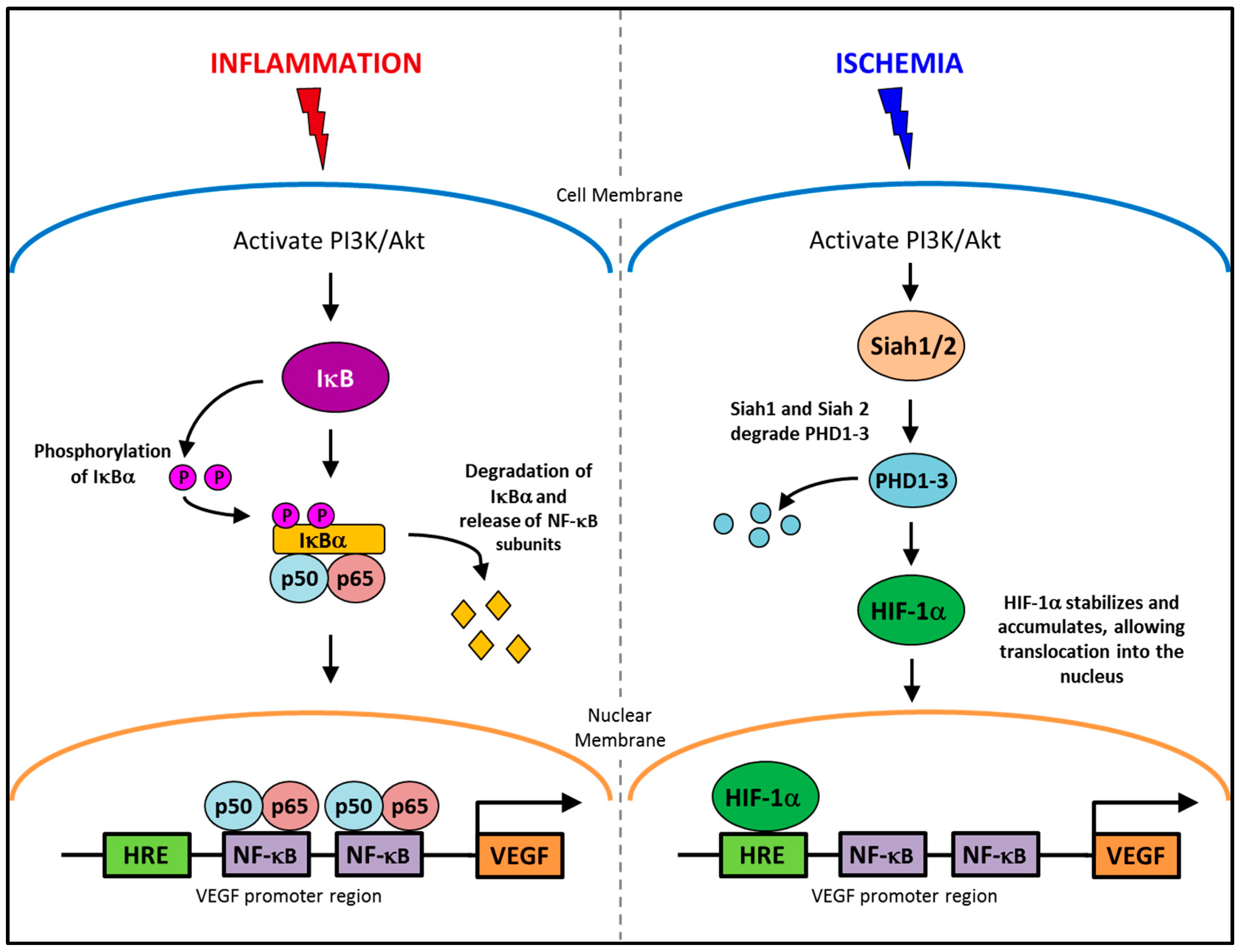
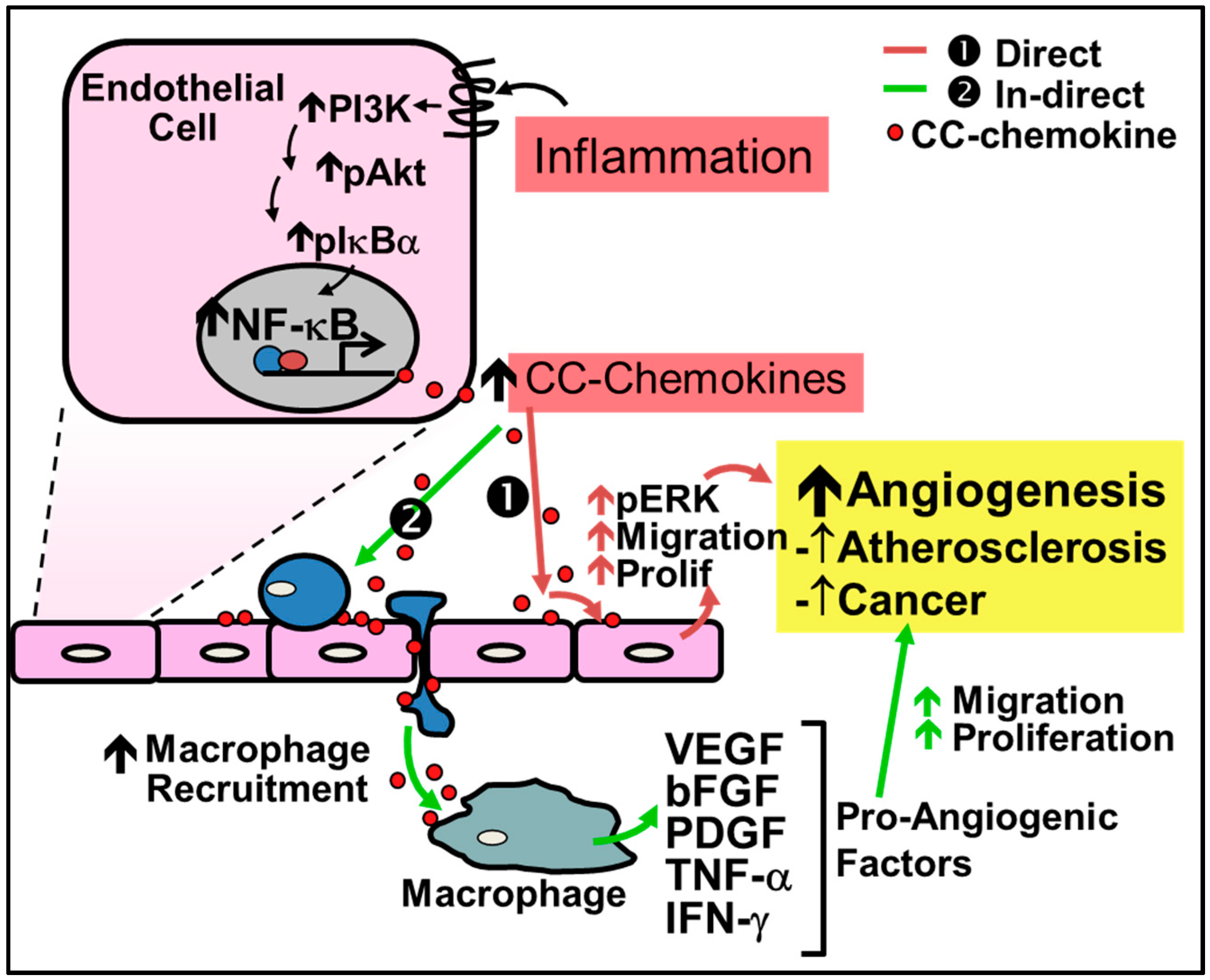
2.1. Pathological Inflammatory Angiogenesis
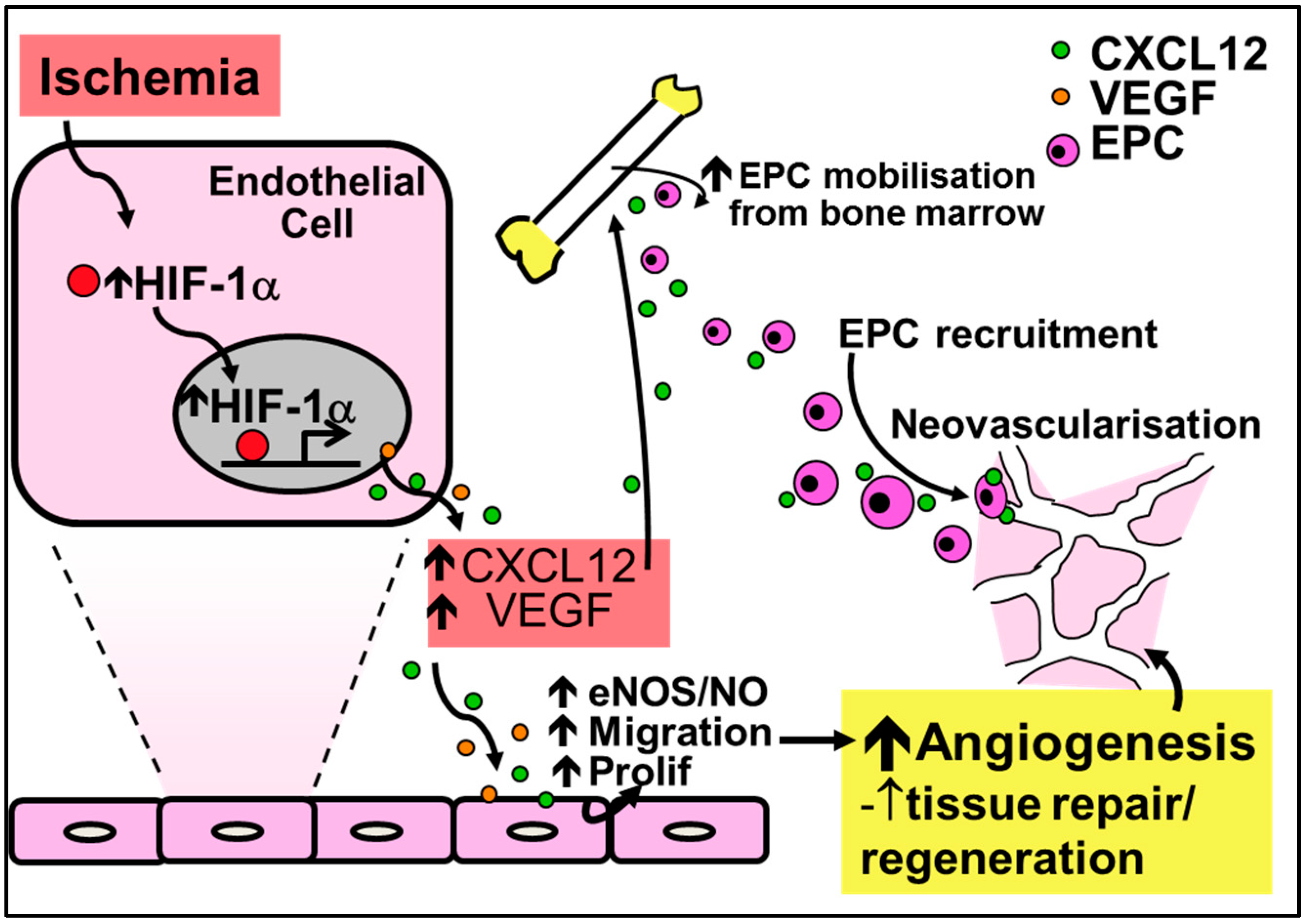
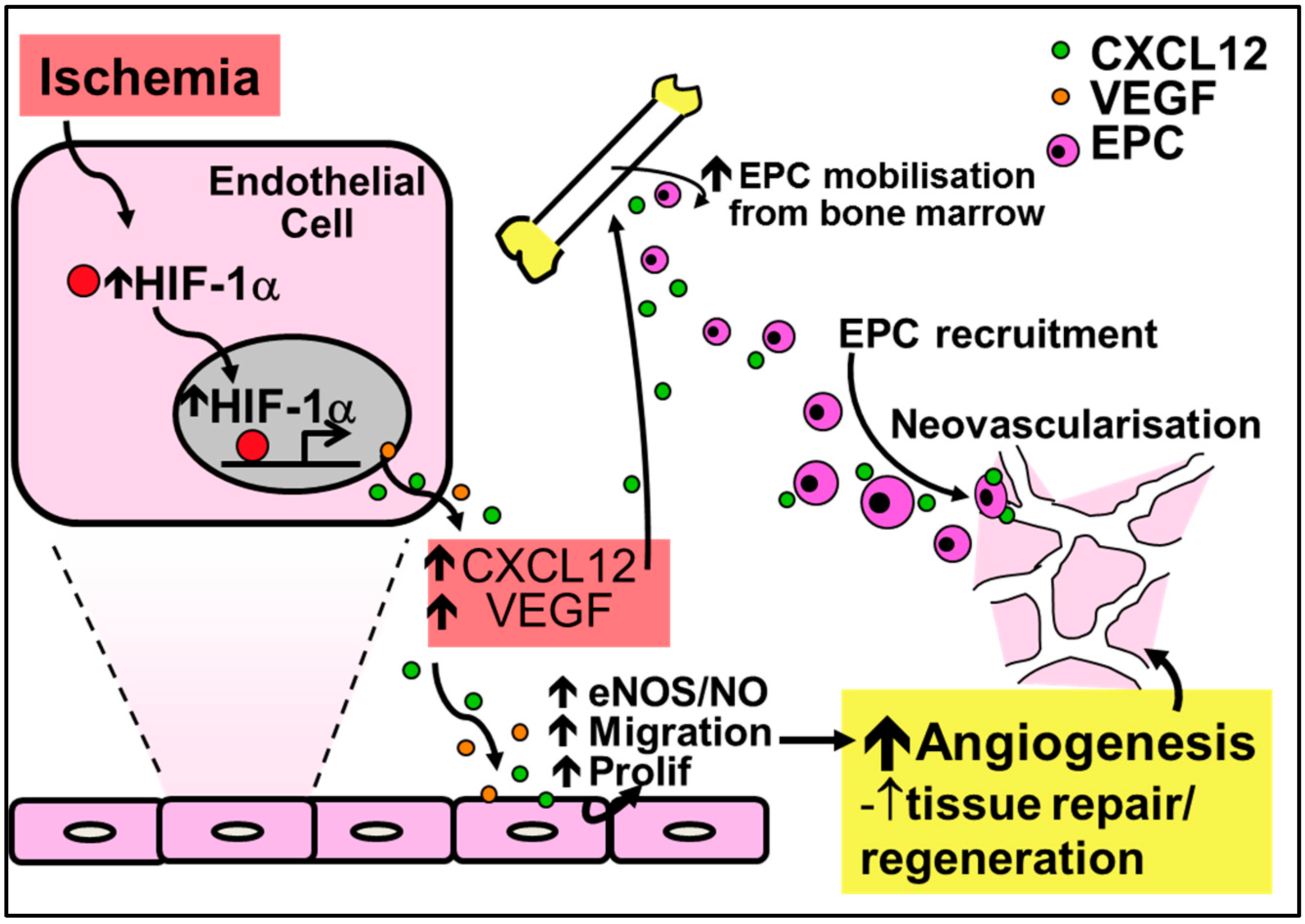
2.2. Physiological Ischemia-Driven Angiogenesis
3. Chemokines
4. Chemokines in Angiogenesis Associated Diseases
4.1. CC-Chemokines in Pathological Inflammation-Driven Angiogenesis
4.1.1. CC-Chemokines in Atherosclerosis
4.1.2. CC-Chemokines in Rheumatoid Arthritis
4.2. Chemokines in Physiological Ischemia-Mediated Angiogenesis
5. CC-Chemokines in Cancer
6. Anti-Angiogenic Therapies
Broad-Spectrum CC-Chemokine Inhibition as a Strategy to Inhibit Pathological Angiogenesis
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Risau, W. Mechanisms of angiogenesis. Nature 1997, 386, 671–674. [Google Scholar] [PubMed]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Dzuiba, J.; van Damme, J.; Walz, A.; Marriott, D.; et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Lasagni, L.; Annunziato, F.; Serio, M.; Romagnani, S. CXC chemokines: The regulatory link between inflammation and angiogenesis. Trends Immunol. 2004, 25, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Namkoong, S.; Kim, Y.-M.; Kim, C.-K.; Lee, H.; Ha, K.-S.; Chung, H.-T.; Kwon, Y.-G.; Kim, Y.-M. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2836–H2846. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Status Report on Noncommunicable Diseases 2014; World Health Organization: Geneva, Switzerland, 2014; p. 298. [Google Scholar]
- Mourad, J.J.; des Guetz, G.; Debbabi, H.; Levy, B.I. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann. Oncol. 2008, 19, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Cabebe, E.; Wakelee, H. Role of anti-angiogenesis agents in treating NSCLC: Focus on bevacizumab and VEGFR tyrosine kinase inhibitors. Curr. Treat. Opt. Oncol. 2007, 8, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Vakili, K.; Zurakowski, D.; Soliman, M.; Butterfield, C.; Sylvin, E.; Lo, K.M.; Gillies, S.; Javaherian, K.; Folkman, J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 4736–4741. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Pakozdi, A.; Szentpetery, A.; Besenyei, T.; Koch, A.E. Chemokines and angiogenesis in rheumatoid arthritis. Front. Biosci. 2009, 1, 44–51. [Google Scholar]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.P.; Levy, N.S.; Wegner, S.; Goldberg, M.A. Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J. Biol. Chem. 1995, 270, 13333–13340. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Marsch, E.; Sluimer, J.C.; Daemen, M.J. Hypoxia in atherosclerosis and inflammation. Curr. Opin. Lipidol. 2013, 24, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.R.; Seed, M.P.; Kircher, C.H.; Willoughby, D.A.; Winkler, J.D. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997, 11, 457–465. [Google Scholar] [PubMed]
- Sunderkotter, C.; Steinbrink, K.; Goebeler, M.; Bhardwaj, R.; Sorg, C. Macrophages and angiogenesis. J. Leukoc. Biol. 1994, 55, 410–422. [Google Scholar] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Purushothaman, K.R.; Fuster, V.; Echeverri, D.; Truszczynska, H.; Sharma, S.K.; Badimon, J.J.; O’Connor, W.N. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: Implications for plaque vulnerability. Circulation 2004, 110, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Oxygen sensing in the hypoxic response pathway: Regulation of the hypoxia-inducible transcription factor. Genes Dev. 2003, 17, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Frew, I.J.; Hagensen, M.; Skals, M.; Habelhah, H.; Bhoumik, A.; Kadoya, T.; Erdjument-Bromage, H.; Tempst, P.; Frappell, P.B.; et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell 2004, 117, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Qi, J.; Ronai, Z. The ubiquitin ligase Siah2 and the hypoxia response. Mol. Cancer Res. 2009, 7, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Kotch, L.E.; Iyer, N.V.; Laughner, E.; Semenza, G.L. Defective vascularization of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev. Biol. 1999, 209, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Licht, A.H.; Müller-Holtkamp, F.; Flamme, I.; Breier, G. Inhibition of hypoxia-inducible factor activity in endothelial cells disrupts embryonic cardiovascular development. Blood 2006, 107, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Munk, V.C.; Miguel, L.S.d.; Humar, R.; Kiefer, F.N.; Butz, N.; Battegay, E.J. INOS is required for in vitro angiogenesis of hypoxic mouse hearts. Semin. Cardiol. 2006, 12, 21–26. [Google Scholar]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 1138–1143. [Google Scholar] [PubMed]
- Phillips, P.G.; Birnby, L.M.; Narendran, A. Hypoxia induces capillary network formation in cultured bovine pulmonary microvessel endothelial cells. Am. J. Physiol. 1995, 268, L789–L800. [Google Scholar] [PubMed]
- Rehman, J.; Li, J.; Orschell, C.M.; March, K.L. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 2003, 107, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; von Ballmoos, M.W.; Faessler, D.; Voelzmann, J.; Ortmann, J.; Diehm, N.; Kalka-Moll, W.; Baumgartner, I.; di Santo, S.; Kalka, C. Paracrine factors secreted by endothelial progenitor cells prevent oxidative stress-induced apoptosis of mature endothelial cells. Atherosclerosis 2010, 211, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Chang, S.-J.; Chueh, Y.-N.; Huang, T.-S.; Huang, P.-H.; Cheng, S.-M.; Tsai, T.-N.; Chen, J.-W.; Wang, H.-W. Distinct angiogenesis roles and surface markers of early and late endothelial progenitor cells revealed by functional group analyses. BMC Genom. 2013, 14, 182. [Google Scholar] [CrossRef] [PubMed]
- Sieveking, D.P.; Buckle, A.; Celermajer, D.S.; Ng, M.K. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: Insights from a novel human angiogenesis assay. J. Am. Coll. Cardiol. 2008, 51, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Mukai, N.; Akahori, T.; Komaki, M.; Li, Q.; Kanayasu-Toyoda, T.; Ishii-Watabe, A.; Kobayashi, A.; Yamaguchi, T.; Abe, M.; Amagasa, T.; et al. A comparison of the tube forming potentials of early and late endothelial progenitor cells. Exp. Cell Res. 2008, 314, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Chan, A. Antiangiogenic therapy for metastatic breast cancer: Current status and future directions. Drugs 2009, 69, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.J.; Lolis, E. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Forster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.D.J. Location, movement and survival: The role of chemokines in haematopoiesis and malignancy. Br. J. Haematol. 2006, 132, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Øynebråten, I.; Bakke, O.; Brandtzaeg, P.; Johansen, F.-E.; Haraldsen, G. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood 2004, 104, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.C.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Kuschert, G.S.; Coulin, F.; Power, C.A.; Proudfoot, A.E.; Hubbard, R.E.; Hoogewerf, A.J.; Wells, T.N. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 1999, 38, 12959–12968. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine: Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Hub, E.; Rot, A. Binding of RANTES, MCP-1, MCP-3, and MIP-1α to cells in human skin. Am. J. Pathol. 1998, 152, 749–757. [Google Scholar] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Mostarica-Stojkovic, M.; Andjelkovic, A.V. CCL2 regulates angiogenesis via activation of ETS-1 transcription factor. J. Immunol. 2006, 177, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Young, H.A.; Ponce, M.L.; Ward, J.M.; Kleinman, H.K.; Murphy, W.J.; Oppenheim, J.J. Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. J. Immunol. 2001, 166, 7571–7578. [Google Scholar] [CrossRef] [PubMed]
- Strasly, M.; Doronzo, G.; Cappello, P.; Valdembri, D.; Arese, M.; Mitola, S.; Moore, P.; Alessandri, G.; Giovarelli, M.; Bussolino, F. CCL16 activates an angiogenic program in vascular endothelial cells. Blood 2004, 103, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Spinetti, G.; Ribatti, D.; Camarda, G.; Morbidelli, L.; Ziche, M.; Santoni, A.; Capogrossi, M.C.; Napolitano, M. I-309 binds to and activates endothelial cell functions and acts as an angiogenic molecule in vivo. Blood 2000, 96, 4039–4045. [Google Scholar] [PubMed]
- Hwang, J.; Kim, C.W.; Son, K.-N.; Han, K.Y.; Lee, K.H.; Kleinman, H.K.; Ko, J.; Na, D.S.; Kwon, B.S.; Gho, Y.S.; et al. Angiogenic activity of human CC chemokine CCL15 in vitro and in vivo. FEBS Lett. 2004, 570, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Möckel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960–1971. [Google Scholar] [CrossRef] [PubMed]
- Galvez, B.G.; Genis, L.; Matias-Roman, S.; Oblander, S.A.; Tryggvason, K.; Apte, S.S.; Arroyo, A.G. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/CCL2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J. Biol. Chem. 2005, 280, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, S.; Amano, K.; Uehira, K.; Yoshida, M.; Nishiwaki, Y.; Nozawa, Y.; Jin, D.; Takai, S.; Miyazaki, M.; Egashira, K.; et al. Bone marrow monocyte lineage cells adhere on injured endothelium in a monocyte chemoattractant protein-1-dependent manner and accelerate reendothelialization as endothelial progenitor cells. Circ. Res. 2003, 93, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; McIntyre, T.M.; McEver, R.P.; Prescott, S.M.; Zimmerman, G.A. Monocyte tethering by p-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-α secretion. Signal integration and NF-κB translocation. J. Clin. Investig. 1995, 95, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Werts, C.; le Bourhis, L.; Liu, J.; Magalhaes, J.G.; Carneiro, L.A.; Fritz, J.H.; Stockinger, S.; Balloy, V.; Chignard, M.; Decker, T.; et al. Nod1 and Nod2 induce CCL5/RANTES through the NF-κB pathway. Eur. J. Immunol. 2007, 37, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Weber, C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc. Res. 2010, 86, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Leech, M.; Bernhagen, J. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 2006, 5, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jiang, D. Fractalkine/CX3CR1 and atherosclerosis. Clin. Chim. Acta 2011, 412, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A.; Cai, S.; Channon, K.M.; Greaves, D.R. Adenoviral-mediated delivery of a viral chemokine binding protein blocks CC-chemokine activity in vitro and in vivo. Immunobiology 2003, 207, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Schober, A.; Zernecke, A. Chemokines: Key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Reape, T.J.; Rayner, K.; Manning, C.D.; Gee, A.N.; Barnette, M.S.; Burnand, K.G.; Groot, P.H. Expression and cellular localization of the CC chemokines PARC and ELC in human atherosclerotic plaques. Am. J. Pathol. 1999, 154, 365–374. [Google Scholar] [CrossRef]
- Berkhout, T.A.; Sarau, H.M.; Moores, K.; White, J.R.; Elshourbagy, N.; Appelbaum, E.; Brawner, T.J.R.; Makwana, J.; Foley, J.J.; Schmidt, D.B.; et al. Cloning, in vitro expression, and functional characterization of a novel human CC chemokine of the monocyte chemotactic protein (MCP) family (MCP-4) that binds and signals through the CC chemokine receptor 2B. J. Biol. Chem. 1997, 272, 16404–16413. [Google Scholar] [CrossRef] [PubMed]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nat. Med. 1998, 394, 894–897. [Google Scholar]
- Gosling, J.; Slaymaker, S.; Gu, L.; Tseng, S.; Zlot, C.H.; Young, S.G.; Rollins, B.J.; Charo, I.F. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J. Clin. Investig. 1999, 103, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Potteaux, S.; Combadiere, C.; Esposito, B.; Lecureuil, C.; Ait-Oufella, H.; Merval, R.; Ardouin, P.; Tedgui, A.; Mallat, Z. Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1858–1863. [Google Scholar] [CrossRef] [PubMed]
- Aiello, R.J.; Bourassa, P.-A.K.; Lindsey, S.; Weng, W.; Natoli, E.; Rollins, B.J.; Milos, P.M. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Koch, A.E. Chemokines and angiogenesis. Curr. Opin. Rheumatol. 2001, 13, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, E.; Struyf, S.; van Damme, J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003, 14, 409–426. [Google Scholar] [CrossRef]
- Horuk, R. Chemokine receptor antagonists: Overcoming developmental hurdles. Nat. Rev. Drug Discov. 2009, 8, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Vergunst, C.E.; Gerlag, D.M.; Lopatinskaya, L.; Klareskog, L.; Smith, M.D.; van den Bosch, F.; Dinant, H.J.; Lee, Y.; Wyant, T.; Jacobson, E.W.; et al. Modulation of CCR2 in rheumatoid arthritis: A double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008, 58, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.F.; Bahnck, K.B.; Blumberg, L.C.; Brissette, W.H.; Burrell, S.A.; Driscoll, J.P.; Fedeles, F.; Fisher, M.B.; Foti, R.S.; Gladue, R.P.; et al. Piperazinyl CCR1 antagonists—Optimization of human liver microsome stability. Bioorg. Med. Chem. Lett. 2007, 17, 3109–3112. [Google Scholar] [CrossRef] [PubMed]
- Fleishaker, D.L.; Garcia Meijide, J.A.; Petrov, A.; Kohen, M.D.; Wang, X.; Menon, S.; Stock, T.C.; Mebus, C.A.; Goodrich, J.M.; Mayer, H.B.; et al. Maraviroc, a chemokine receptor-5 antagonist, fails to demonstrate efficacy in the treatment of patients with rheumatoid arthritis in a randomized, double-blind placebo-controlled trial. Arthritis Res. Ther. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Balanescu, A.; Tseluyko, V.; Bojin, S.; Drescher, E.; Dairaghi, D.; Miao, S.; Marchesin, V.; Jaen, J.; Schall, T.J.; et al. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: Carat-2, a randomised, placebo controlled clinical trial. Ann. Rheum. Dis. 2013, 72, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Karshovska, E.; Zernecke, A.; Weber, C. SDF-1α-mediated tissue repair by stem cells: A promising tool in cardiovascular medicine? Trends Cardiovasc. Med. 2006, 16, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.P.; Youn, S.W.; Cho, H.J.; Li, L.; Kim, T.Y.; Yook, H.S.; Chung, J.W.; Hur, J.; Yoon, C.H.; Park, K.W.; et al. Integrin-linked kinase, a hypoxia-responsive molecule, controls postnatal vasculogenesis by recruitment of endothelial progenitor cells to ischemic tissue. Circulation 2006, 114, 150–159. [Google Scholar] [CrossRef] [PubMed]
- De Falco, E.; Porcelli, D.; Torella, A.R.; Straino, S.; Iachininoto, M.G.; Orlandi, A.; Truffa, S.; Biglioli, P.; Napolitano, M.; Capogrossi, M.C.; et al. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood 2004, 104, 3472–3482. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, Z.H.; Li, Z.J.; Yang, R.C.; Qian, G.Q.; Han, Z.C. Enhancement of neovascularization with cord blood CD133+ cell-derived endothelial progenitor cell transplantation. Thromb. Haemost. 2004, 91, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Liekens, S.; Schols, D.; Hatse, S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 2010, 16, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.T.; Unzek, S.; Popovic, Z.B.; Goldman, C.K.; Forudi, F.; Kiedrowski, M.; Rovner, A.; Ellis, S.G.; Thomas, J.D.; di Corleto, P.E.; et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 2003, 362, 697–703. [Google Scholar] [CrossRef]
- Wang, Y.; Haider, H.; Ahmad, N.; Zhang, D.; Ashraf, M. Evidence for ischemia induced host-derived bone marrow cell mobilization into cardiac allografts. J. Mol. Cell. Cardiol. 2006, 41, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, K.; Gupta, S.K. Cloning and relative expression analysis of rat stromal cell derived factor-1 (SDF-1)1: SDF-1 α mRNA is selectively induced in rat model of myocardial infarction. Inflammation 2001, 25, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ge, J.; Zhang, S.; Sun, A.; Shen, J.; Chen, L.; Wang, K.; Zou, Y. Time course of myocardial stromal cell-derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats with experimental myocardial infarction. Basic Res. Cardiol. 2005, 100, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fu, G.; Dai, T.; Huang, H. Migration of endothelial progenitor cells mediated by stromal cell-derived factor-1α/CXCR4 via PI3K/AKT/eNOS signal transduction pathway. J. Cardiovasc. Pharmacol. 2007, 50, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Molecular mechanisms of tumor angiogenesis and tumor progression. J. Neuro-Oncol. 2000, 50, 63–70. [Google Scholar] [CrossRef]
- Zachary, I. Vascular endothelial growth factor. Int. J. Biochem. Cell Biol. 1998, 30, 1169–1174. [Google Scholar] [CrossRef]
- Van Hinsbergh, V.W.; Collen, A.; Koolwijk, P. Angiogenesis and anti-angiogenesis: Perspectives for the treatment of solid tumors. Ann. Oncol. 1999, 10, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, Y.; Pienta, K.J. Multiple roles of chemokine (C-C MOTIF) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer Inst. 2010, 102, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Bai, Z.; Srinoulprasert, Y.; Yang, B.; Hayasaka, H.; Miyasaka, M. Chemokines in tumor progression and metastasis. Cancer Sci. 2005, 96, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.C.; Scott, K.A.; Wilson, J.L.; Thompson, R.G.; Proudfoot, A.E.I.; Balkwill, F.R. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003, 63, 8360–8365. [Google Scholar] [PubMed]
- Vaupel, P. The role of hypoxia-induced factors in tumor progression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lange, A.; Mottram, P.; Alvarez, X.; Cheng, P.; Hogan, M.; Moons, L.; Wei, S.; Zou, L.; Machelon, V.; et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005, 65, 465–472. [Google Scholar] [PubMed]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galun, E.; et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.J. Targeting angiogenesis in cancer: Clinical development of bevacizumab. Nat. Clin. Pract. Oncol. 2004, 1, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, F.; Mazzoni, F.; Micol Mela, M.; Antonuzzo, L.; Checcacci, D.; Saggese, M. Bevacizumab in non-small cell lung cancer. Drugs 2008, 68, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Borgstrom, P.; Bourdon, M.A.; Hillan, K.J.; Sriramarao, P.; Ferrara, N. Neutralizing anti-vascular endothelial growth factor antibody completely inhibits angiogenesis and growth of human prostate carcinoma micro tumors in vivo. Prostate 1998, 35, 1–10. [Google Scholar] [CrossRef]
- Robinson, E.S.; Khankin, E.V.; Karumanchi, S.A.; Humphreys, B.D. Hypertension induced by VEGF signaling pathway inhibition: Mechanisms and potential use as a biomarker. Semin. Nephrol. 2010, 30, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.W.; Hoekman, K.; Lupu, F.; Broxterman, H.J.; van der Valk, P.; Kakkar, A.K.; Pinedo, H.M. Platelet and coagulation activation with vascular endothelial growth factor generation in soft tissue sarcomas. Am. Assoc. Cancer Res. 2000, 6, 166–171. [Google Scholar]
- Verheul, H.M.; van Erp, K.; Homs, M.Y.; Yoon, G.S.; van der Groep, P.; Rogers, C.; Hansel, D.E.; Netto, G.J.; Pili, R. The relationship of vascular endothelial growth factor and coagulation factor (fibrin and fibrinogen) expression in clear cell renal cell carcinoma. Urology 2010, 75, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Larrivee, B.; Pollet, I.; Karsan, A. Activation of vascular endothelial growth factor receptor-2 in bone marrow leads to accumulation of myeloid cells: Role of granulocyte-macrophage colony-stimulating factor. J. Immunol. 2005, 175, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Satchi-Fainaro, R.; Puder, M.; Davies, J.W.; Tran, H.T.; Sampson, D.A.; Greene, A.K.; Corfas, G.; Folkman, J. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat. Med. 2004, 10, 255–261. [Google Scholar] [CrossRef]
- Chen, H.X.; Cleck, J.N. Adverse effects of anticancer agents that target the VEGF pathway. Nat. Rev. Clin. Oncol. 2009, 6, 465–477. [Google Scholar]
- Shih, T.; Lindley, C. Bevacizumab: An angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 2006, 28, 1779–1802. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vela, M.; Aris, M.; Llorente, M.; Garcia-Sanz, J.A.; Kremer, L. Chemokine receptor-specific antibodies in cancer immunotherapy: Achievements and challenges. Front. Immunol. 2015, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Devalaraja, M.N.; Richmond, A. Multiple chemotactic factors: Fine control or redundancy? Trends Pharmacol. Sci. 1999, 20, 151–156. [Google Scholar] [CrossRef]
- Mantovani, A. The chemokine system: Redundancy for robust outputs. Immunol. Today 1999, 20, 254–257. [Google Scholar] [CrossRef]
- Smith, P.; Fallon, R.E.; Mangan, N.E.; Walsh, C.M.; Saraiva, M.; Sayers, J.R.; McKenzie, A.N.J.; Alcami, A.; Fallon, P.G. Schistosoma mansoni secretes a chemokine binding protein with antiinflammatory activity. J. Exp. Med. 2005, 202, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, P.; Power, C.A.; Proudfoot, A.E.I. Evasins: Therapeutic potential of a new family of chemokine-binding proteins from ticks. Front. Immunol. 2016, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Brett, J.M.; Fremont, D.H. Dual GPCR and GAG mimicry by the M3 chemokine decoy receptor. J. Exp. Med. 2007, 204, 3157–3172. [Google Scholar] [CrossRef] [PubMed]
- Parry, C.M.; Simas, J.P.; Smith, V.P.; Stewart, C.A.; Minson, A.C.; Efstathiou, S.; Alcami, A. A broad-spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 2000, 191, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Webb, L.M.C.; Smith, V.P.; Alcami, A. The gammaherpesvirus chemokine binding protein can inhibit the interaction of chemokines with glycosaminoglycans. J. Fed. Am. Soc. Exp. Biol. 2004, 18, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Symons, J.A.; Collins, P.D.; Williams, T.J.; Smith, G.L. Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J. Immunol. 1998, 160, 624–633. [Google Scholar] [PubMed]
- Graham, K.A.; Lalani, A.S.; Macen, J.L.; Ness, T.L.; Barry, M.; Liu, L.Y.; Lucas, A.; Clark-Lewis, I.; Moyer, R.W.; McFadden, G. The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology 1997, 229, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Smith, T.D.; Smolak, P.J.; Friend, D.; Hagen, H.; Gerhart, M.; Park, L.; Pickup, D.J.; Torrance, D.; Mohler, K.; et al. Poxvirus genomes encode a secreted, soluble protein that preferentially inhibits β chemokine activity yet lacks sequence homology to known chemokine receptors. Virology 1997, 236, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Derider, M.; McCornack, M.A.; Jao, S.C.; Isern, N.; Ness, T.; Moyer, R.; LiWang, P.J. Solution structure of the complex between poxvirus-encoded CC chemokine inhibitor vCCI and human MIP-1β. Proc. Natl. Acad. Sci. USA 2006, 103, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.G.; Studer, C.; Zuber, J.F.; Demange, B.J.; Manning, U.; Urfer, R. The viral CC chemokine-binding protein vCCI inhibits monocyte chemoattractant protein-1 activity by masking its CCR2B-binding site. J. Biol. Chem. 2001, 276, 43270–43276. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.A.; Bursill, C.A.; Hu, Y.; Choudhury, R.P.; Xu, Q.; Greaves, D.R.; Channon, K.M. Gene transfer of a broad-spectrum CC-chemokine inhibitor reduces vein graft atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005, 112, I235–I241. [Google Scholar] [PubMed]
- Bursill, C.A.; Choudhury, R.P.; Ali, Z.; Greaves, D.R.; Channon, K.M. Broad-spectrum CC-chemokine blockade by gene transfer inhibits macrophage recruitment and atherosclerotic plaque formation in apolipoprotein e-knockout mice. Circulation 2004, 110, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A.; McNeill, E.; Wang, L.; Hibbitt, O.C.; Wade-Martins, R.; Paterson, D.J.; Greaves, D.R.; Channon, K.M. Lentiviral gene transfer to reduce atherosclerosis progression by long-term CC-chemokine inhibition. Gene Ther. 2009, 16, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A.; Cash, J.L.; Channon, K.M.; Greaves, D.R. Membrane-bound CC chemokine inhibitor 35K provides localized inhibition of CC chemokine activity in vitro and in vivo. J. Immunol. 2006, 177, 5567–5573. [Google Scholar] [CrossRef]
- Seki, E.; Minicis, S.D.; Gwak, G.-Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic firbosis in mice. J. Clin. Investig. 2009, 119, 1858–1870. [Google Scholar]
- Dabbagh, K.; Xiao, Y.; Smith, C.; Stepick-Biek, P.; Kim, S.G.; Lamm, W.J.E.; Liggitt, D.H.; Lewis, D.B. Local blockade of allergic airway hyperreactivity and inflammation by the poxvirus-derived pan-CC-chemokine inhibitor vCCI. J. Immunol. 2000, 165, 3418–3422. [Google Scholar]
- Barcelos, L.S.; Talvani, A.; Teixeira, A.S.; Cassali, G.D.; Andrade, S.P.; Teixeira, M.M. Production and in vivo effects of chemokines CXCL1–3/KC and CCL2/JE in a model of inflammatory angiogenesis in mice. Inflamm. Res. 2004, 53, 576–584. [Google Scholar] [PubMed]
- Goede, V.; Brogelli, L.; Ziche, M.; Augustin, H.G. Induction of inflammatory angiogenesis by monocyte chemoattractant protein-1. Int. J. Cancer 1999, 82, 765–770. [Google Scholar] [CrossRef]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Charo, D.N.; Wang, R.; Charo, I.F.; Messina, L. CCR2−/− knockout mice revascularize normally in response to severe hindlimb ischemia. J. Vasc. Surg. 2004, 40, 786–795. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of CC-Chemokines in the Regulation of Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1856. https://doi.org/10.3390/ijms17111856
Ridiandries A, Tan JTM, Bursill CA. The Role of CC-Chemokines in the Regulation of Angiogenesis. International Journal of Molecular Sciences. 2016; 17(11):1856. https://doi.org/10.3390/ijms17111856
Chicago/Turabian StyleRidiandries, Anisyah, Joanne T. M. Tan, and Christina A. Bursill. 2016. "The Role of CC-Chemokines in the Regulation of Angiogenesis" International Journal of Molecular Sciences 17, no. 11: 1856. https://doi.org/10.3390/ijms17111856