1. Introduction
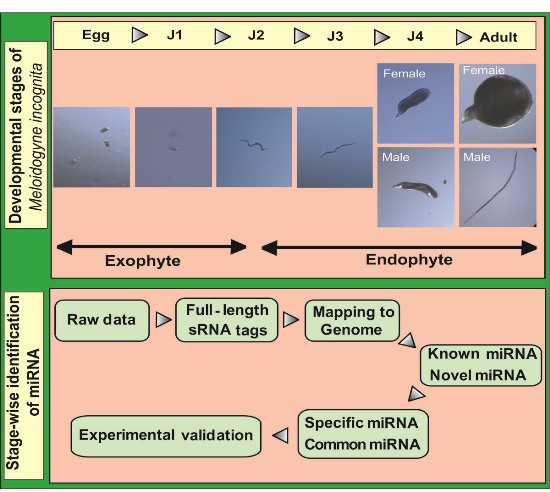
Root-knot nematodes (RKNs) belong to the group of plant-parasitic nematodes (PPN), which are detrimental to diverse plant species [
1]. Members of genus
Meloidogyne constitute the RKNs, among which
Meloidogyne incognita (
M. incognita) is an important agricultural pest [
2]. The life cycle of this nematode can be divided into five stages beginning with eggs that develop into root-infecting J2 juveniles. Upon invading the host, the J2 worms attach to the root and develop permanent feeding sites. Molting under favorable conditions leads to the formation of sedentary third (J3) and fourth (J4) stage juveniles. In the next stage, mature male and female adults are formed. Male adults, however, are not required for reproduction and saccate female worms produce eggs that hatch to release J2 worms into the soil to continue infecting neighboring plant roots.
M. incognita can affect a wide range of hosts and commonly results in stunted plants, loss of vigor, and wilting, symptoms that occur due to the disruption of nutrient and water uptake [
1,
2]. Conventional control methods against RKNs employ fumigant nematicides and non-fumigant chemicals including carbamates and organophosphates, which are effective but have several drawbacks such as direct environmental pollution, indirect pollution due to mobility from the root zone, and reduction of soil health by adhering to organic matter [
3,
4]. Currently emerging methods report on the use of RNA interference (RNAi) as an eco-sustainable strategy to control root-knot nematodes from infecting plants.
MicroRNAs (miRNAs) are short endogeneous RNA molecules (length ~22 nt) that are involved in the post-transcriptional regulation of genes in living organisms [
5]. Since their discovery, a huge amount of research has been carried out on sequencing miRNAs from several organisms including plants, insects, and higher animals. These RNAs do not code for proteins but are involved in the regulation of mRNA by binding to their 3′ untranslated region (UTR) and causing the phenomenon of RNA interference (RNAi) [
6]. As mentioned earlier, RNAi has been widely proposed as a potential strategy to control nematodes in plants [
7,
8,
9,
10]. Each miRNA contains a “seed sequence” at its 5′ end that aids target recognition and a stem-loop structure for further processing by enzymes Drosha or Dicer to release a precursor miRNA (pre-miRNA), both properties currently used to identify putative miRNAs [
11,
12]. Although updated miRNA databases contain huge collections of miRNA from viruses, microalgae, worms, flies, and mammals, their expression in cells and the exact targets of several of these miRNAs remain to be validated [
12].
The nematode
Caenorhabditis elagans provides an excellent model for miRNA-based studies that can be employed to control other parasitic nematodes [
13]. Several studies hitherto have been conducted on the development of strategies and identification of miRNA targets to control plant-parasitic nematodes [
14,
15,
16]. In the context of PPNs, the availability of the whole genome sequence of
M. incognita [
17] has led to several investigations focusing on elucidating potential miRNAs that can be used to control the pest in plants. The currently available literature provides us with information on miRNAs in the egg and J2 stages of
M. incognita [
15,
18]. Comprehensive information on miRNA of
M. incognita from all five life stages (egg, J2, J3, J4, and female) would be very useful to understand the physiological processes occurring in the nematode at various stages of its growth and also develop stage-specific markers or control strategies. To our knowledge, stage-wise exploration of miRNAs in
M. incognita has not been carried out before and this work presents information on the nematode miRNAs expressed at each stage of its life cycle.
3. Discussion
Reports on the sRNAs in
M. incognita are currently emerging that indicate its importance as a plant parasitic nematode, and this report aims to study the stage-wise expression of miRNA across various stages of its development. This is the first summary of information on its sRNA across all stages and can serve as a potential resource for miRNA-mediated control studies on
M. incognita in the future. Reports of miRNA-mediated regulation of
C. elegans genes across developmental stages are available that can provide a model for other nematodes [
13,
19,
20]. However, a comprehensive report on miRNAs from plant parasitic nematodes such as
M. incognita can be helpful to identify and study the regulation of plant parasitism-related genes in nematodes.
Earlier studies in miRNA from
M. incognita provide an excellent model to identify and study miRNAs of the RKN but are limited by the stages covered across the nematode life cycle [
15,
18]. In the present study, we sequenced small RNAs from all five stages of the
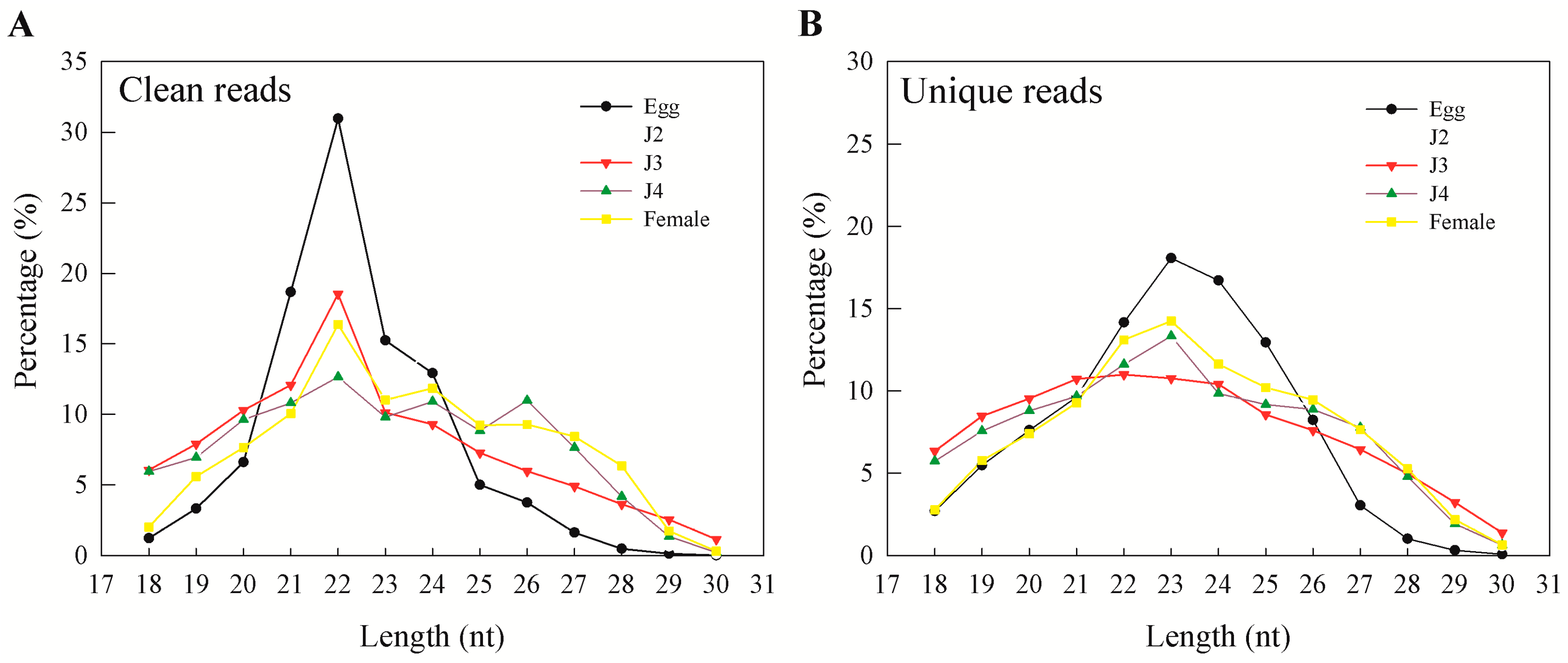
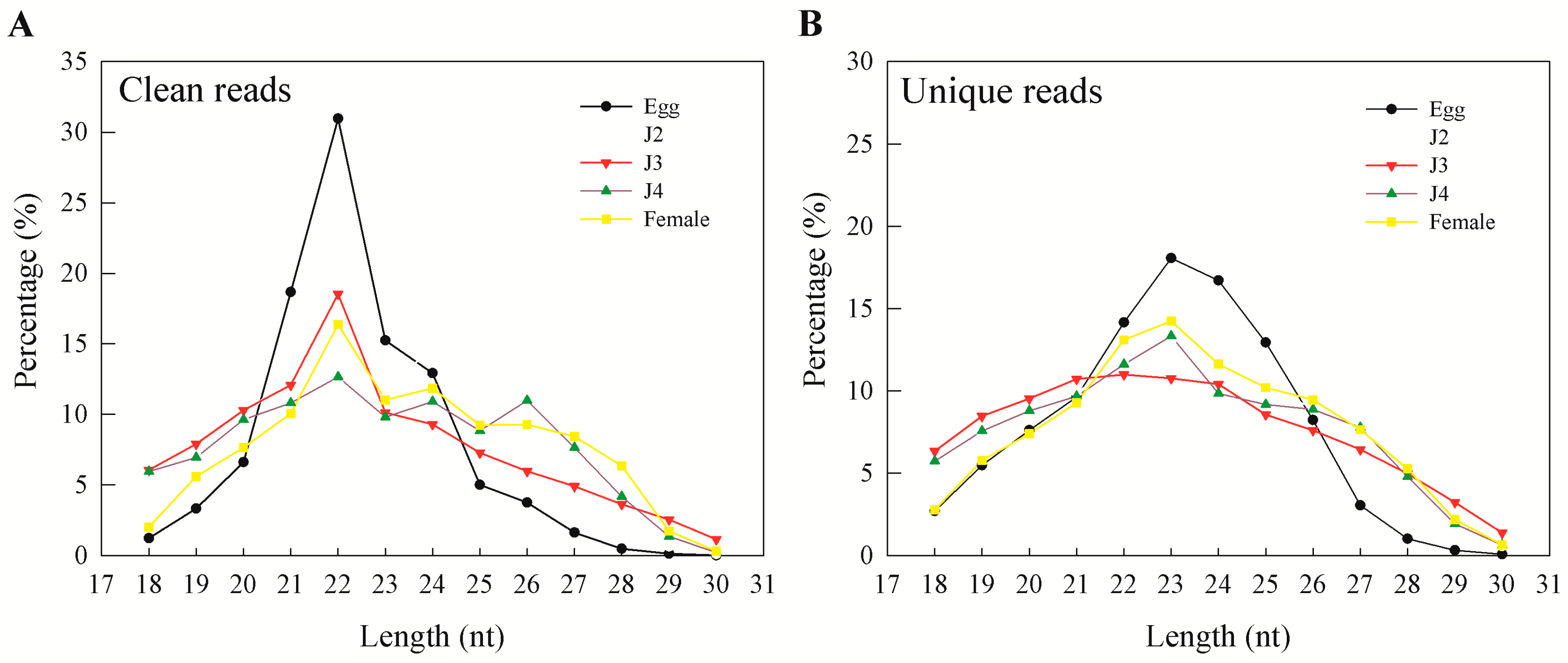
M. incognita life cycle. During our analysis, preliminary mapping of known sequence reads showed a high percentage of mapping that was greatly reduced when unique sequences were mapped to the genome. This was also observed earlier and reported by Wang and coworkers, who explained genetic polymorphisms, the incompleteness of the genome, and sequencing errors as reasons for the phenomenon [
15]. In our results, more than 95% of clean reads obtained from sequencing were longer than 18 nt. In general, the majority of the sRNAs range from 21 to 24 nt in size—information that is particularly helpful during initial predictions [
17]. Also, an sRNA length of 23 nt was found to be dominant in unique sequence reads, which is consistent with earlier reports on
M. incognita sRNAs [
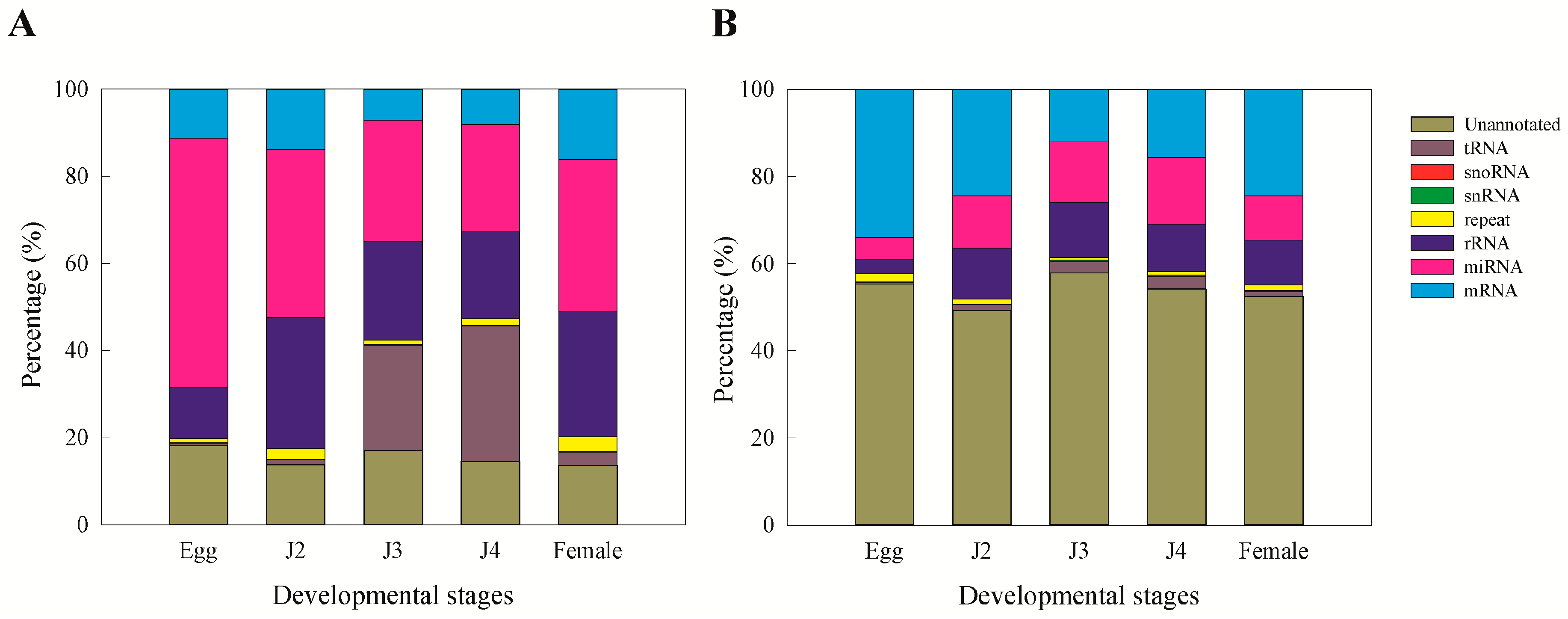
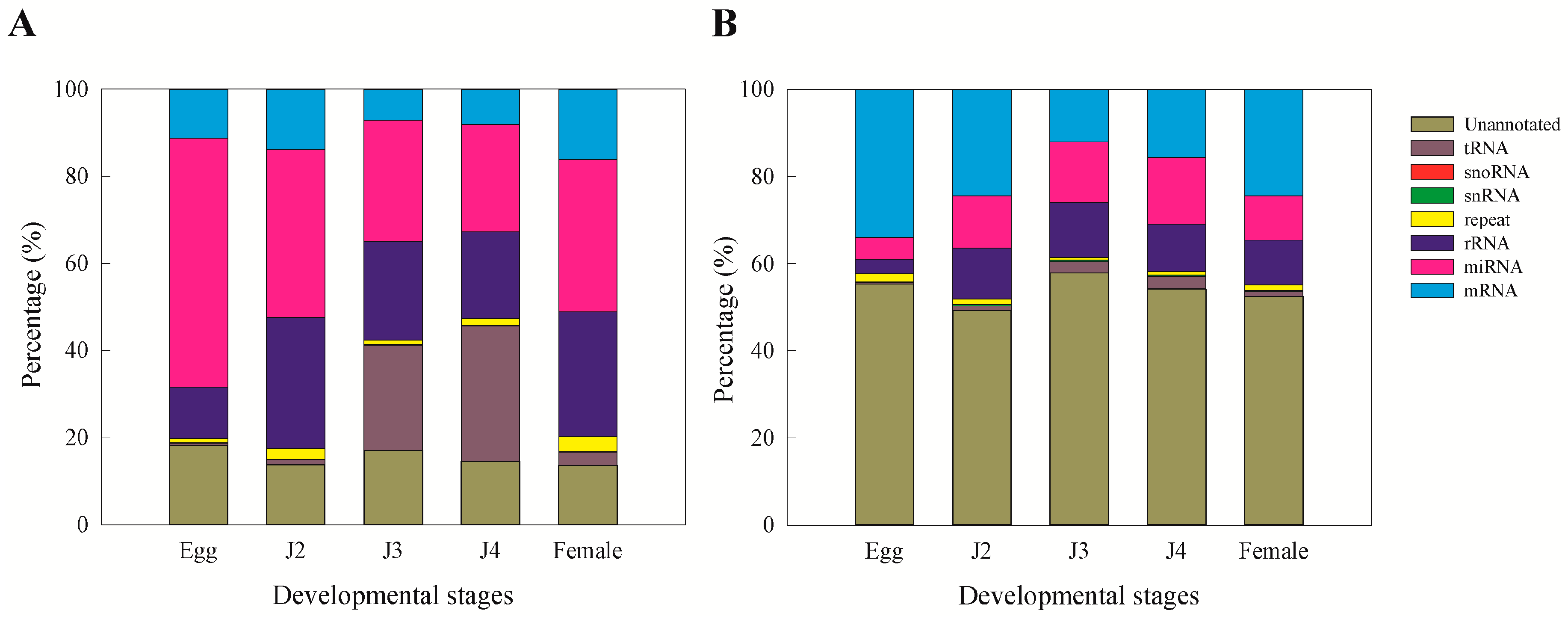
15]. Moreover, we observed that miRNAs and unannotated RNAs were the dominant classes of sRNAs, as also observed in a previous study involving sRNAs from the J2 stage of the nematode
M. incognita [
15].
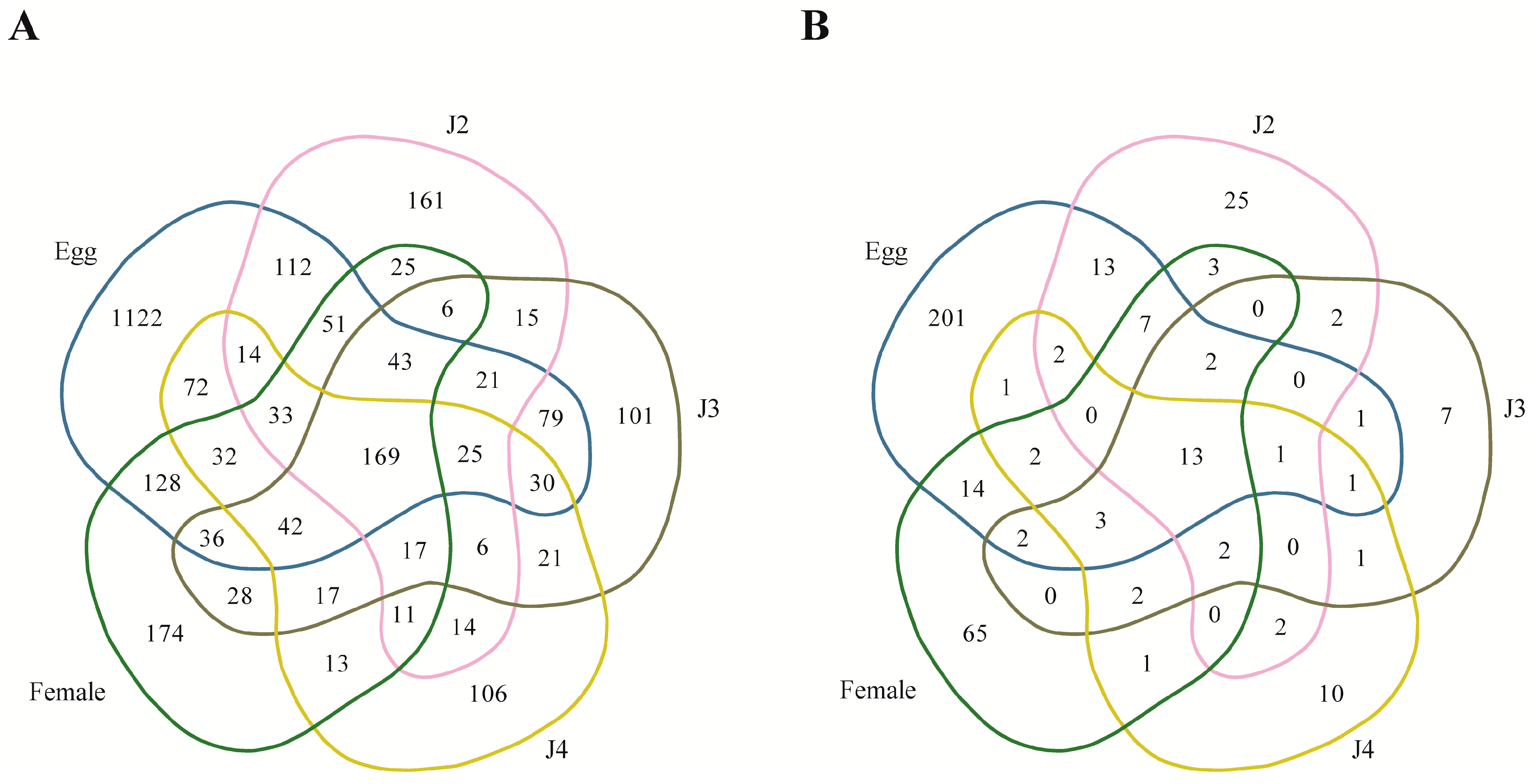
A total of 9325 known microRNA families and 1407 novel families (read count > 3) were identified in our study. In both cases, the egg stage contributed to the highest amount of microRNAs (known—37.8%; novel—46.1%). Highly expressed known miRNA families include miR-58, miR-1c, miR-124b, miR-71, miR-7062, miR-30e, miR-228, miR-100a, miR-6763, and let-7. Although the exact function of these miRNAs in the
M. incognita metabolism remains to be studied, we found reports on the probable function of these miRNAs in other species. The microRNA miR-1 has been known to target heat shock protein HSP60, causing glucose-mediated apoptosis in cardiac muscle cells [
21]. miR-124 has been reported to be expressed in neuronal cells and target anti-neural function protein SCP1 and splicing repressor PTBP1 [
22,
23]. In the case of miR-30, the targets include an integrin ITGB3, ubiquitin-conjugating E2 enzyme UBC9, and tumor protein p53 [
24,
25]. The microRNA families miR-58 and miR-71 have been extensively reported in
Caenorhabditis elegans, where miR-58 has been found to regulate the Transforming Growth Factor (TGF) pathway [
26,
27], and miR-71 functions in the control of aging in the model nematode [
28]. Cluster analysis showed that the miRNA families, both known and novel, were localized to 91 clusters in the genome of
M. incognita, where each cluster contained at least one gene of miRNA. We were able to confirm all clusters previously reported in
M. incognita [
15] except for the novel cluster consisting of NOVEL-1-1/NOVEL-39. Several of the clusters have also been found in other organisms, including
Drosophila, mouse, other nematodes, and mammals (
Table S7). MicroRNA families from
C. remanei (miR-239b),
Brugia malayi (miR-228), and
Ascaris suum (miR-234) have hitherto not been reported in
M. incoginta but reported in other nematodes (
Table 6). We were able to map 2147 known and 464 novel miRNA to scaffold assembly set sequences (ASM18041v1a) of
M. incognita. Mapping the microRNAs from several organisms showed that they are widely found to originate from the intergenic region or introns [
26,
29]. Our mapping data also show that our miRNAs were localized to introns and intergenic regions.
The number of known as well as novel miRNA in the present study were higher than the number of miRNAs predicted from
M. incognita based from prior studies [
15,
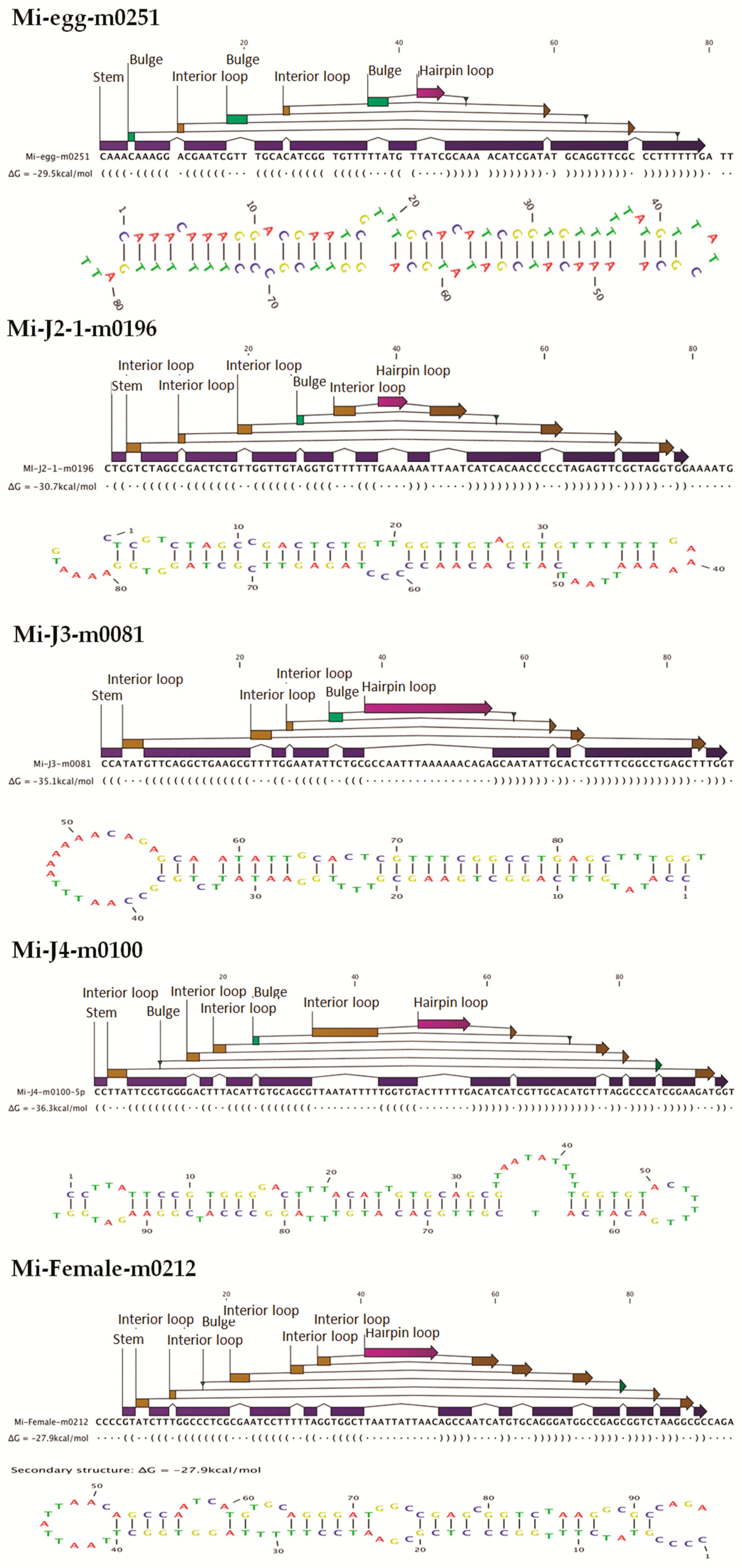
30]. This is because earlier reports used no mismatches and gaps as conditions for miRNA prediction. However, in our study, we matched sequences with no more than two mismatches and free gaps around the seed region to be considered as candidate miRNAs. This is because, as explained earlier, the available genome data is a draft genome and may not be perfect. Our conditions helped to identify a high number of probable miRNA candidates that may have been lost by the stringent conditions used in the prior studies. MIREAP makes use of the sRNA secondary structure, dicer cleavage site, and the minimum free energy to predict novel miRNAs. Candidate novel miRNAs must satisfy two major criteria, namely the presence of a ∼22-nt miRNA sequence contained in one arm of the hairpin, which forms a fold-back precursor structure, and conservation of that ∼22-nt miRNA sequence as well as its secondary structure. Further, we also used the minimal free folding energy index (MFEI) values to confirm the pre-miRNA sequences [
30]. In our studies, 41.7% of the novel miRNAs had negative MFEI values of over 0.85. The range of MFEI values for
M. incognita miRNAs was reported to be 0.38 to 2.4 [
18]. In our study, the average MFEI value was found to be 0.87, which suggests that the majority of the predicted novel miRNA are valid as miRNAs. Results of prior studies also showed miRNAs with MFEI values less than 0.85 [
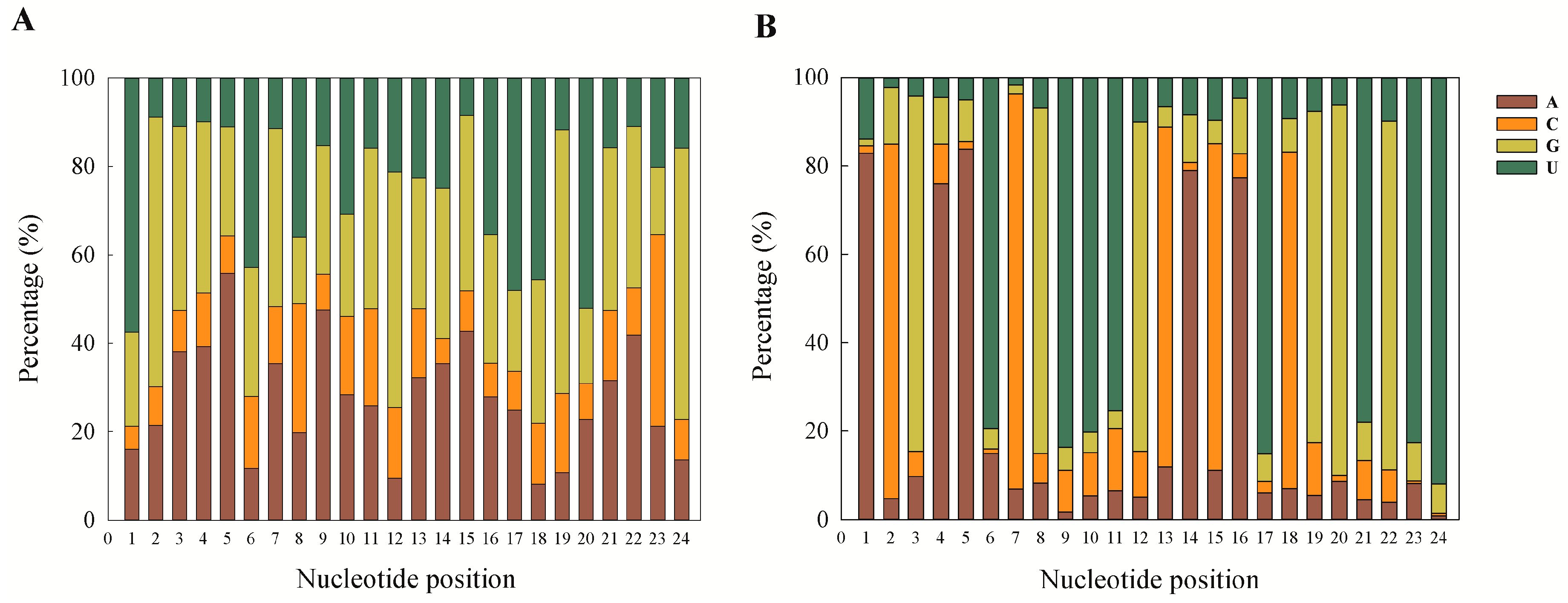
18]. The first nucleotide position of the known and novel mature miRNAs in the present study was found to be dominated by A and U. This is a ubiquitous phenomenon observed in miRNA studies [
15,
18,
31,
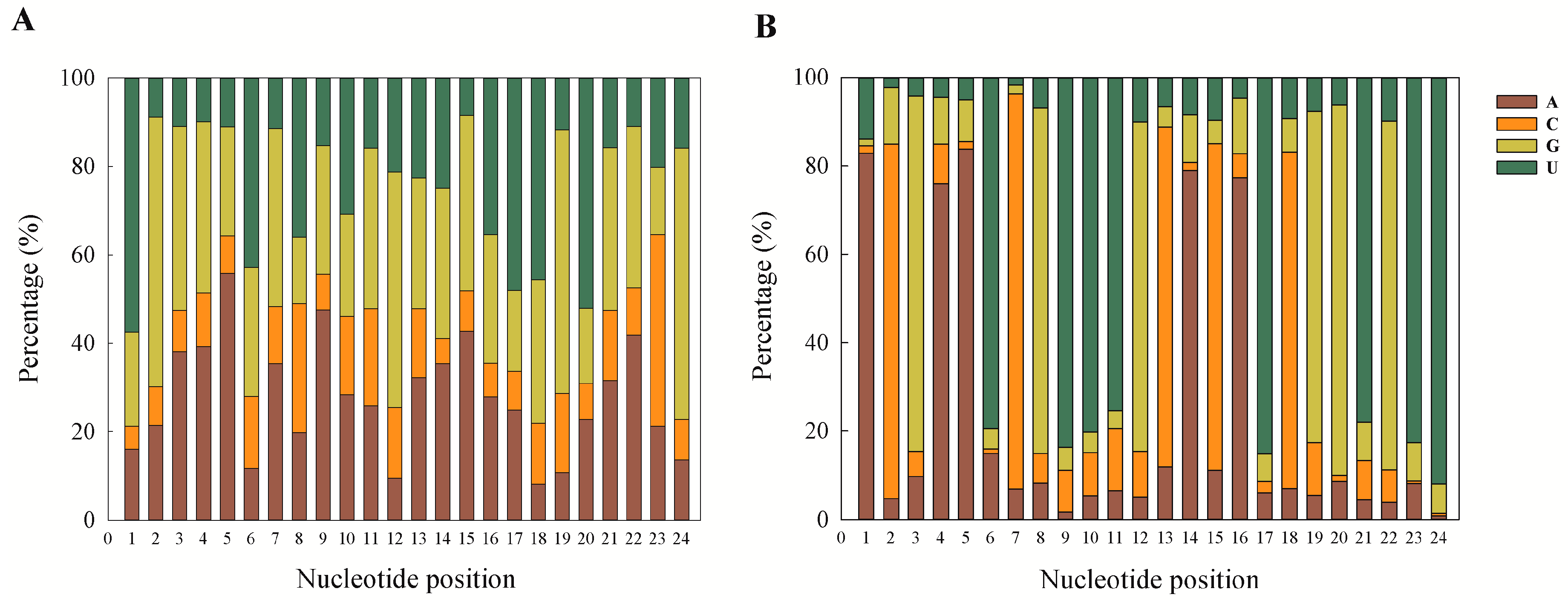
32]. This can also be taken as a putative marker for confirmation of sRNA sequences as miRNAs, and our results are also in accordance with previous reports.
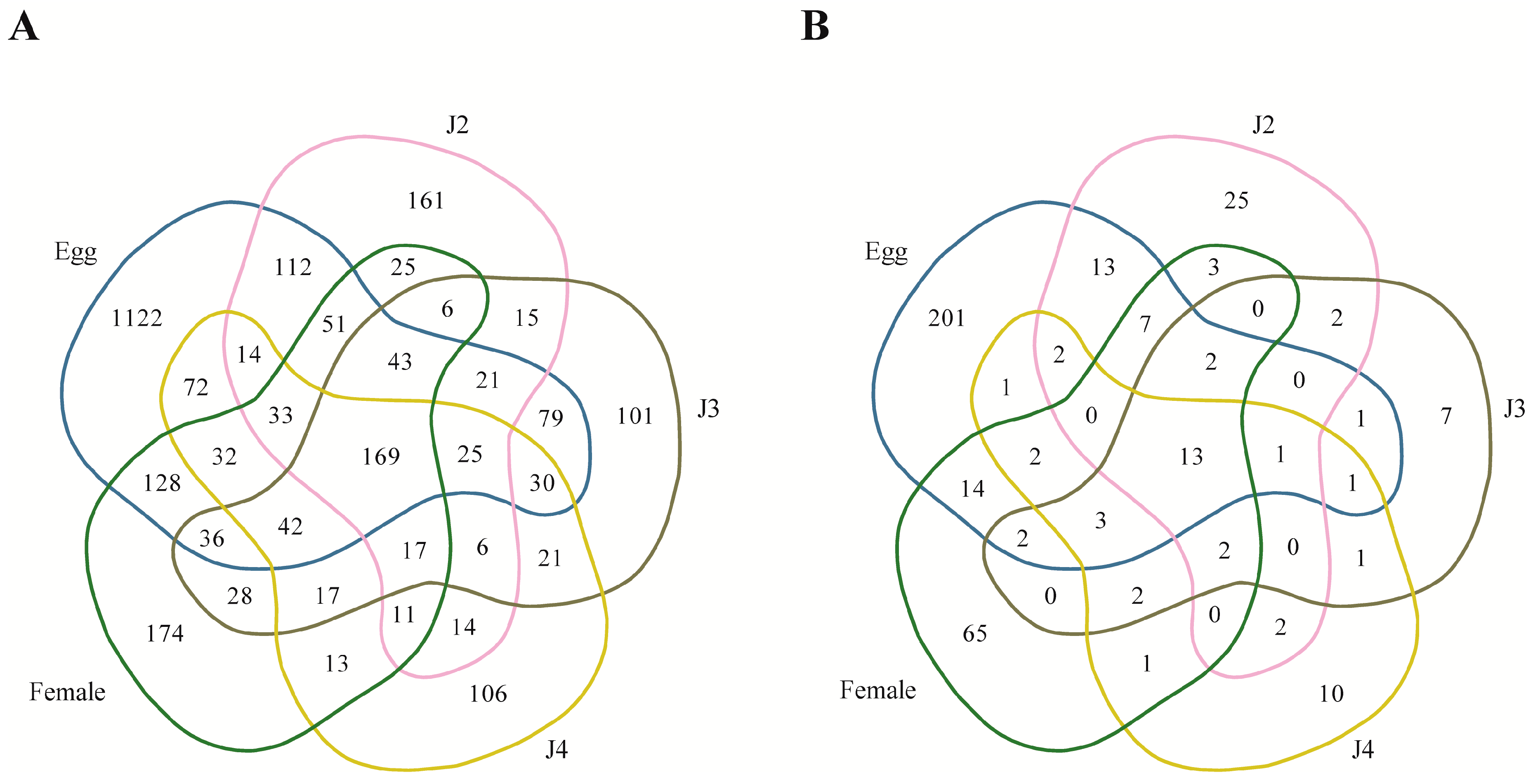
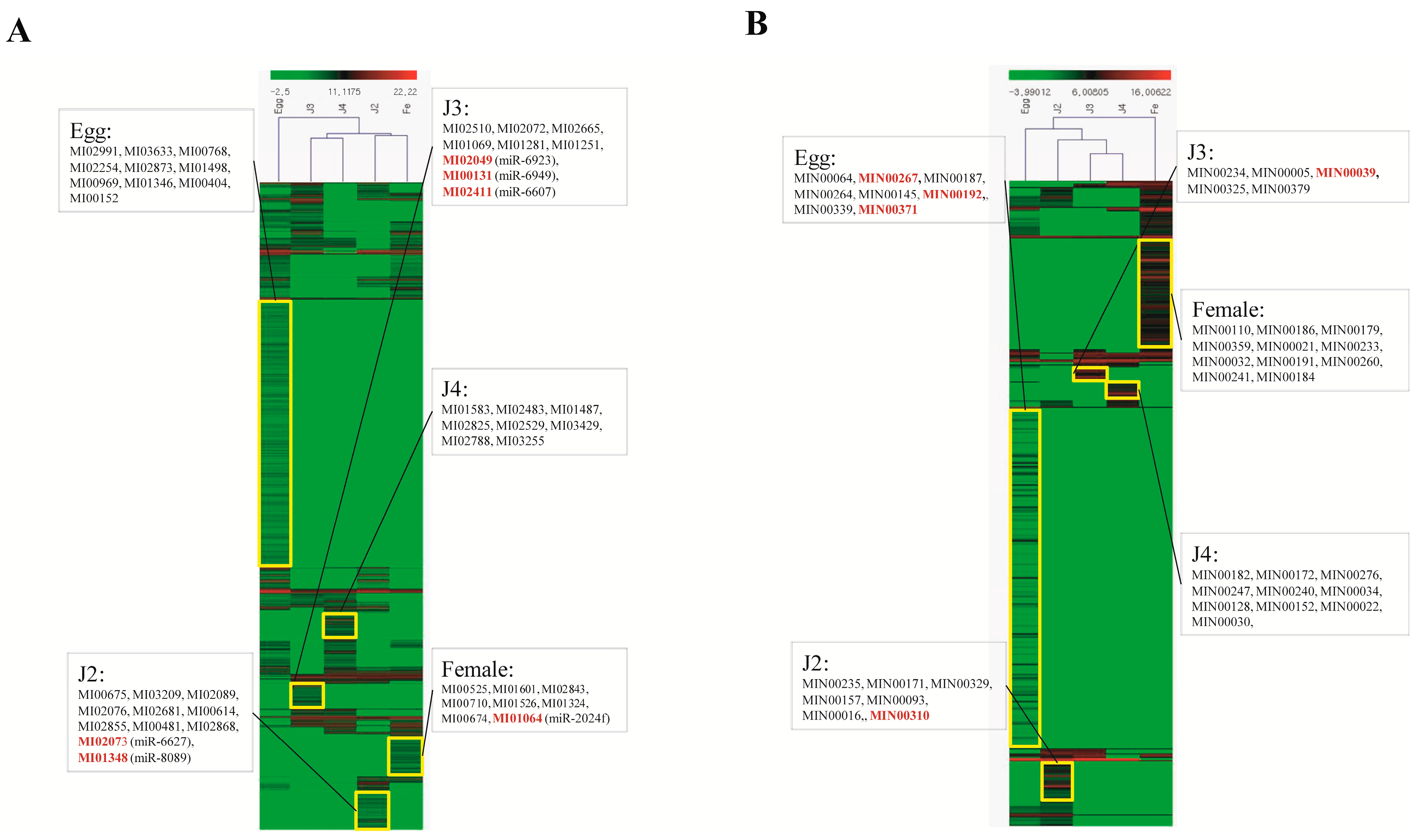
Among the stage-specific miRNAs, known miRNAs specific to the egg stage include miR-286, which is highly specific to the embryo stages of
Drosophila melanogaster [
33] (
Table 8). Other stage-specific miRNA families in the egg stage include miR-493, which is involved in the inhibition of liver metastasis in the mouse, and miR-27, which regulates cholesterol homeostasis and fatty acid metabolism [
34,
35]. Specific miRNA families from the J2 stage include miR-2401, miR-4451, and miR-2a, which have been reported to be synthesized by bovine animals in response to viral infection, occurring in normal and malignant human B cells, and involved in the repression of
Drosophila pro-apoptotic factors, respectively [
36,
37,
38]. In the J3 stage, specific miRNAs included miR-8 and miR-187, which are involved in the regulation of atrophin and the control of ovarian cancer, respectively [
39,
40]. Highly expressed specific miRNA families in the J4 stage included miR-6736, miR-2072-3, miR-7348, miR-2032a, and miR-2163. Specific miRNA families in the female stage are miR-17 and miR-3180, which are involved in cancers, and miR-133, which is reported to be expressed in muscle tissues [
41,
42,
43].
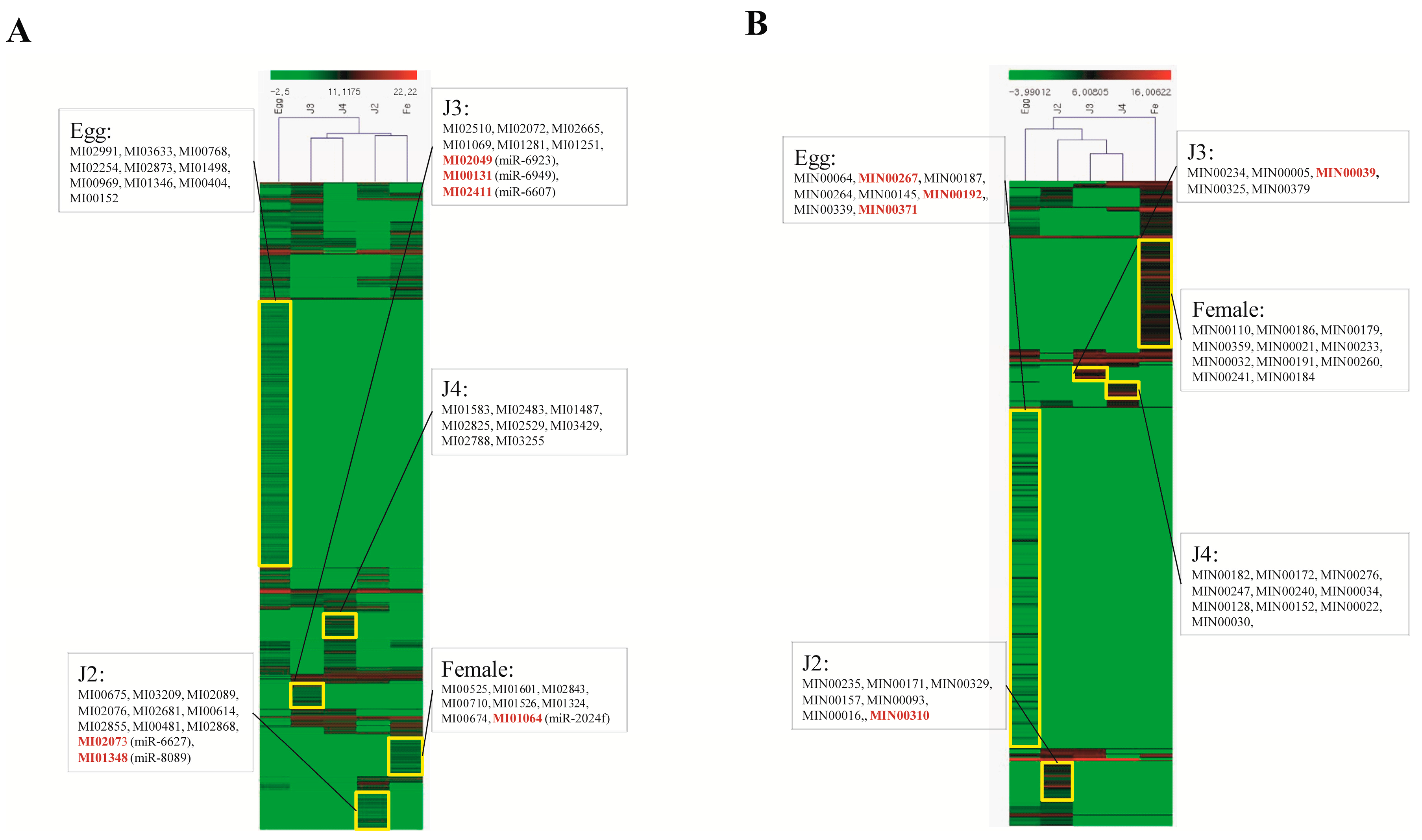
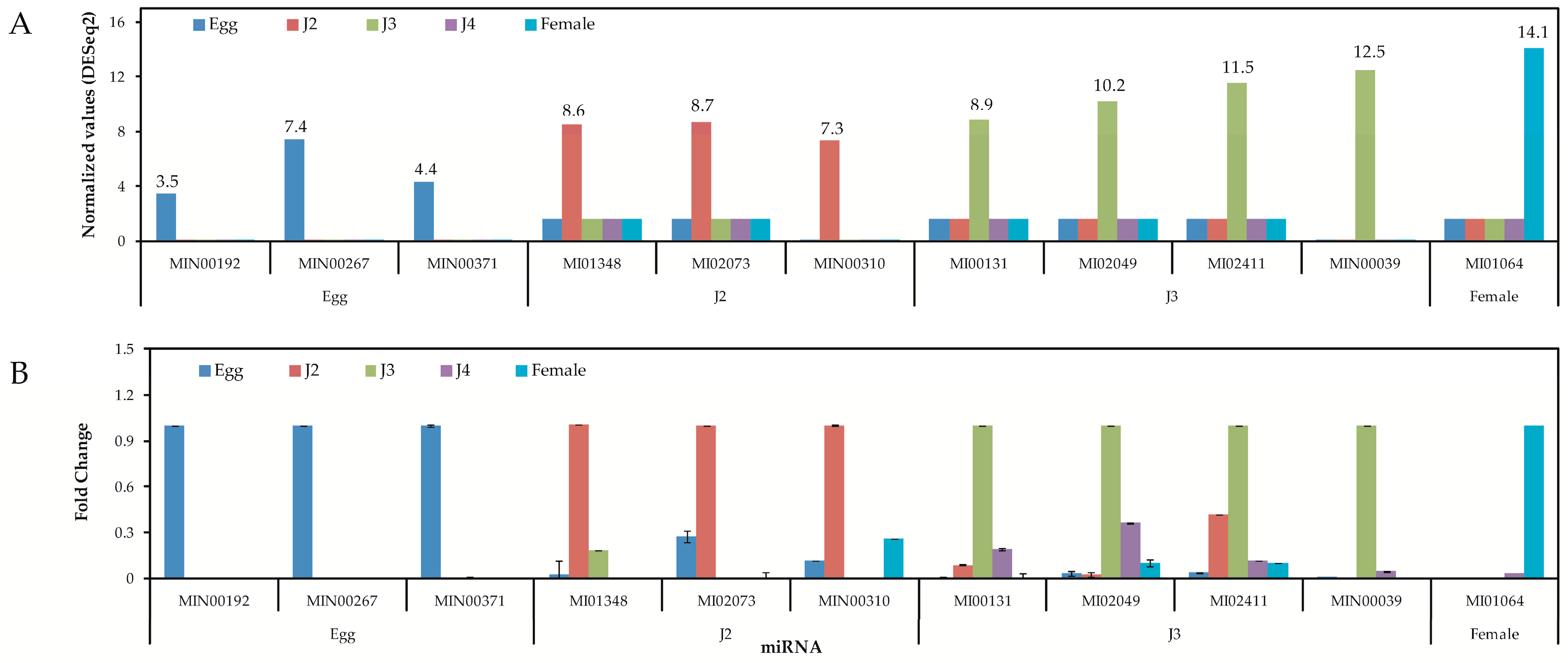
Quantitative analysis of expression using PCR showed that the stage-specific miRNAs observed in the sequencing based studies were truly expressed in the nematode. We were able to confirm that these miRNAs were authentic and 14 of the 40 known and novel miRNAs in the egg, J2, J3, J4, and female exhibited stage-specific high expression patterns in both sequencing reads and quantitative PCR analyses. However, though the stage-specific miRNA candidate from the J4 stage was highly expressed in quantitative analyses, its expression was found to be high in the J3 stage when studied using PCR. Also, a few miRNAs that were shown to be stage-specific in digital expression studies were found to be expressed in other stages also, albeit in much lower quantities. Among the stage-specific known microRNAs taken for validation, MI01348 has been reported earlier in humans [
44,
45]. Other known stage-specific microRNAs were reported to be expressed in the mouse (MI02073), the chicken (MI00131), and chordate and cnidarian species (MI01064) [
44,
46,
47,
48]. However, the functions of these microRNAs in
M. incognita remain to be elucidated and hitherto are only collected in an miRNA database—miRBase. In summary, through the present study, we have tried to amend the available data on
M. incognita miRNAs to include miRNAs from all stages of development. This information can be very useful for any studies on the stage-specific control of the nematode or stage-specific functions in the future.
4. Materials and Methods
4.1. Sample Preparation
The nematode M. incognita was maintained in RKN-susceptible tomato plant roots (Solanum lycopersicum var. Rutgers) in a greenhouse at 25 °C. A fresh batch of plants was prepared for collection of M. incognita at various stages. Nematode samples were collected quickly at each developmental stage, i.e., egg, J2, J3, J4, and female, and snap frozen using liquid nitrogen, followed by temporary storage at −70 °C. Total RNA from each sample was extracted using CoreZol reagent (Corebio, Seoul, Korea) as per the manufacturer′s instructions.
4.2. Small RNA Library Construction
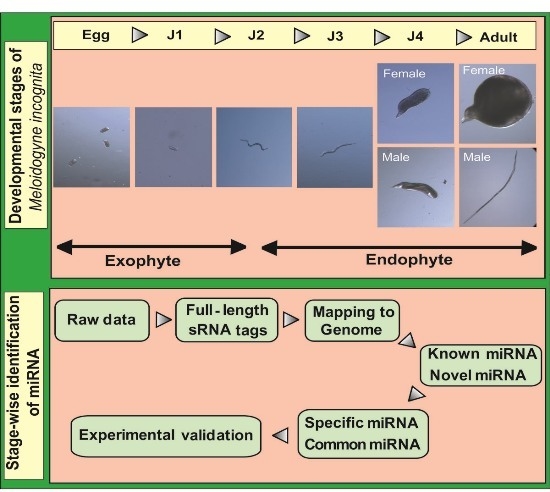
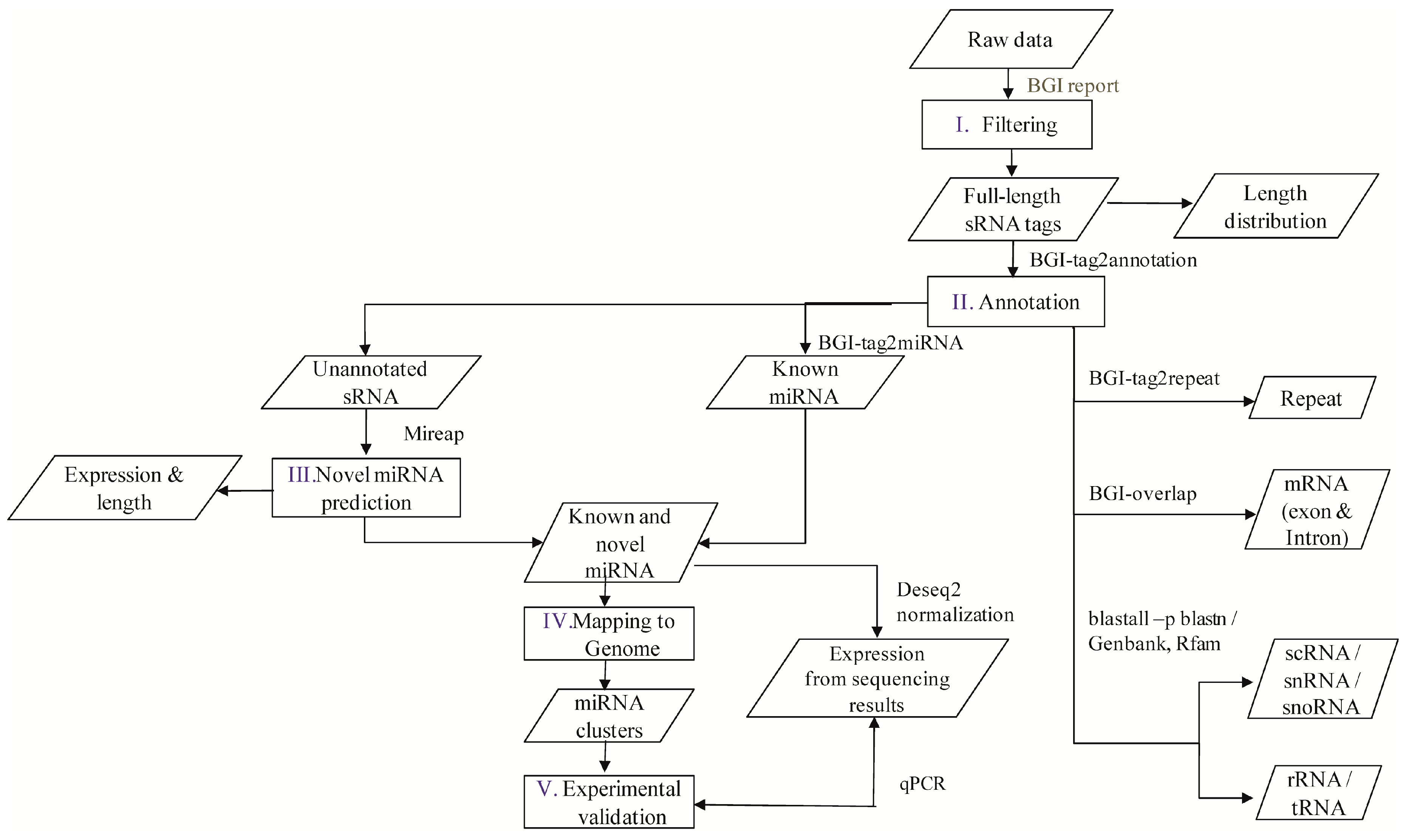
A flowchart depicting the flow of experiments carried out is explained in the abstract graphic. Isolation of small RNAs and library construction were performed as described by [
49]. In brief, from the isolated total RNA, nucleotides of length 18–30 bp were obtained by gel separation and the purified short nucleotide RNA sequences were ligated with 3′ adapter 6–20 nt and 5′ adapter 12–25 nt at both ends, which was followed by purification according to the manufacturer’s protocol (T4 RNA Ligase, 200 U, 30 U/µL, Takara: D2050, Seoul, Korea). Ligated small RNAs were reverse transcribed with the complimentary sequence of 3′ adaptor (SuperScriptH III Reverse Transcriptase, Invitrogen: 18064-014, Carlsbad, CA, USA), and amplified. Finally, the PCR amplification products were sequenced at BGI (Beijing Genome Institute, Beijing, China) using Illumina HiSeq 2000 (Illumina Inc., San Diego, CA, USA) [
50].
4.3. Annotation of M. incognita sRNAs
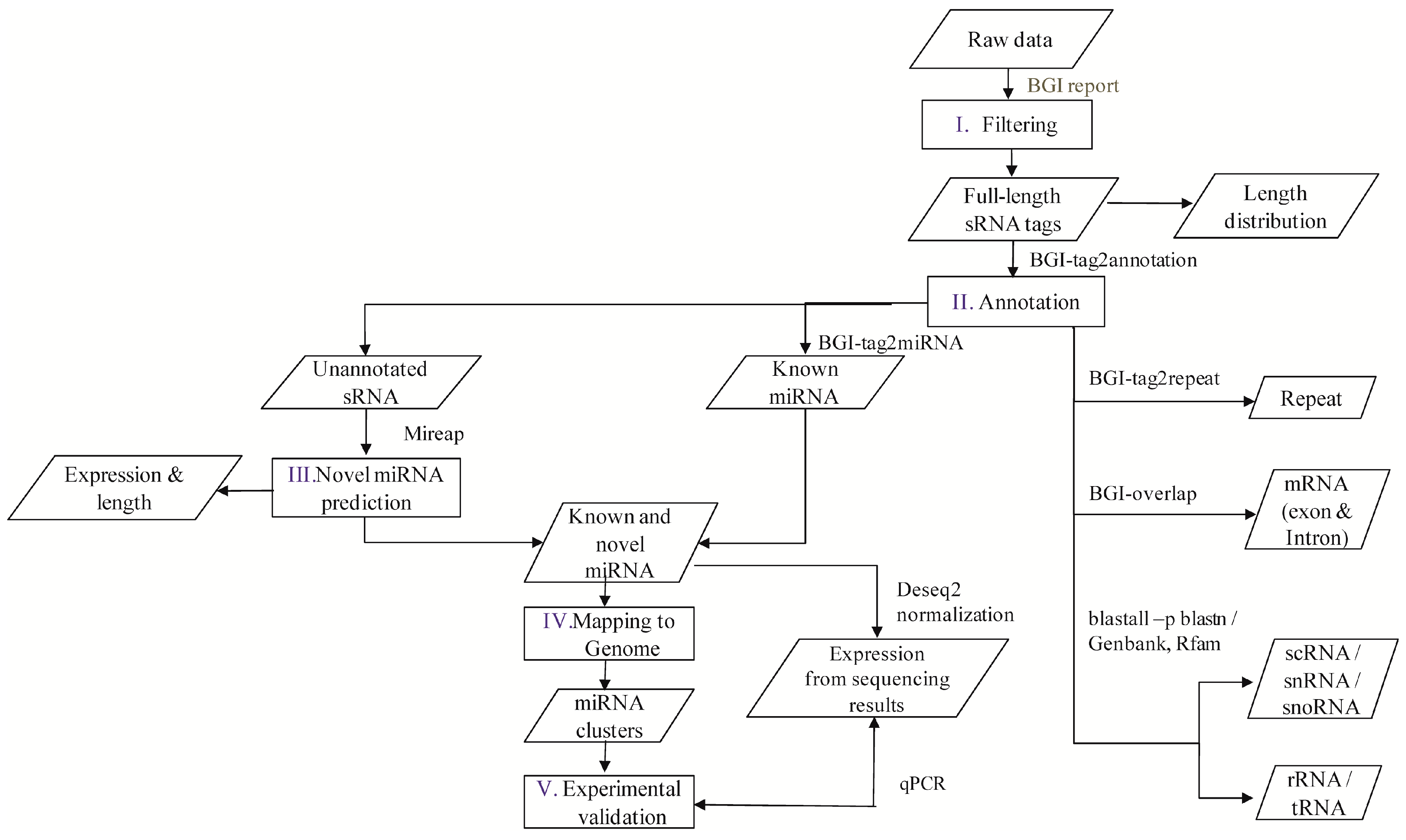
The software developed and provided by BGI was used to profile the data from high-throughput sequencing. After obtaining the FASTQ format files, the data were processed as per the following steps: (1) exclusion of low quality reads; (2) elimination of reads with 5′ primer contaminants; (3) elimination of reads without 3′ primer; (4) removal of reads without the insert tag; (5) removal of reads containing poly A and (6) elimination of reads shorter than 18 nt. The final clean reads were obtained and their length distribution was summarized. These clean read raw data are available at National Center for Biotechnology Information-Sequence Read Archive (NCBI-SRA) with accession numbers SRP076824, SRP077023, SRP077024, SRP077025, and SRP077026. In the present study, two versions of the reference genome were used, i.e., the Contig assembly sequence set (ASM18041v1) from NCBI was used for annotation and miRNA prediction, whereas scaffold assembly set sequences (ASM18041v1a) from the WormBase database (Available online:
http://www.wormbase.org/) were used for miRNA mapping.
Clean reads of length ranging from 18 to 30 nt were analyzed to classify the small RNAs. First, the purified tags from the short clean reads were compared to the M. incognita genome sequence from NCBI (ASM18041v1). To find repeat associated RNA, small RNA tags were aligned to the reference genome using tag2repeat (BGI). The ribosomal RNA (rRNA), small nucleolar RNA (snoRNA), small nuclear RNA (snRNA), and transfer RNA (tRAN) were identified by Basic Local Alignment Search Tool (BLAST-NCBI) against the Genbank and Rfam databases. Small RNA tags were aligned to the reference genome, particularly the gene co-ordinates (exons and introns), to find the degraded fragments of mRNA by BGI-overlap software (Genome Institute, Beijing, China). To make every unique small RNA is mapped to only one annotation, the classification was followed by BGI small RNA pipeline (tag2annotation-BGI) such as rRNA, small cytoplasmic RNA (scRNA), small nucleolar RNA (snoRNA), small nuclear RNA (snRNA), tRNA, repeat sequences, and degradation mRNA, i.e., exons (sense and antisense) and introns (sense and antisense); the remaining reads were classified as unannotated small RNAs.
4.4. M. incognita miRNA Analysis
Known miRNAs were identified by matching the sRNAs to miRBase (v. 18) animal mature miRNA database using Tag2miRNA (BGI). Matched sequences with no more than two mismatches and two gaps around the seed region were considered as candidate conserved miRNAs (mismatches ≤ 2, and gaps ≤ 2 around the seed region). A small RNA around the location of an annotated pre-miRNA had at least 16 nt overlap, aligning to known miRNAs in miRBase. Also, they were aligned to the pre-miRNA and mature miRNA of all plants and animals in miRNAs, allowing two mismatches and free gaps. Any candidate small RNA sequence with more than three reads was considered a reliable representation of a miRNA molecule. Unannotated sRNAs that did not belong to any class of RNA but could be aligned to the reference genome were subjected to novel miRNA prediction using MIREAP software (Available online:
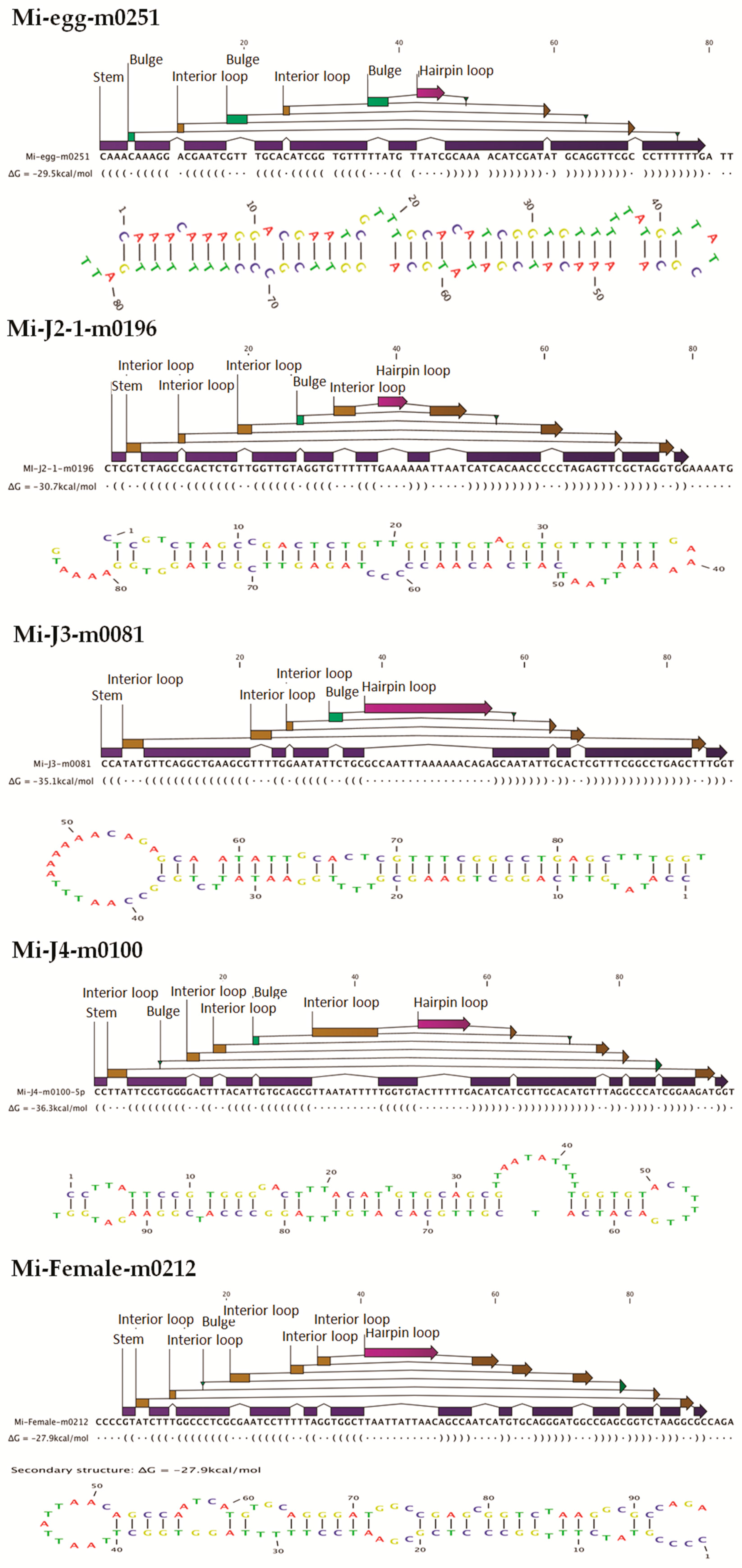
http://sourceforge.net/projects/mireap) with default parameters. Candidate novel miRNAs satisfying two major criteria, namely presence of a ~22-nt miRNA sequence contained in one arm of the hairpin which when predicted forms a fold-back precursor structure and conservation of that ~22-nt miRNA sequence and its secondary structure were selected. The number of mature miRNAs with predicted hairpin was not less than five in the alignment result. Secondary structures of the miRNAs were confirmed for the presence of stem-loop structure. Pre-miRNA sequences from MIREAP results were used to calculate the minimal folding energy index (MFEI) [
30].
The family distribution of the miRNAs and their nucleotide bias were obtained statistically to reduce any bias in the expression and coding of the miRNAs. For cluster analysis, miRNAs were grouped into clusters if they were located within a 2000 bp threshold in the genome during mapping [
15]. Known and novel miRNAs with a read count >10 were mapped with reference to the
M. incognita genome assembly database (ASM18041v1a; WormBase) to study their location in the genome. Stage-specific miRNAs (read count ≥ 3) were normalized using the DESeq2 package [
51]. The DESeq2 normalized values of the miRNAs were taken as an estimation of the specific miRNA expression. For experimental validation we chose 40 stage-specific miRNAs combining all stages and three commonly expressed miRNAs as candidates for internal standards (
Tables S13 and S14).
4.5. Experimental Validation of M. incognita miRNA Expression
Quantitative PCR analysis was used to experimentally validate the expression of miRNAs. Samples of the nematode M. incognita were taken at different developmental stages (egg, J2, J3, J4, and female) and used to isolate total RNA. Nematodes of each developmental stage were carefully collected and total RNA was extracted, followed by storage at −70 °C. Synthesis of cDNA from total RNA samples was carried out using Universal cDNA synthesis kit II (Exiqon, Woburn, MA, USA). For qPCR ExiLENT SYBR® Green kit from Exiqon (Woburn, MA, USA) was used and qPCR was performed on ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with S.D.S 2.2.2 software, as follows: 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 60 s with three independent replications containing three technical repeats each time. Negative control sets with no cDNA and no master mix were also tested. Three miRNAs, MI02950 (miR-71-5p), MI03018 (miR-58-3p), and MI02743 (miR-7904-3p), which consistently had analogous expression profiles at all stages of the nematode life cycle, were analyzed to be used as internal control for qPCR studies. The real-time qPCR studies were carried out as two independent experiments with at least three technical repeats. We calculated the fold changes in gene expression from the ΔΔCt values for all the selected miRNAs.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}