
Anti-Oncogenic gem-Dihydroperoxides Induce Apoptosis in Cancer Cells by Trapping Reactive Oxygen Species

 ,
,  ,
, 
Abstract
:
1. Introduction
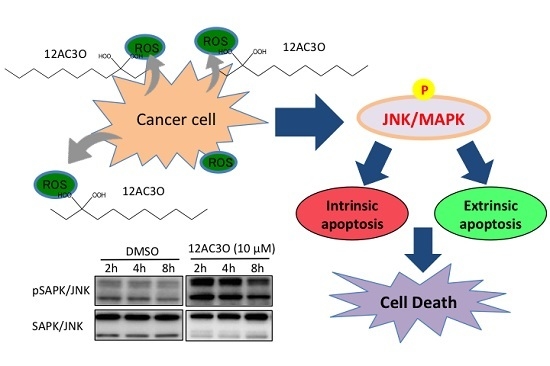
2. Results
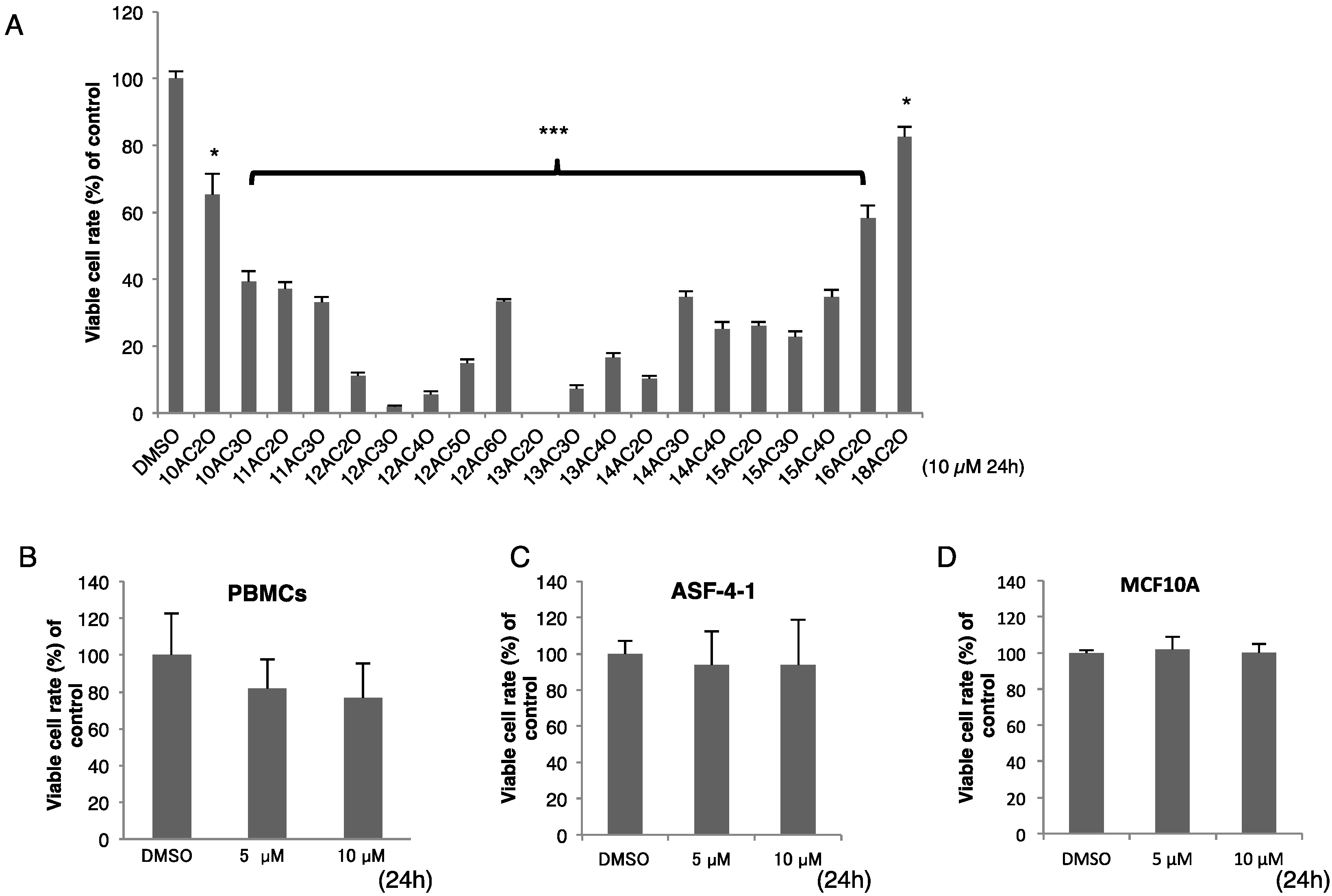
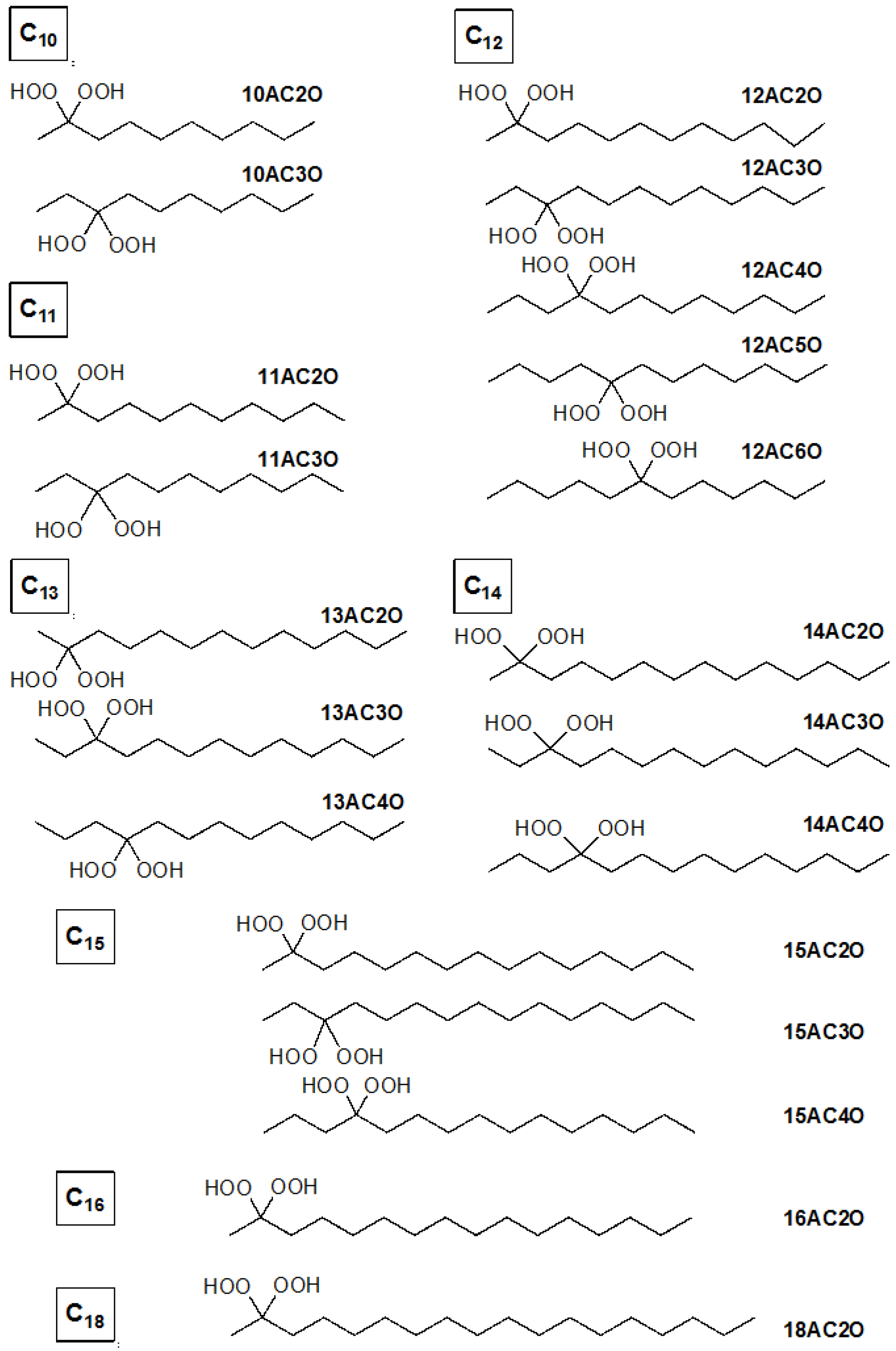
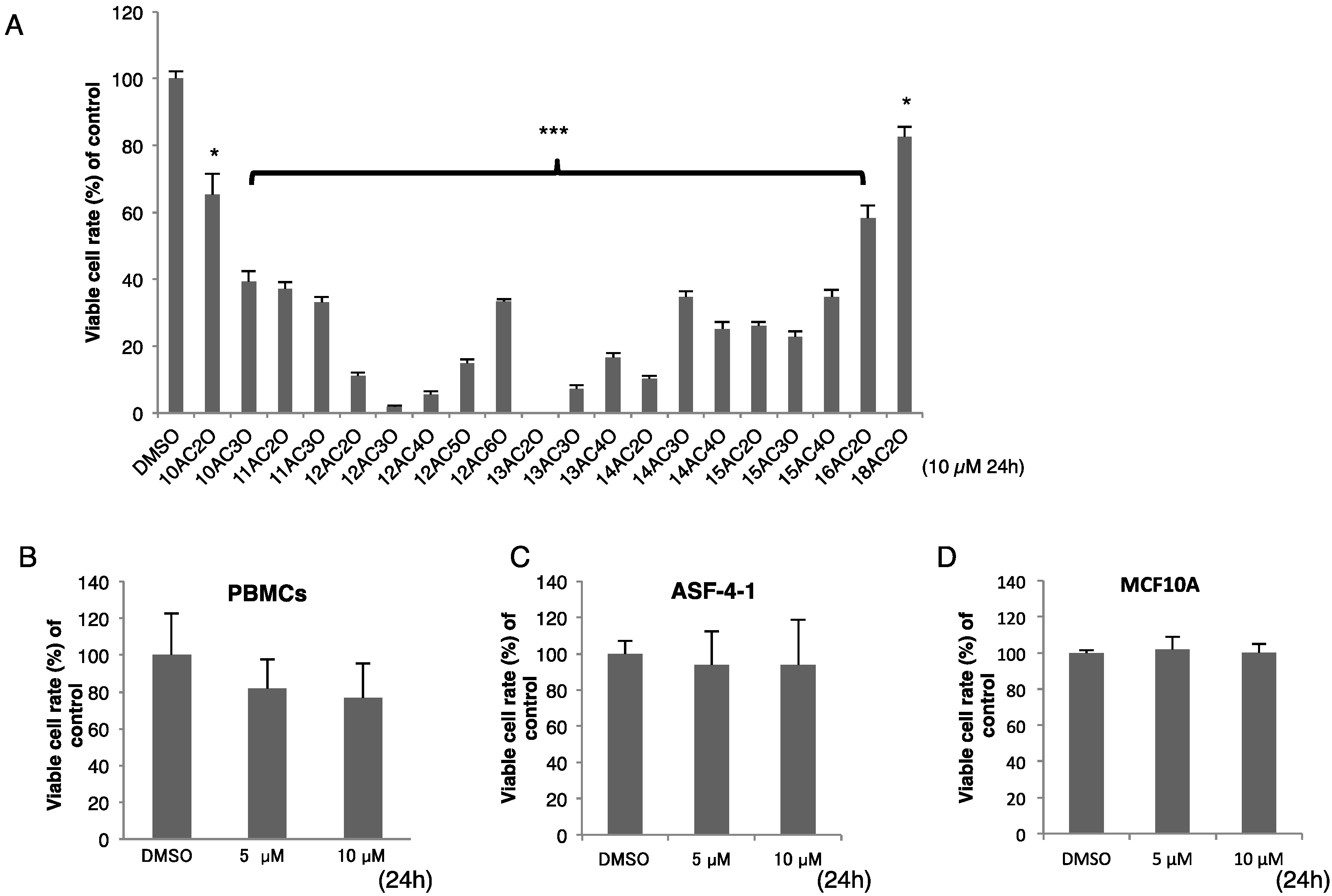
2.1. Effects of DHPs on Cell Growth in Human Leukemia K562 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | IC50 Value (µM) |
|---|---|
| 12AC2O | 1.65 |
| 12AC3O | 0.81 |
| 12AC4O | 1.69 |
| 12AC5O | 2.14 |
| 12AC6O | 3.83 |
| Etoposide | 5.30 |
| Cytarabine | 2.10 |
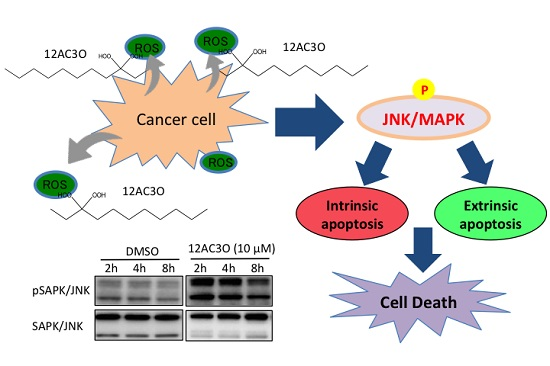
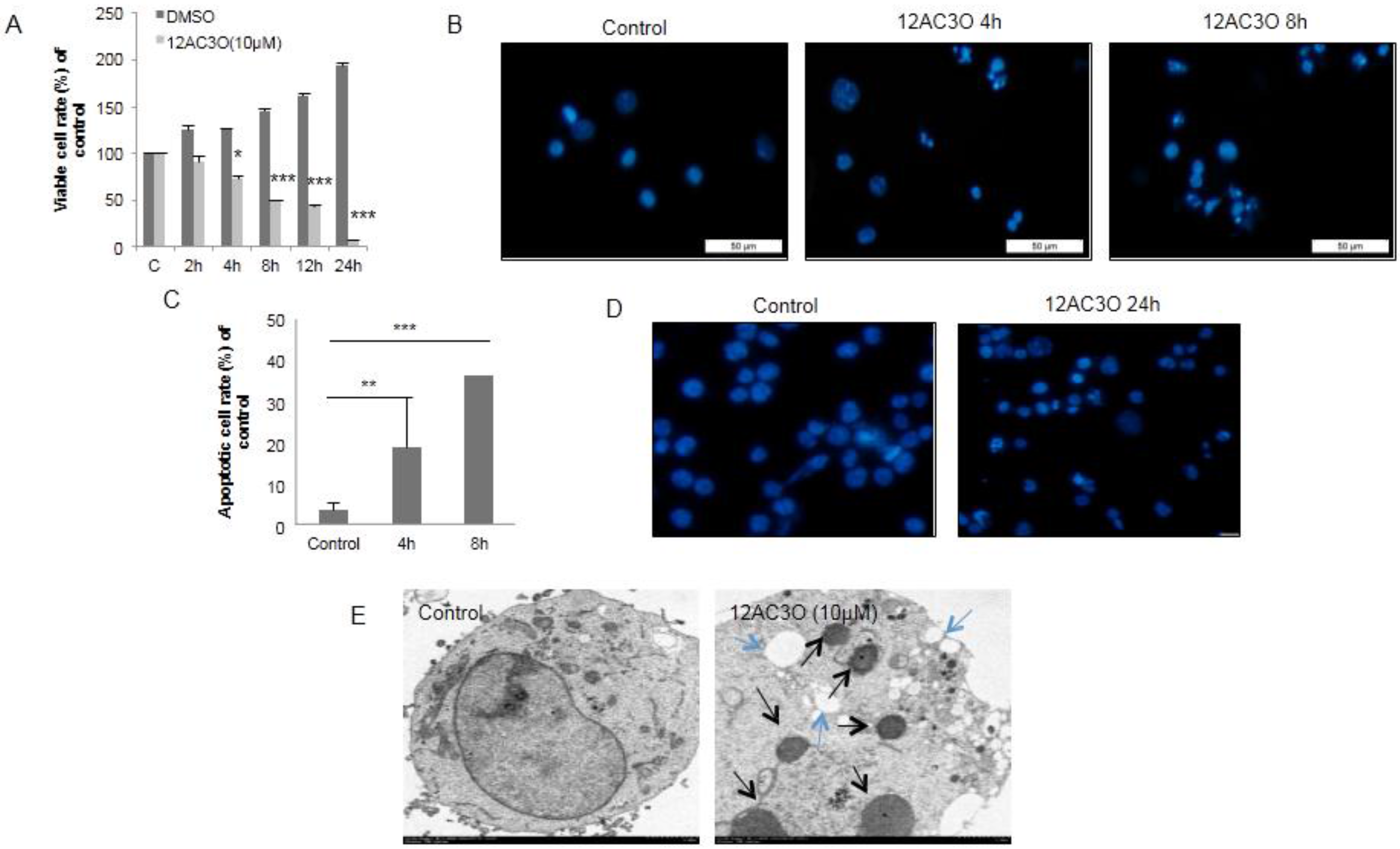
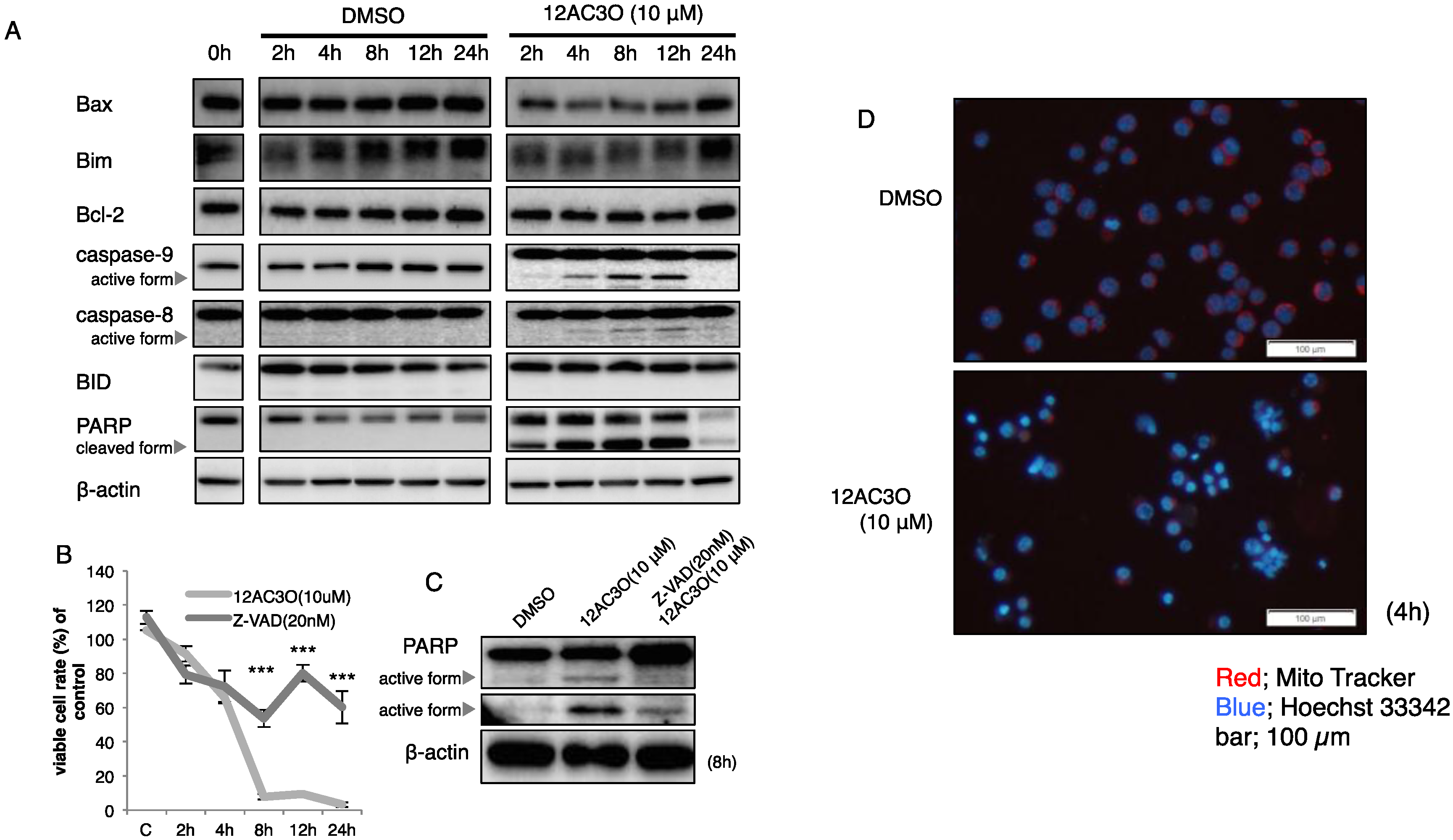
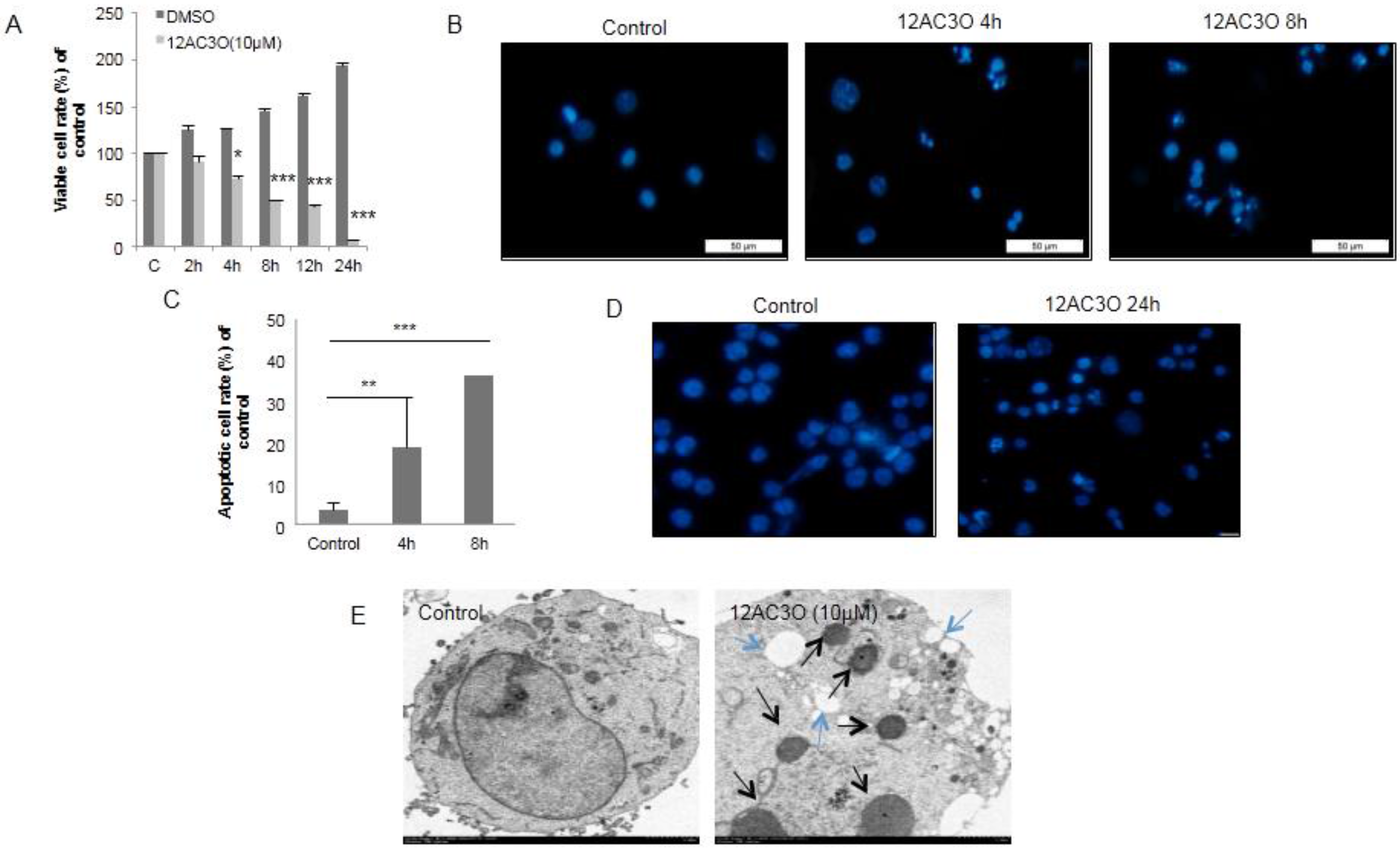
2.2. 12AC3O Induced Apoptotic Cell Death

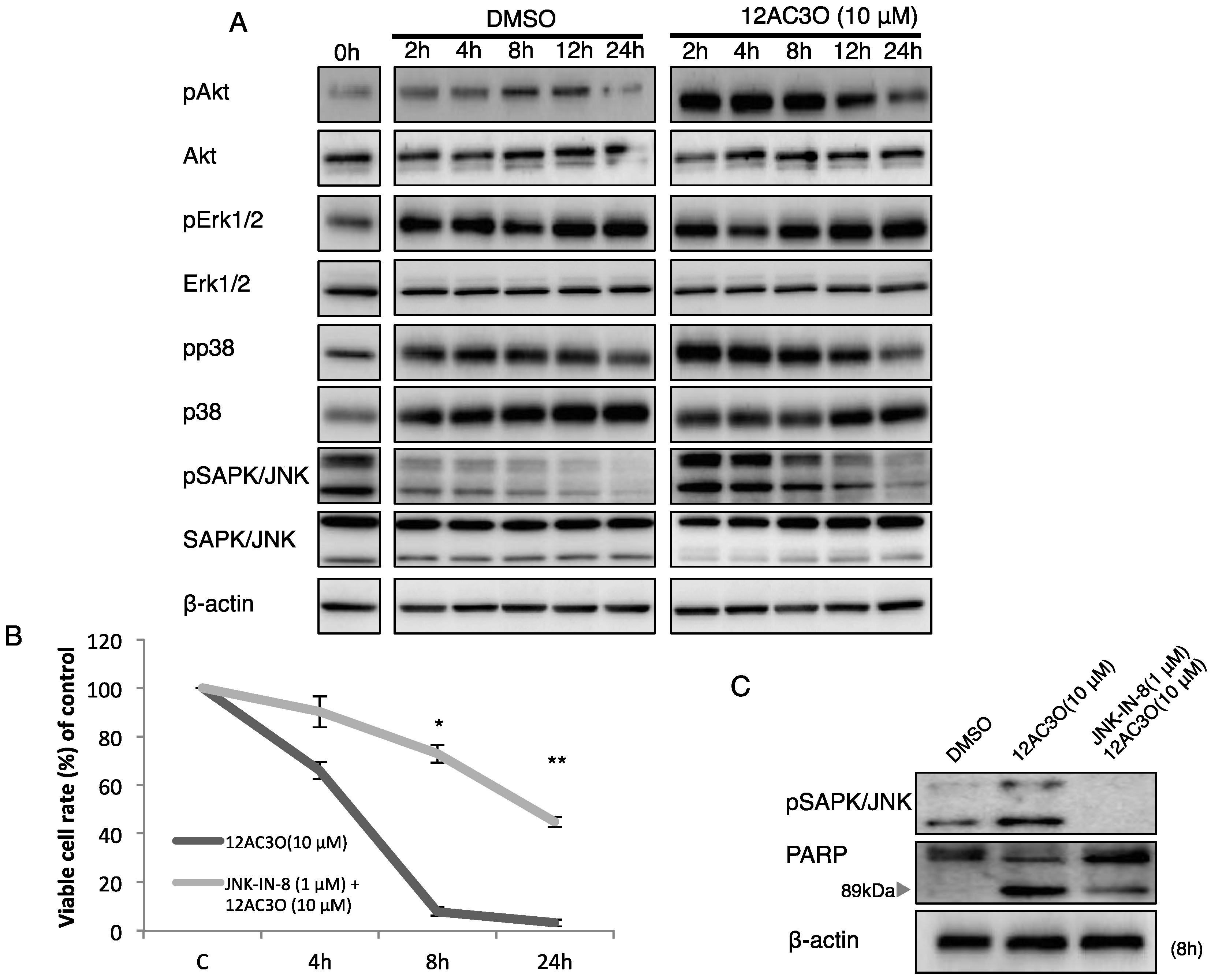
2.3. The SAPK/JNK Was Up-Regulated in the Early Phase of Treatment with 12AC3O
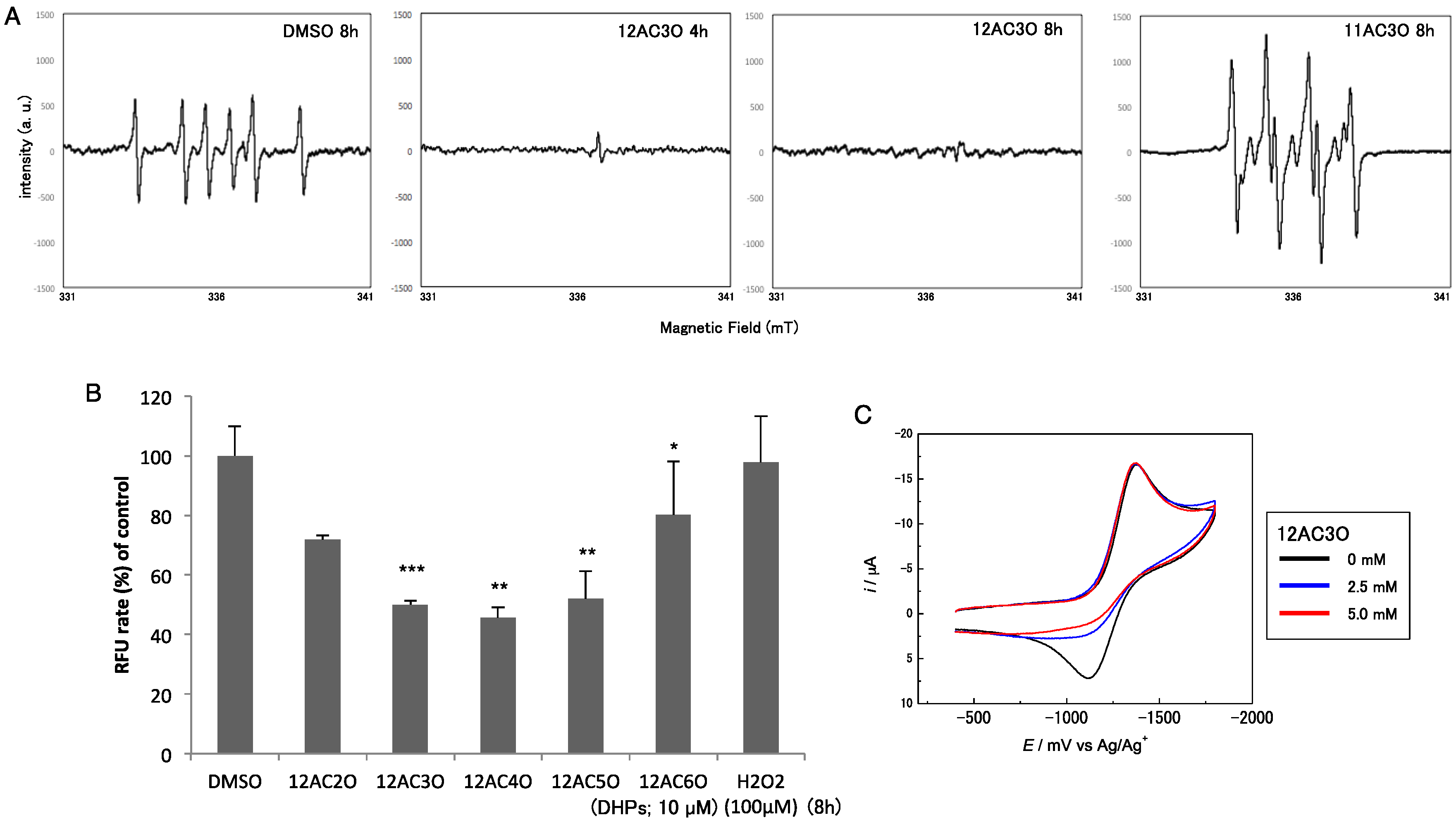
2.4. Superoxide-Scavenging Effect of 12AC3O on K562 Cells
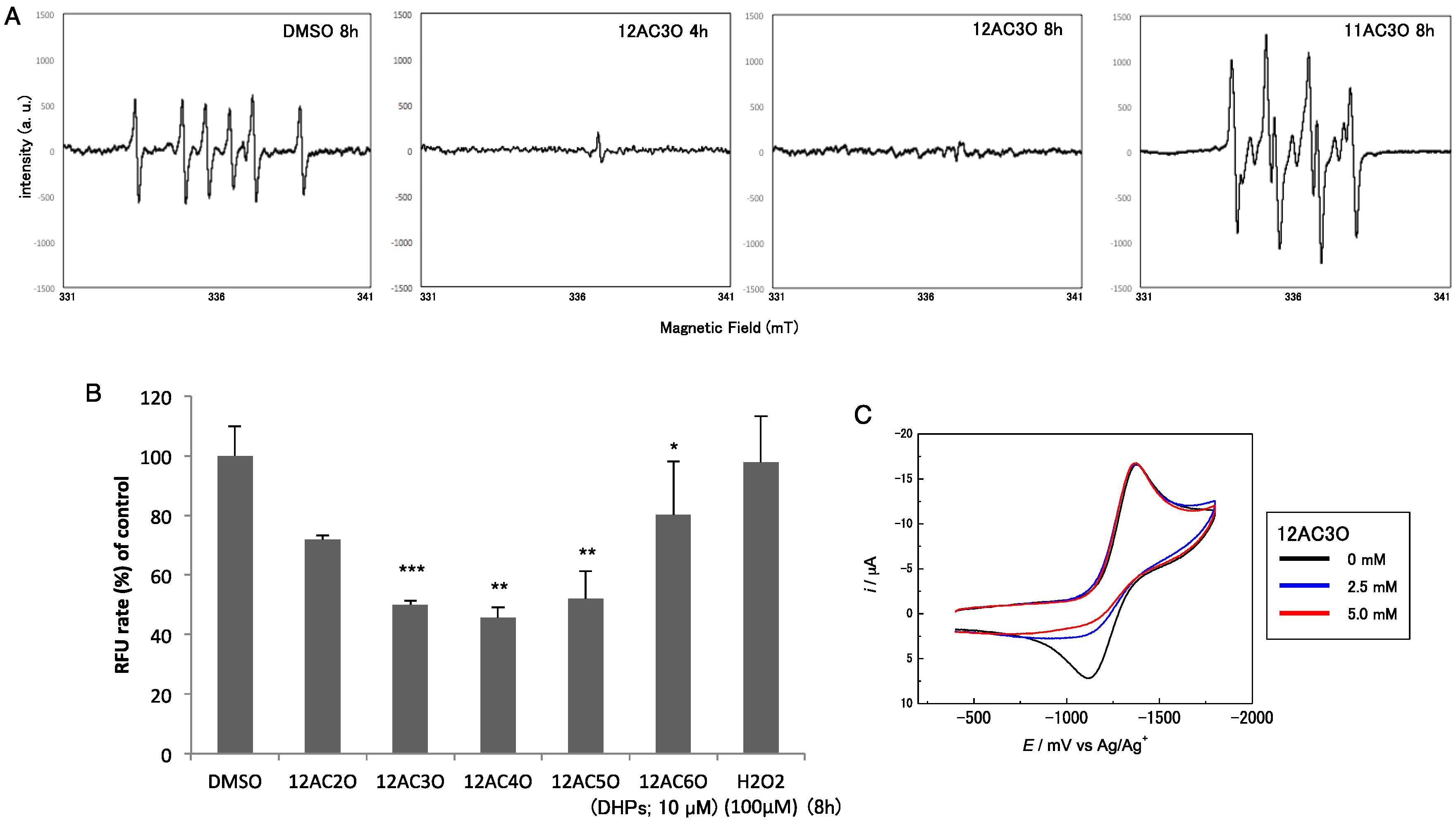
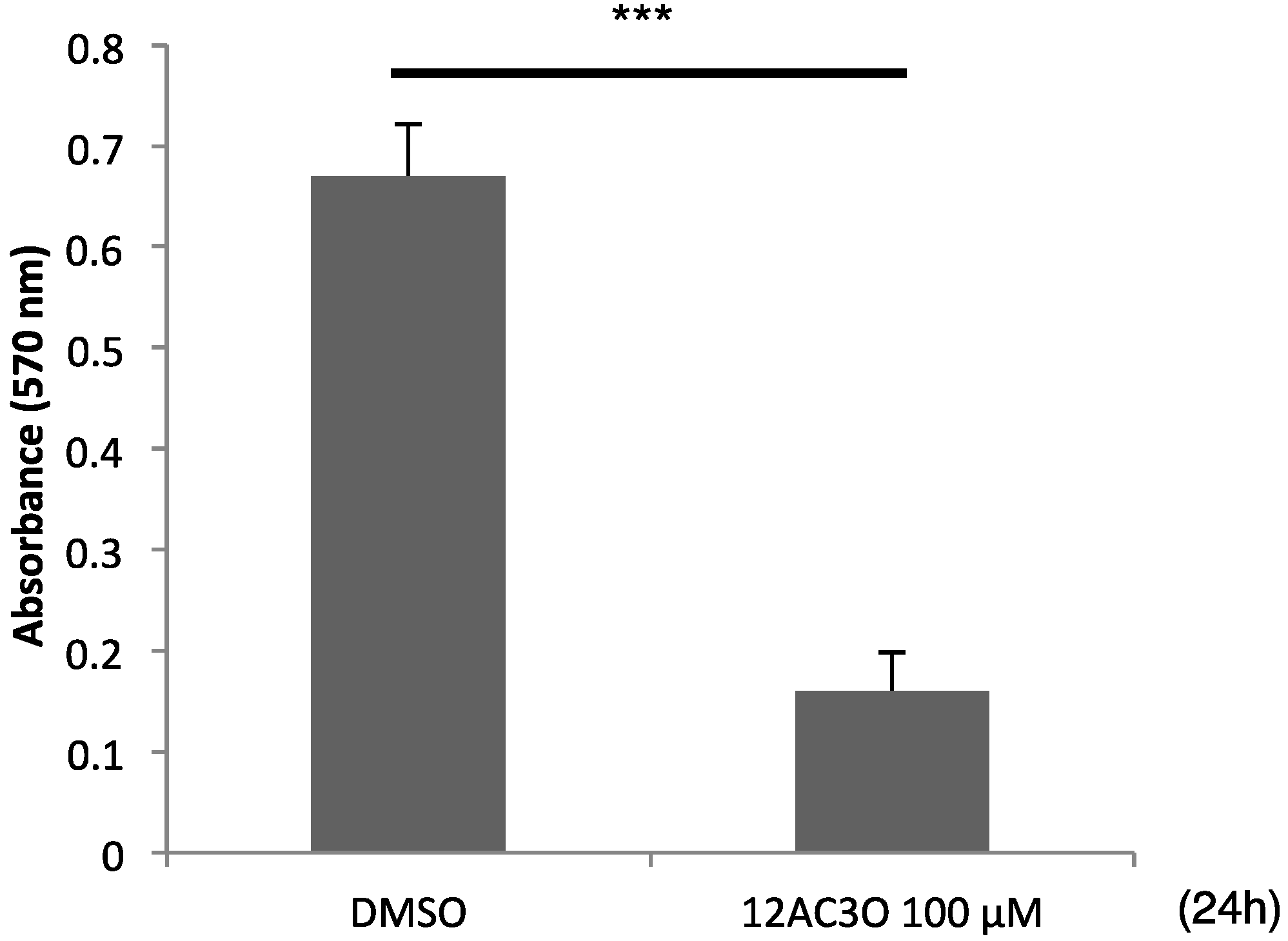
2.5. 12AC3O Inhibited Tumor Progression in Vitro
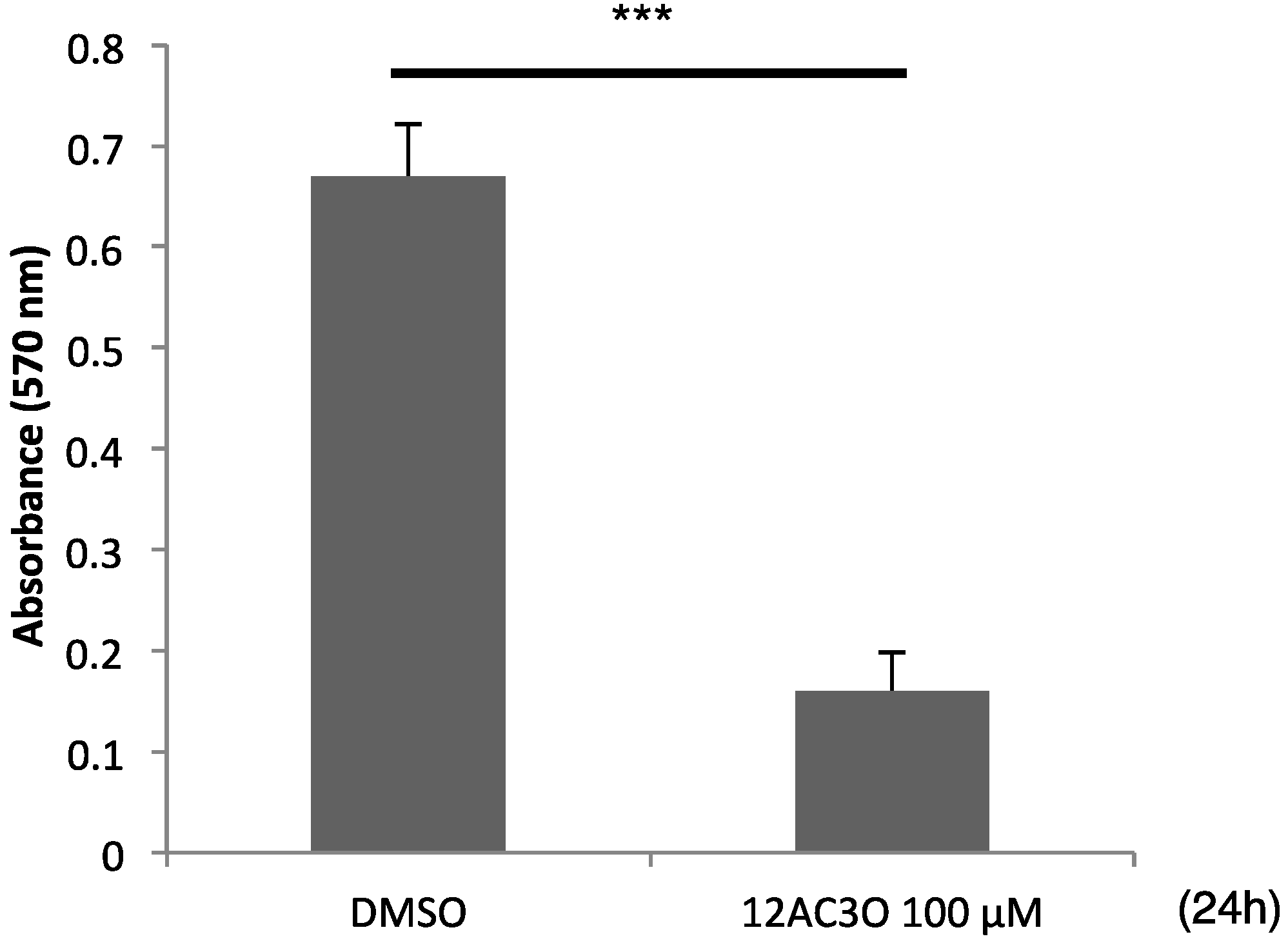
3. Discussion
4. Materials and Methods
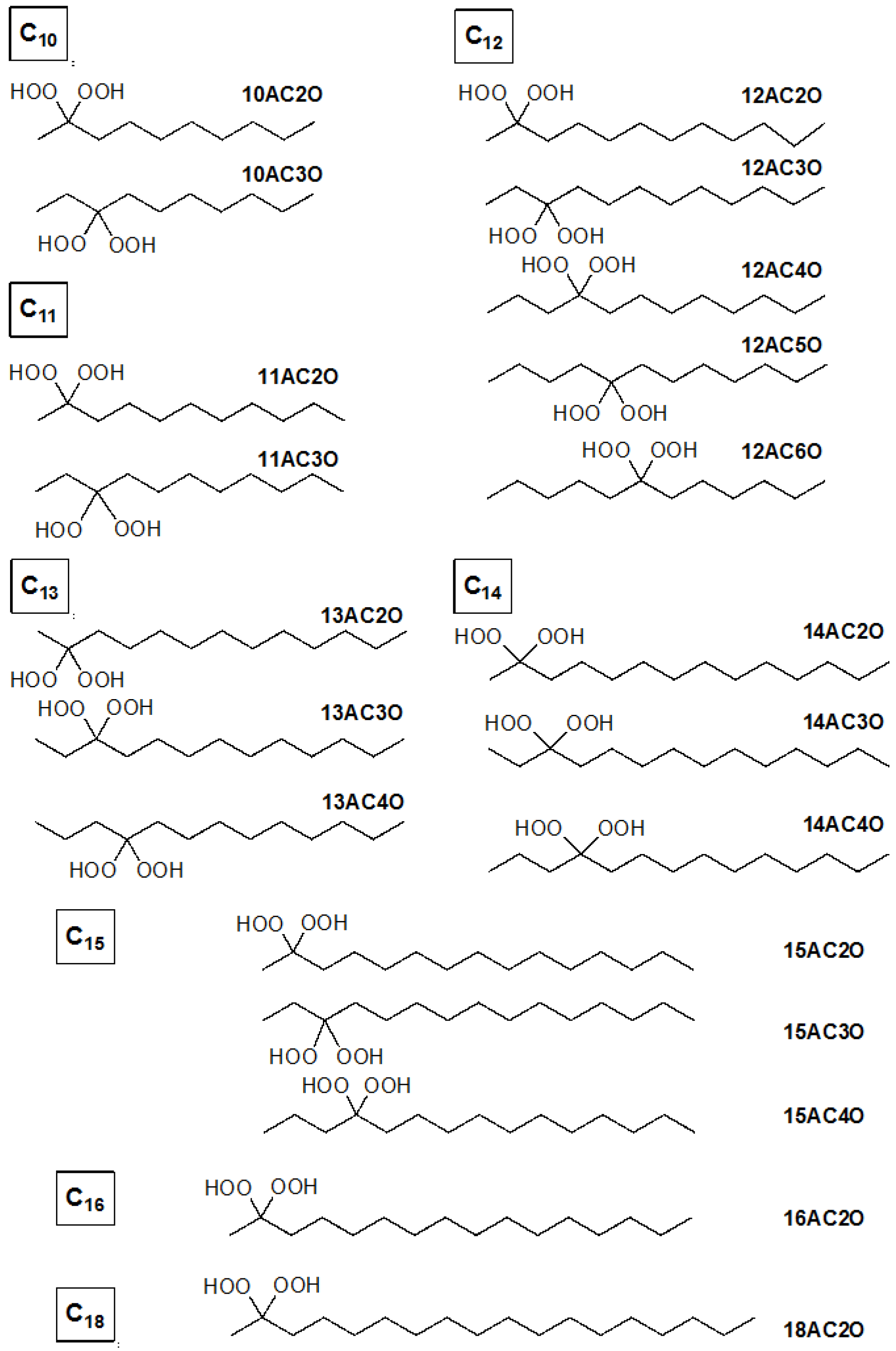
4.1. Synthesis of Germinal (gem)-Dihydroperoxides
4.2. Cell Culture and Cell Viability
4.3. Purification of Peripheral Blood Monocytes (PBMCs)
4.4. Morphological Assessment of Apoptosis
4.5. Inhibitor Agents
4.6. Protein Extraction and Western Blotting
4.7. Electron Microscopic Study
4.8. Electron Spin Resonance (ESR) Spectral Measurements
4.9. Intracellular ROS Assay
4.10. Electrochemical Measurements
4.11. 3D Spheroid Colorimetric Proliferation/Viability Assay
4.12. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Cadenas, E.; Packer, L. Methods in Enzymology. Hydrogen peroxide and cell signaling, part C. Preface. Methods Enzymol. 2013, 528, xv. [Google Scholar] [PubMed]
- Shinohara, H.; Taniguchi, K.; Kumazaki, M.; Yamada, N.; Ito, Y.; Otsuki, Y.; Uno, B.; Hayakawa, F.; Minami, Y.; Naoe, T.; et al. Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronicmyeloid leukemia. Cancer Lett. 2015, 360, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Harashima, N.; Moritani, T.; Huang, W.; Harada, M. The roles of ROS and caspases in TRAIL-induced apoptosis and necroptosis in human pancreatic cancer cells. PLoS ONE 2015, 10, e0127386. [Google Scholar] [CrossRef] [PubMed]
- Tada, N.; Cui, L.; Okubo, H.; Miura, T.; Itoh, A. A facile catalyst-free synthesis of gem-dihydroperoxides with aqueous hydrogen peroxide. Chem. Commun. 2010, 46, 1772–1774. [Google Scholar] [CrossRef] [PubMed]
- Zmitek, K.; Zupan, M.; Stavber, S.; Iskra, J. Iodine as a catalyst for efficient conversion of ketones to gem-dihydroperoxides by aqueous hydrogen peroxide. Org. Lett. 2006, 8, 2491–2494. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Ortmann, R.; Jomaa, H.; Schlitzer, M. New antimalarial drugs. Angew. Chem. Int. Ed. 2003, 42, 5274–5293. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Dunstan, H.; Sauerbrey, A.; Miyachi, H.; Chitambar, C.R. The anti-malarial artesunate is also active against cancer. Int. J. Oncol. 2001, 18, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, M.; Zhang, R.; Wang, H. Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy. J. Cell. Mol. Med. 2009, 13, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Y.; Zhang, J.; Sun, L.-L.; Li, B.-H.; Gao, H.-L.; Xie, T.; Zhang, N.; Ye, Z.-M. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: An in vitro and in vivo study. Cell Death Dis. 2015, 6, e1604. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Noguchi, S.; Kumazaki, M.; Yamada, N.; Ito, T.; Oyama, M.; Ito, Y.; Otsuki, Y.; Naito, S.; Iinuma, M. 3-Decenoic acid derivatives induce autophagic cell death through the down-regulation of E2F1. J. Toxicol. Res. 2013, 3, 29–34. [Google Scholar]
- Sawyer, D.T.; Chiericato, G.; Angelis, C.T.; Nanni, E.J.; Tsuchiya, T. Effects of media and electrode materials on the electrochemical reduction of dioxygen. Anal. Chem. 1982, 54, 1720–1724. [Google Scholar] [CrossRef]
- Andrieux, C.P.; Hapiot, P.; Saveant, J.M. Mechanism of superoxide ion disproportionation in aprotic solvents. J. Am. Chem. Soc. 1987, 109, 3768–3775. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Quinone–hydroquinone π-conjugated redox reaction involving proton-coupled electron transfer plays an important role in scavenging superoxide by polyphenolic antioxidants. Chem. Lett. 2010, 39, 162–164. [Google Scholar] [CrossRef]
- Fleury, C.; Mignotte, B.; Vayssière, J.-L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Qin, G.; Wu, L.; Liu, H.; Pang, Y.; Zhao, C.; Wu, S.; Wang, X.; Chen, T. Artesunate induces apoptosis via a ROS-independent and Bax-mediated intrinsic pathway in HepG2 cells. Exp. Cell Res. 2015, 336, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, 5896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Li, S.-P.; Westermarck, J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008, 22, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cells proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Seki, N.; Nakagawa, Y.; Yi, H.; Matsumoto, K.; Ito, Y.; Ito, K.; Funaoka, M.; Maruyama, W.; Naoi, M.; et al. A highly bioactive lignophenol derivative from bamboo lignin exhibits a potent activity to suppress apoptosis induced by oxidative stress in human neuroblastoma SH-SY5Y cells. Bioorg. Med. Chem. 2004, 12, 4791–4801. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuranaga, Y.; Yamada, N.; Kashiwaya, M.; Nakamura, M.; Cui, L.; Kumazaki, M.; Shinohara, H.; Sugito, N.; Taniguchi, K.; Ito, Y.; et al. Anti-Oncogenic gem-Dihydroperoxides Induce Apoptosis in Cancer Cells by Trapping Reactive Oxygen Species. Int. J. Mol. Sci. 2016, 17, 71. https://doi.org/10.3390/ijms17010071
Kuranaga Y, Yamada N, Kashiwaya M, Nakamura M, Cui L, Kumazaki M, Shinohara H, Sugito N, Taniguchi K, Ito Y, et al. Anti-Oncogenic gem-Dihydroperoxides Induce Apoptosis in Cancer Cells by Trapping Reactive Oxygen Species. International Journal of Molecular Sciences. 2016; 17(1):71. https://doi.org/10.3390/ijms17010071
Chicago/Turabian StyleKuranaga, Yuki, Nami Yamada, Maiko Kashiwaya, Moeko Nakamura, Lei Cui, Minami Kumazaki, Haruka Shinohara, Nobuhiko Sugito, Kohei Taniguchi, Yuko Ito, and et al. 2016. "Anti-Oncogenic gem-Dihydroperoxides Induce Apoptosis in Cancer Cells by Trapping Reactive Oxygen Species" International Journal of Molecular Sciences 17, no. 1: 71. https://doi.org/10.3390/ijms17010071
APA StyleKuranaga, Y., Yamada, N., Kashiwaya, M., Nakamura, M., Cui, L., Kumazaki, M., Shinohara, H., Sugito, N., Taniguchi, K., Ito, Y., Nakayama, T., Uno, B., Itoh, A., & Akao, Y. (2016). Anti-Oncogenic gem-Dihydroperoxides Induce Apoptosis in Cancer Cells by Trapping Reactive Oxygen Species. International Journal of Molecular Sciences, 17(1), 71. https://doi.org/10.3390/ijms17010071