
2,6-Bis(1,4,7,10-tetraazacyclododecan-1-ylmethyl)pyridine and Its Benzene Analog as Nonmetallic Cleaving Agents of RNA Phosphodiester Linkages


Abstract
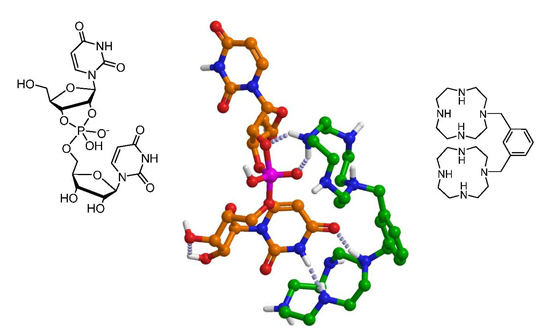
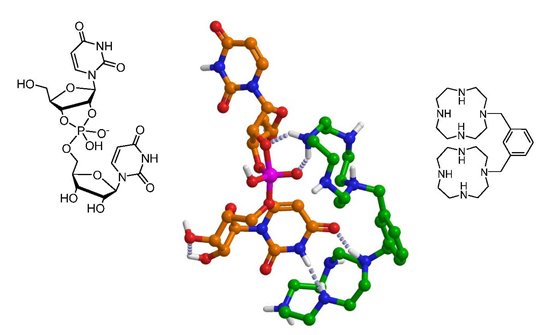
:
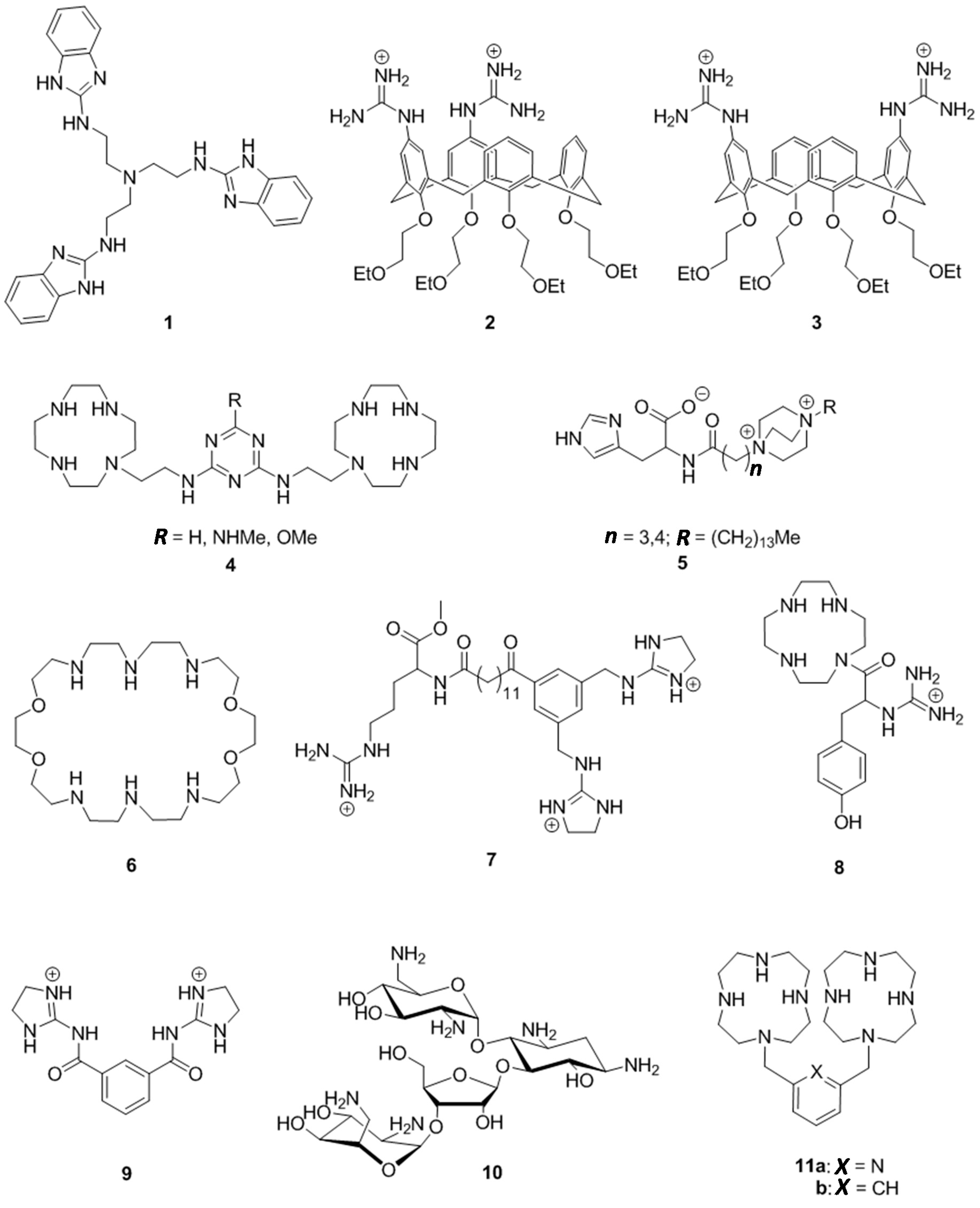
1. Introduction
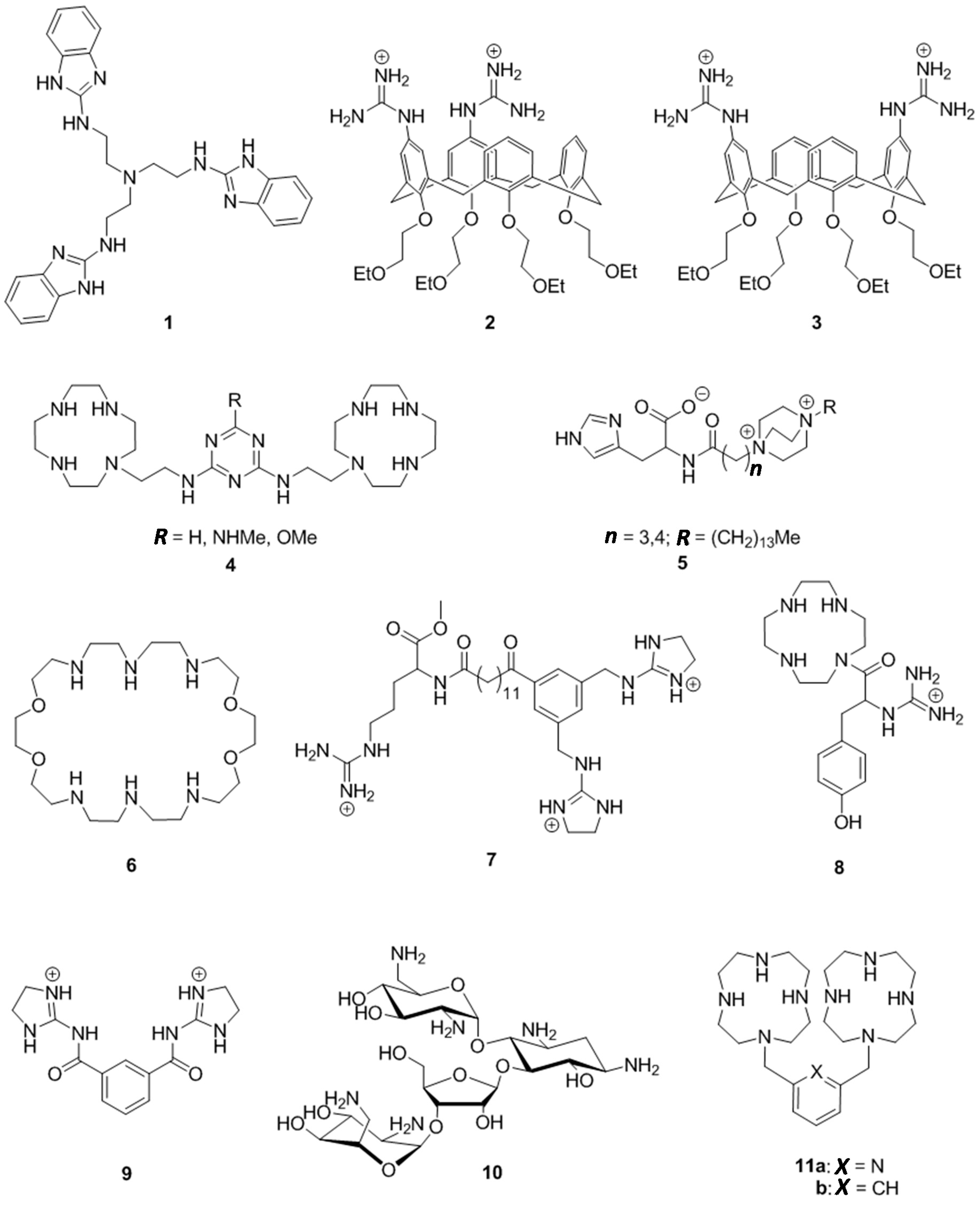
2. Results
2.1. Synthesis of Cleaving Agents 11a and 11b
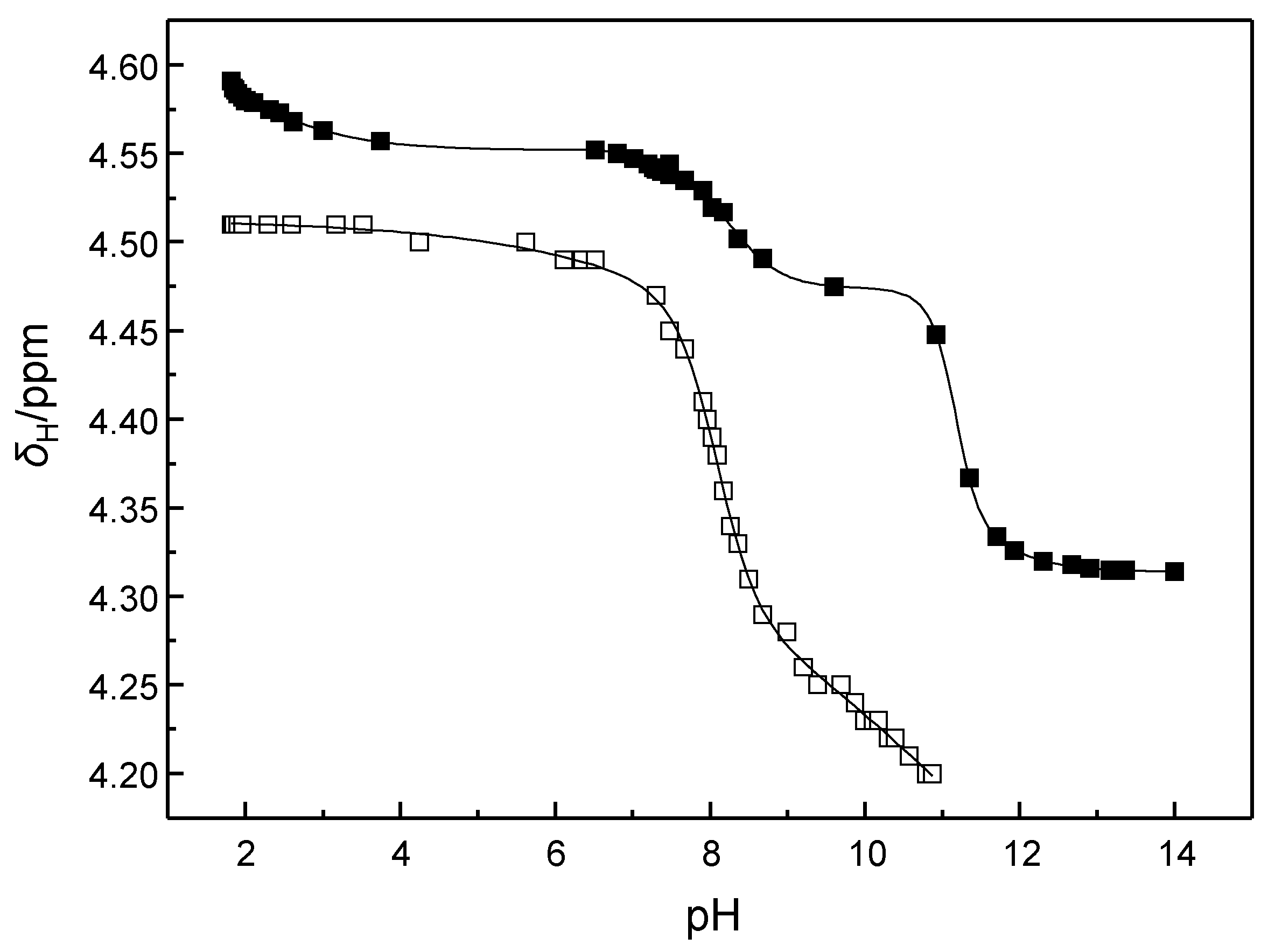
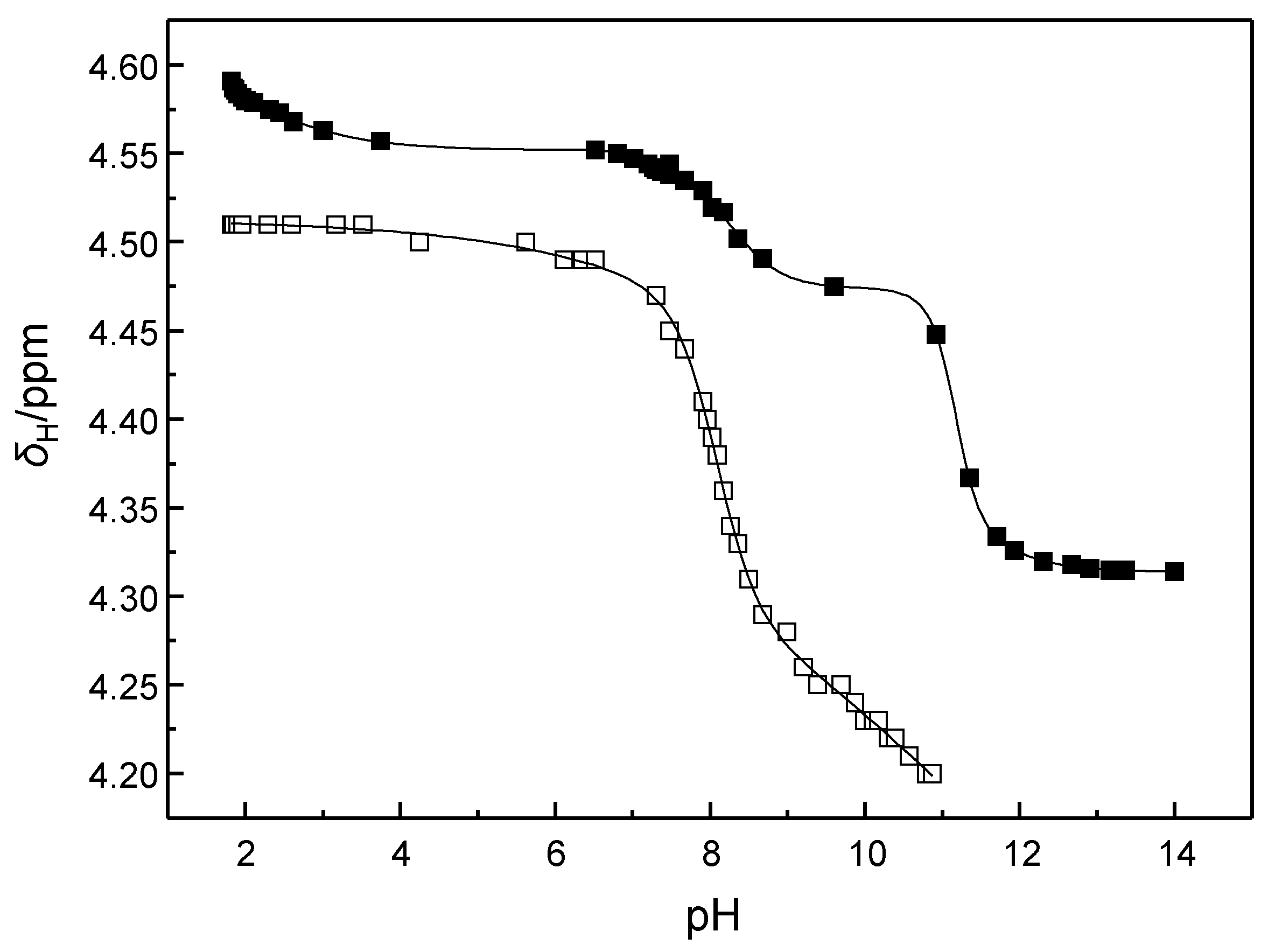
2.2. Determination of the pKa Values of the Cleaving Agents
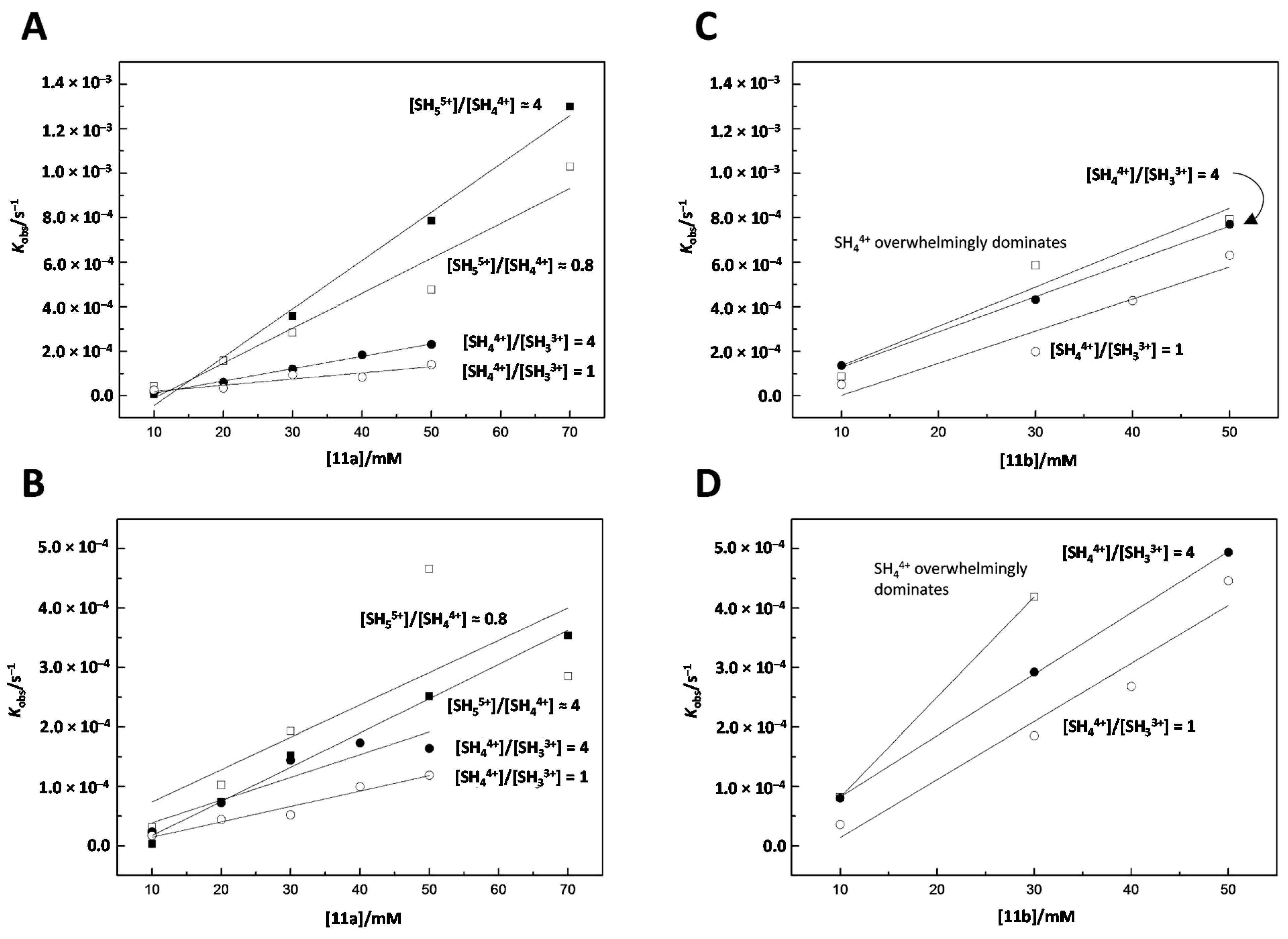
2.3. Kinetic Measurements



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rate Constant | 11a | 11b | ||
|---|---|---|---|---|
| Cleavage | Isomerization | Cleavage | Isomerization | |
| k5/10−3 L·mol−1·s−1 | 26.1 ± 0.2 | 6.2 ± 0.2 | N/A a | N/A a |
| k4/10−3 L·mol−1·s−1 | 7.37 ± 0.06 | 4.7 ± 0.1 | 17.5 ± 0.4 | 15 ± 3 |
| k3/10−3 L·mol−1·s−1 | N/A b | 0.5 ± 0.2 | 11.1 ± 0.8 | 4 ± 6 |
3. Discussion
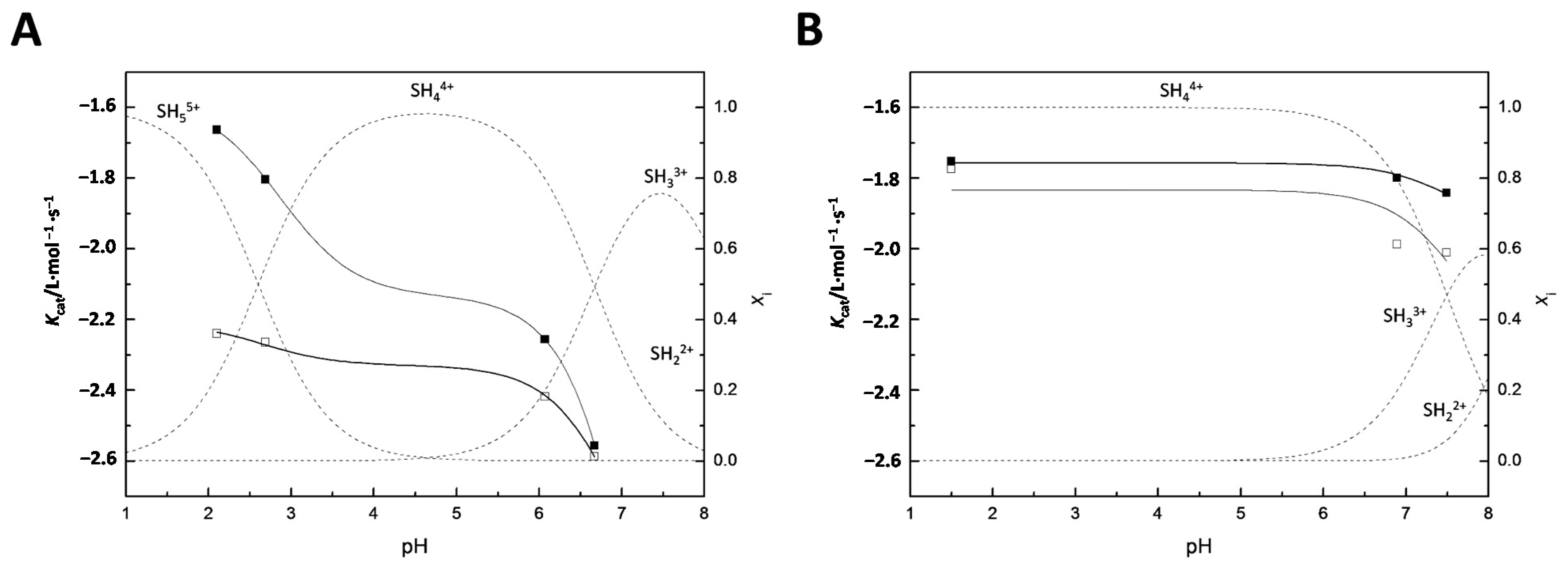
3.1. Base-Selectivity of 11a

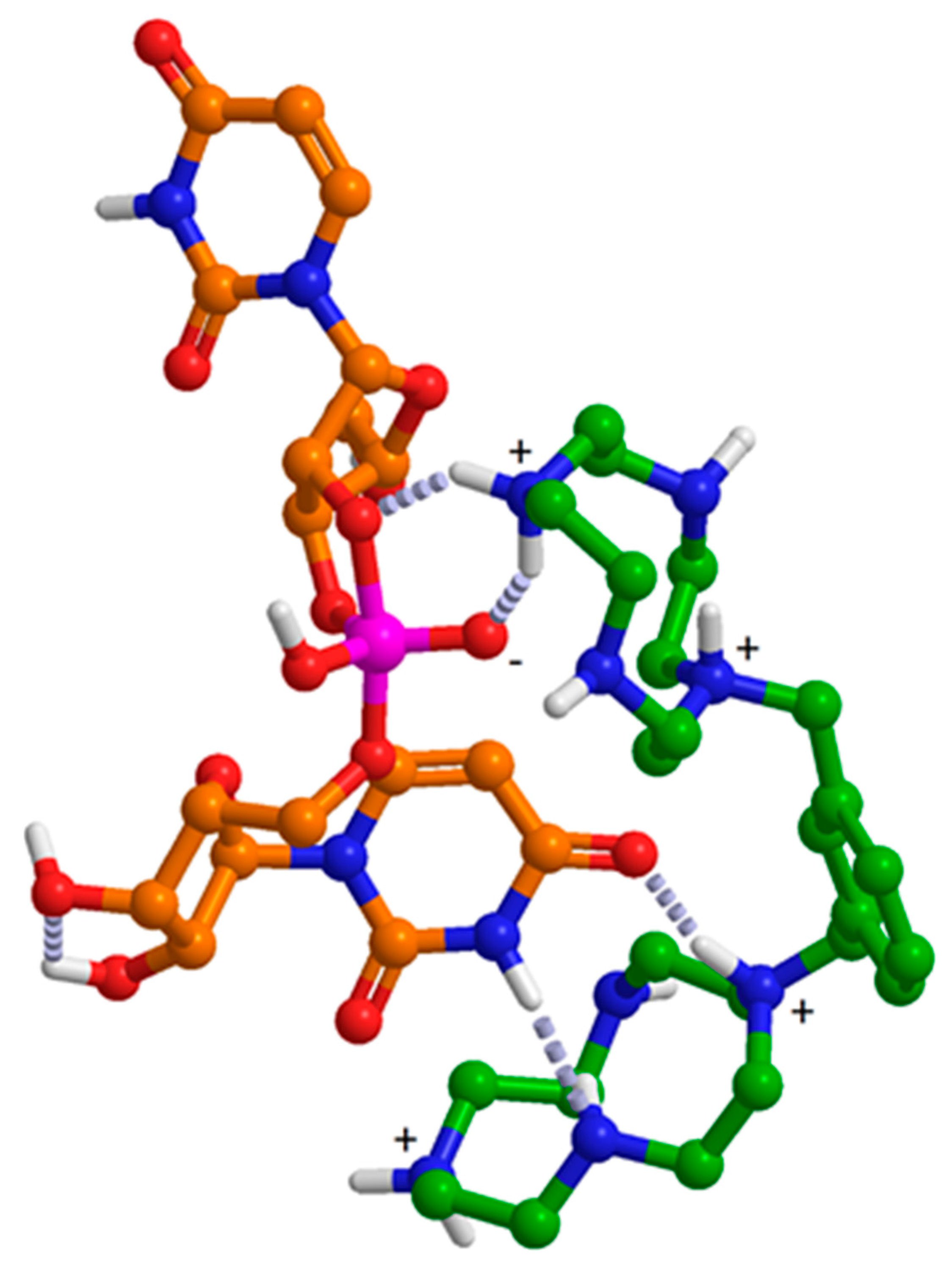
3.2. Mechanism of the Catalysis by 11a and 11b
4. Experimental Section
4.1. General Methods
4.2. 2,6-Bis(1,4,7,10-tetraazacyclododecan-1-ylmethyl)pyridine (11a)
4.3. 1,3-Bis(1,4,7,10-tetraazacyclododecan-1-ylmethyl)benzene (11b)
4.4. Determination of the pKa Values
4.5. Kinetic Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Morrow, J.R.; Iranzo, O. Synthetic metallonucleases for RNA cleavage. Curr. Opin. Chem. Biol. 2004, 8, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Mancin, F.; Tecilla, P. Zinc(II) complexes as hydrolytic catalysts of phosphate diester cleavage: From model substrates to nucleic acids. New J. Chem. 2007, 31, 800–817. [Google Scholar] [CrossRef]
- Lönnberg, H. Cleavage of RNA phosphodiester bonds by small molecular entities: A mechanistic insight. Org. Biomol. Chem. 2011, 9, 1687–1703. [Google Scholar] [CrossRef] [PubMed]
- Desbouis, D.; Troitsky, I.P.; Belousoff, M.J.; Spiccia, L.; Graham, B. Copper(II), zinc(II) and nickel(II) complexes as nuclease mimetics. Coord. Chem. Rev. 2012, 256, 897–937. [Google Scholar] [CrossRef]
- Linjalahti, H.; Feng, G.; Mareque-Rivas, J.C.; Mikkola, S.; Williams, N.H. Cleavage and isomerization of UpU promoted by dinuclear metal ion complexes. J. Am. Chem. Soc. 2008, 130, 4232–4233. [Google Scholar] [CrossRef] [PubMed]
- Nwe, K.; Andolina, C.M.; Morrow, J.R. Tethered dinuclear europium(III) macrocyclic catalysts for the cleavage of RNA. J. Am. Chem. Soc. 2008, 130, 14861–14871. [Google Scholar] [CrossRef] [PubMed]
- Niittymäki, T.; Lönnberg, H. Artificial ribonucleases. Org. Biomol. Chem. 2006, 4, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Murtola, M.; Wenska, M.; Strömberg, R. PNAzymes that are artificial RNA restriction enzymes. J. Am. Chem. Soc. 2010, 132, 8984–8990. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, U.; Strick, A.; Ludwig, V.; Peter, S.; Kalden, E.; Göbel, M.W. Metal-free catalysts for the hydrolysis of RNA derived from guanidines, 2-aminopyridines, and 2-aminobenzimidazoles. J. Am. Chem. Soc. 2005, 127, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Gnaccarini, C.; Peter, S.; Scheffer, U.; Vonhoff, S.; Klussmann, S.; Göbel, M.W. Site-specific cleavage of RNA by a metal-free artificial nuclease attached to antisense oligonucleotides. J. Am. Chem. Soc. 2006, 128, 8063–8067. [Google Scholar] [CrossRef] [PubMed]
- Salvio, R.; Cacciapaglia, R.; Mandolini, L.; Sansone, F.; Casnati, A. Diguanidinocalix[4]arenes as effective and selective catalysts of the cleavage of diribonucleoside monophosphates. RSC Adv. 2014, 4, 34412–34416. [Google Scholar] [CrossRef]
- Lönnberg, T.A.; Helkearo, M.; Jancsó, A.; Gajda, T. Mimics of small ribozymes utilizing a supramolecular scaffold. Dalton Trans. 2012, 41, 3328–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lönnberg, T.; Luomala, M. Intracomplex general acid/base catalyzed cleavage of RNA phosphodiester bonds: The leaving group effect. Org. Biomol. Chem. 2012, 10, 6785–6791. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.L.; Zenkova, M.A.; Gross, H.J.; Vlassov, V.V. Enhanced RNA cleavage within bulge-loops by an artificial ribonuclease. Nucleic Acids Res. 2005, 33, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Bencini, A.; Berni, E.; Bianchi, A.; Giorgi, C.; Valtancoli, B. ApA cleavage promoted by oxa-aza macrocycles and their Zn(II) complexes. The Role of pH and metal coordination in the hydrolytic mechanism. Supramol. Chem. 2001, 13, 489–497. [Google Scholar] [CrossRef]
- Kurz, K.; Göbel, M.W. Hydrolytical cleavage of TAR-RNA, the trans-activation responsive region of HIV-1, by a bis(guanidinium) catalyst attached to arginine. Helv. Chim. Acta 1996, 79, 1967–1979. [Google Scholar] [CrossRef]
- Michaelis, K.; Kalesse, M. Selective cleavage of unpaired uridines with a tyrosine–cyclen conjugate. ChemBioChem 2001, 2, 79–83. [Google Scholar] [CrossRef]
- Göbel, M.W.; Roussev, C.D.; Scheffer, U. RNA cleavage catalyzed by amphoteric bis(acyl)guanidinium derivatives. Helv. Chim. Acta 2014, 97, 215–227. [Google Scholar] [CrossRef]
- Belousoff, M.J.; Graham, B.; Spiccia, L.; Tor, Y. Cleavage of RNA oligonucleotides by aminoglycosides. Org. Biomol. Chem. 2009, 7, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, V.V.; Zuber, G.; Felden, B.; Behr, J.-P.; Giegè, R. Cleavage of tRNA with imidazole and spermine imidazole constructs: A new approach for probing RNA structure. Nucleic Acids Res. 1995, 23, 3161–3167. [Google Scholar] [CrossRef] [PubMed]
- Konevetz, D.A.; Beck, I.E.; Beloglazova, N.G.; Sulimenkov, I.V.; Silnikov, V.N.; Zenkova, M.A.; Shishkin, G.V.; Vlassov, V.V. Artificial ribonucleases: Synthesis and RNA cleaving properties of cationic conjugates bearing imidazole residues. Tetrahedron 1999, 55, 503–512. [Google Scholar] [CrossRef]
- Fouace, S.; Gaudin, C.; Picard, S.; Corvaisier, S.; Renault, J.; Carboni, B.; Felden, B. Polyamine derivatives as selective RNaseA mimics. Nucleic Acids Res. 2004, 32, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giegé, R.; Felden, B.; Zenkova, M.A.; Silnikov, V.N.; Vlassov, V.V. Cleavage of RNA with synthetic ribonuclease mimics. In Methods in Enzymology; Academic Press: Waltham, MA, USA, 2000; Volume 318, pp. 147–165. [Google Scholar]
- Shinozuka, K.; Nakashima, Y.; Shimizu, K.; Sawai, H. Synthesis and characterization of polyamine-based biomimetic catalysts as artificial ribonuclease. Nucleosides Nucleotides Nucleic Acids 2001, 20, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, N.A.; Medvedeva, D.A.; Zenkova, M.A.; Vlassov, V.V. Cleavage of RNA by an amphiphilic compound lacking traditional catalytic groups. Bioorg. Chem. 2008, 36, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Burakova, E.; Kovalev, N.; Zenkova, M.; Vlassov, V.; Silnikov, V. Structure–activity relationships in new polycationic molecules based on two 1,4-diazabicyclo[2.2.2]octanes as artificial ribonucleases. Bioorg. Chem. 2014, 57, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.-X.; Zhang, J.; Liu, P.-Y.; Xia, C.-Q.; Zhou, Z.-Y.; Xie, R.-G.; Yu, X.-Q. Dinuclear macrocyclic polyamine zinc(II) complexes: Syntheses, characterization and their interaction with plasmid DNA. J. Inorg. Biochem. 2005, 99, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Delepine, A.-S.; Tripier, R.; Handel, H. Cyclen-based bismacrocycles for biological anion recognition. A potentiometric and NMR study of AMP, ADP and ATP nucleotide complexation. Org. Biomol. Chem. 2008, 6, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.A.; Stokes, R.H. Electrolyte Solutions; Butterworth & Co.: London, UK, 1959. [Google Scholar]
- Järvinen, P.; Oivanen, M.; Lönnberg, H. Interconversion and phosphoester hydrolysis of 2′,5′- and 3′,5′-dinucleoside monophosphates: Kinetics and mechanisms. J. Org. Chem. 1991, 56, 5396–5401. [Google Scholar] [CrossRef]
- Wang, Q.; Lönnberg, H. Simultaneous interaction with base and phosphate moieties modulates the phosphodiester cleavage of dinucleoside 3′,5′-monophosphates by dinuclear Zn2+ complexes of di(azacrown) ligands. J. Am. Chem. Soc. 2006, 128, 10716–10728. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Leino, E.; Jancsó, A.; Szilágyi, I.; Gajda, T.; Hietamäki, E.; Lönnberg, H. Zn2+ complexes of di- and tri-nucleating azacrown ligands as base-moiety-selective cleaving agents of RNA 3′,5′-phosphodiester bonds: Binding to guanine base. ChemBioChem 2008, 9, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Laine, M.; Ketomäki, K.; Poijärvi-Virta, P.; Lönnberg, H. Base moiety selectivity in cleavage of short oligoribonucleotides by di- and tri-nuclear Zn(II) complexes of azacrown-derived ligands. Org. Biomol. Chem. 2009, 7, 2780–2787. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lain, L.; Lahdenpohja, S.; Lönnberg, H.; Lönnberg, T. 2,6-Bis(1,4,7,10-tetraazacyclododecan-1-ylmethyl)pyridine and Its Benzene Analog as Nonmetallic Cleaving Agents of RNA Phosphodiester Linkages. Int. J. Mol. Sci. 2015, 16, 17798-17811. https://doi.org/10.3390/ijms160817798
Lain L, Lahdenpohja S, Lönnberg H, Lönnberg T. 2,6-Bis(1,4,7,10-tetraazacyclododecan-1-ylmethyl)pyridine and Its Benzene Analog as Nonmetallic Cleaving Agents of RNA Phosphodiester Linkages. International Journal of Molecular Sciences. 2015; 16(8):17798-17811. https://doi.org/10.3390/ijms160817798
Chicago/Turabian StyleLain, Luigi, Salla Lahdenpohja, Harri Lönnberg, and Tuomas Lönnberg. 2015. "2,6-Bis(1,4,7,10-tetraazacyclododecan-1-ylmethyl)pyridine and Its Benzene Analog as Nonmetallic Cleaving Agents of RNA Phosphodiester Linkages" International Journal of Molecular Sciences 16, no. 8: 17798-17811. https://doi.org/10.3390/ijms160817798