
Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2

Abstract
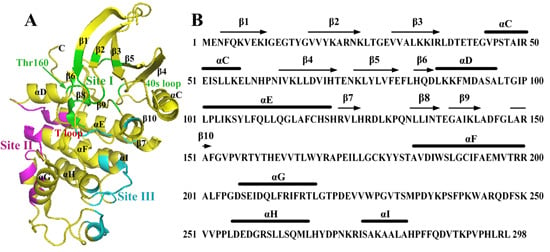
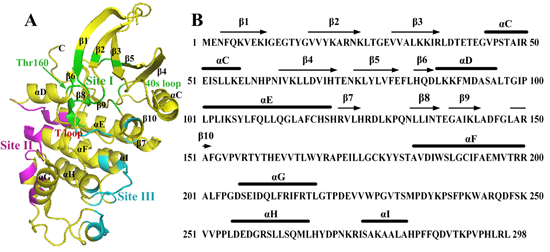
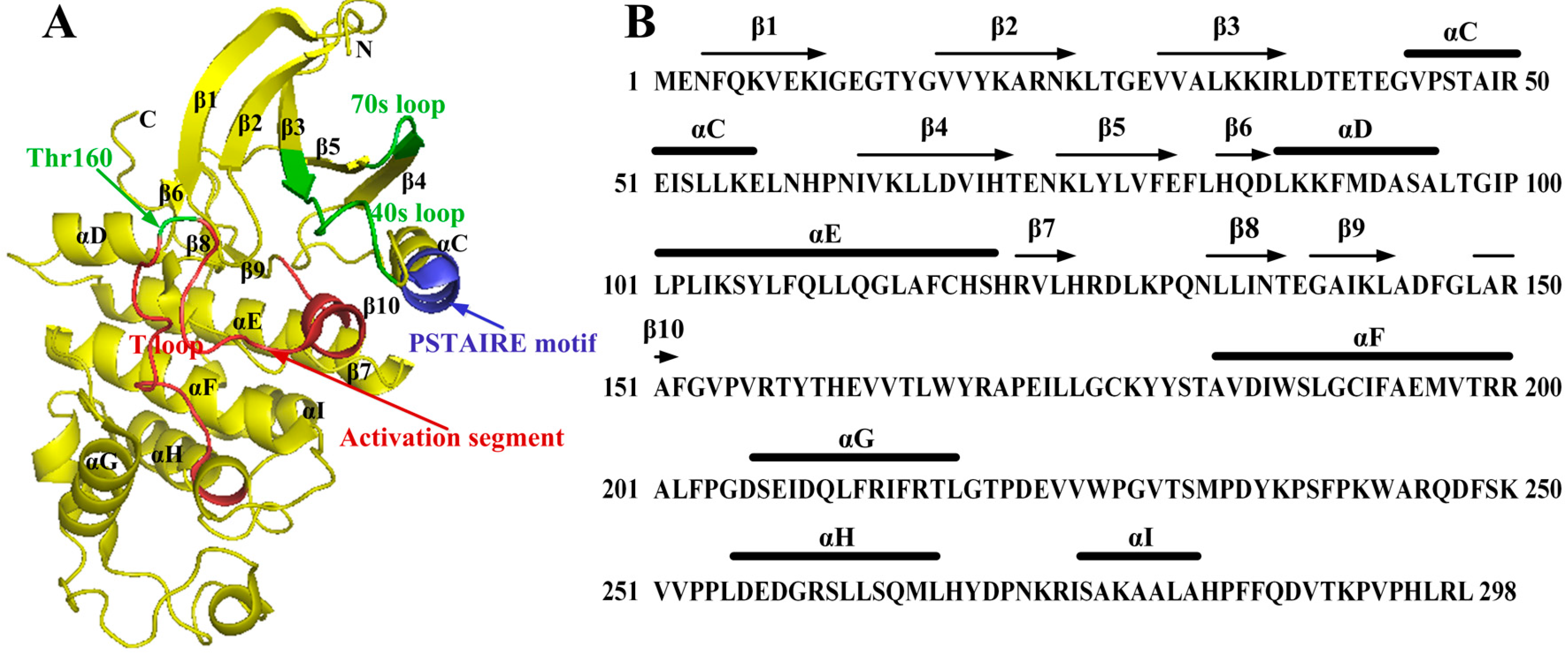
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
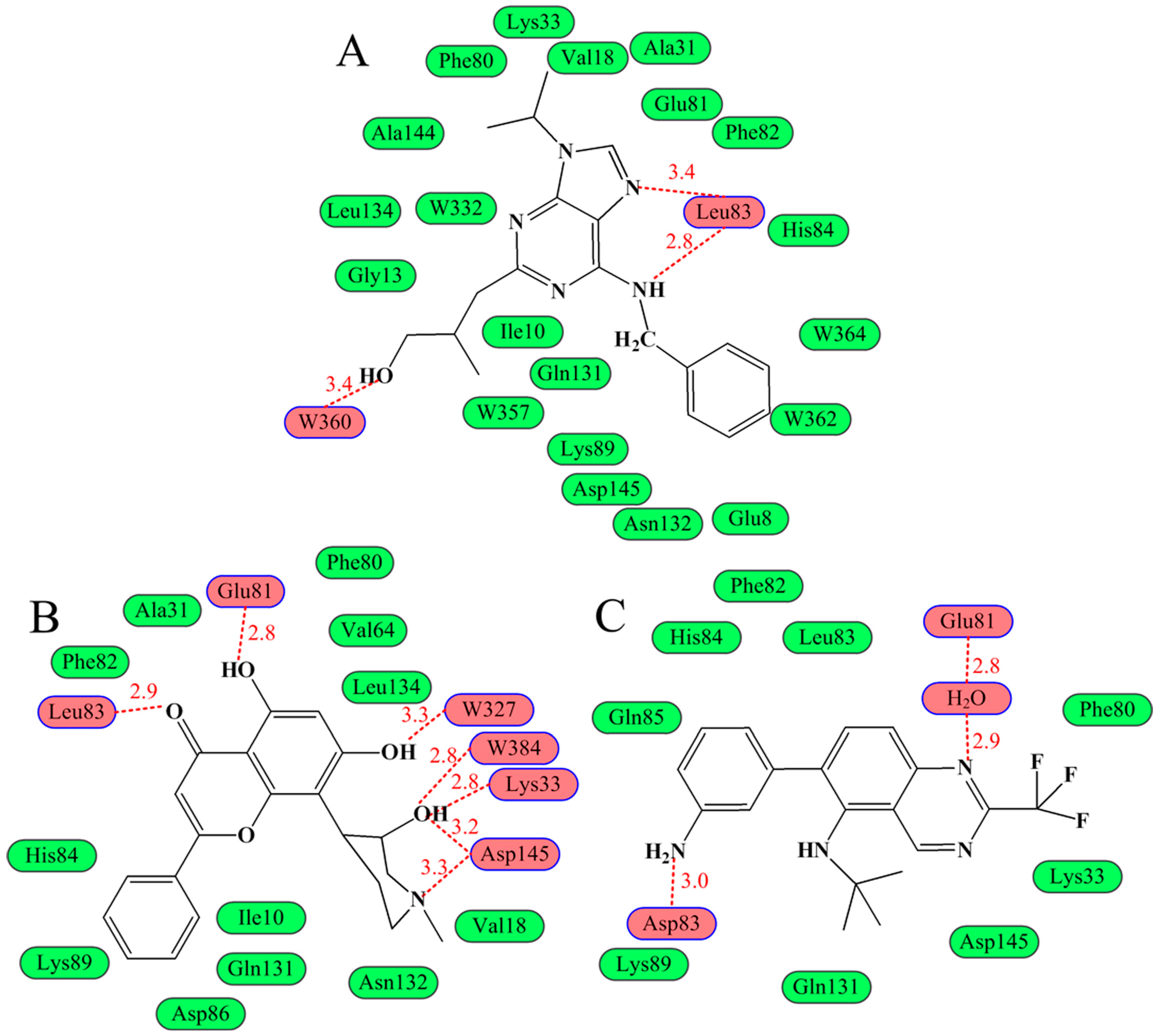{kind=link}
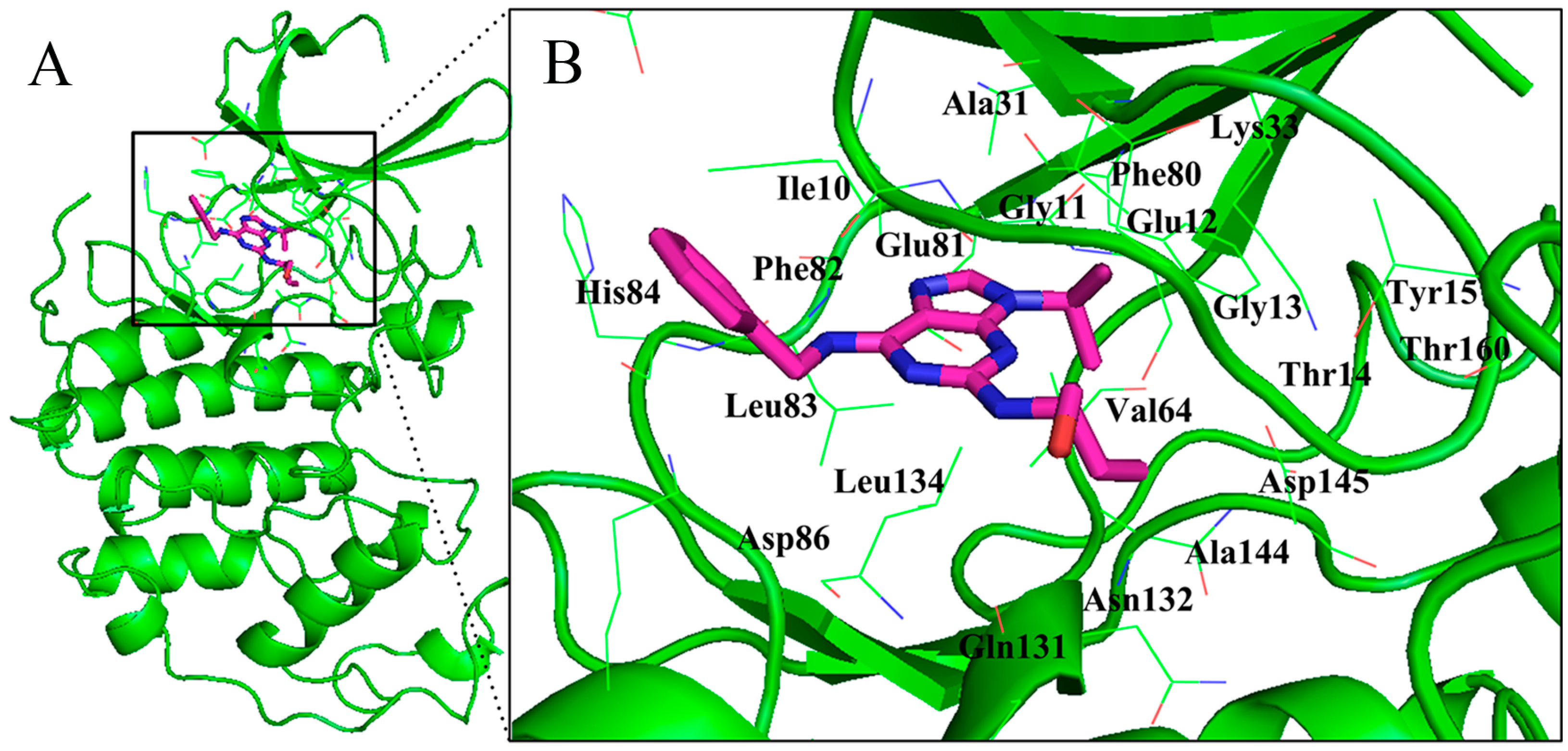{kind=link}
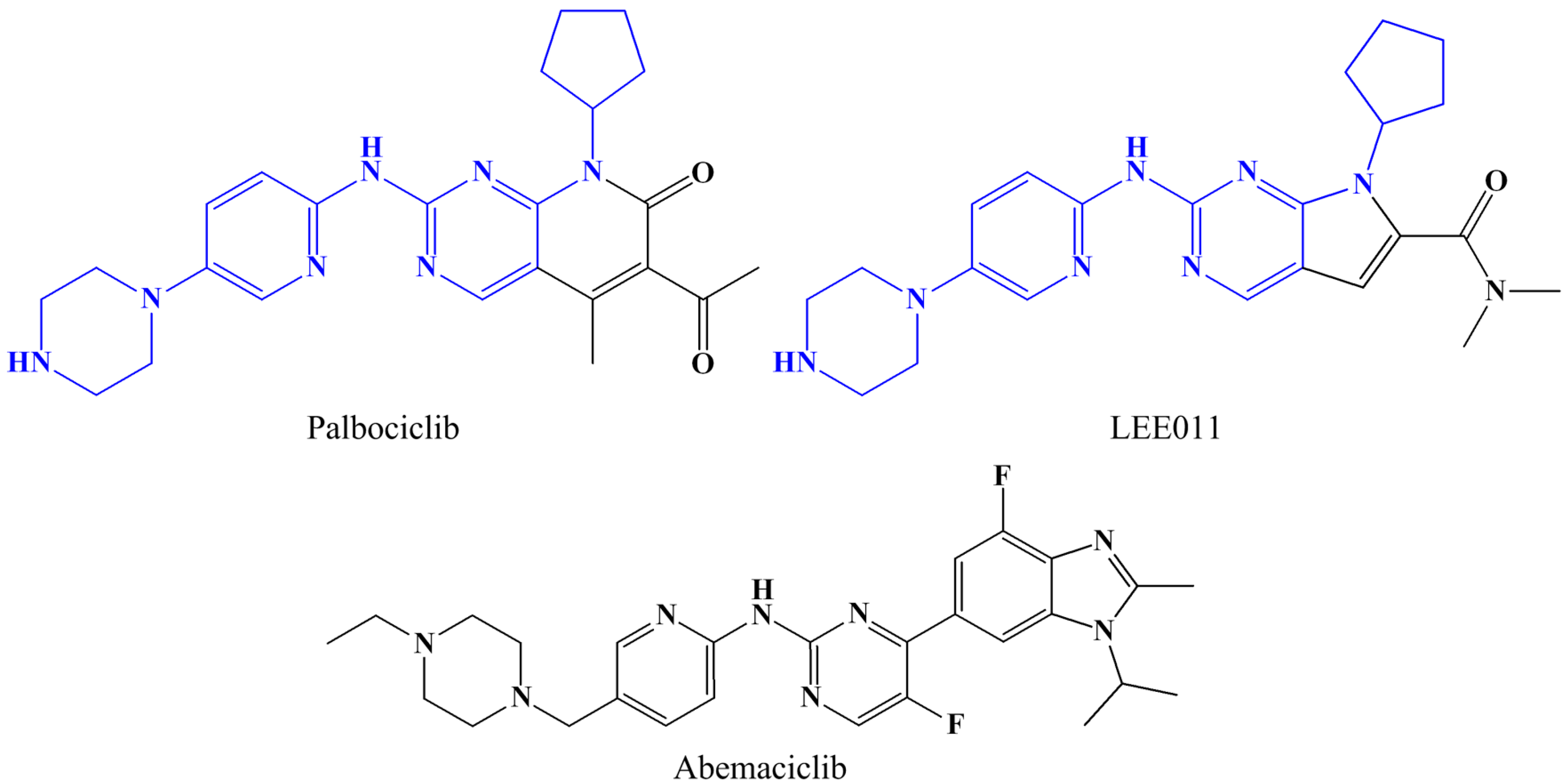{kind=link}
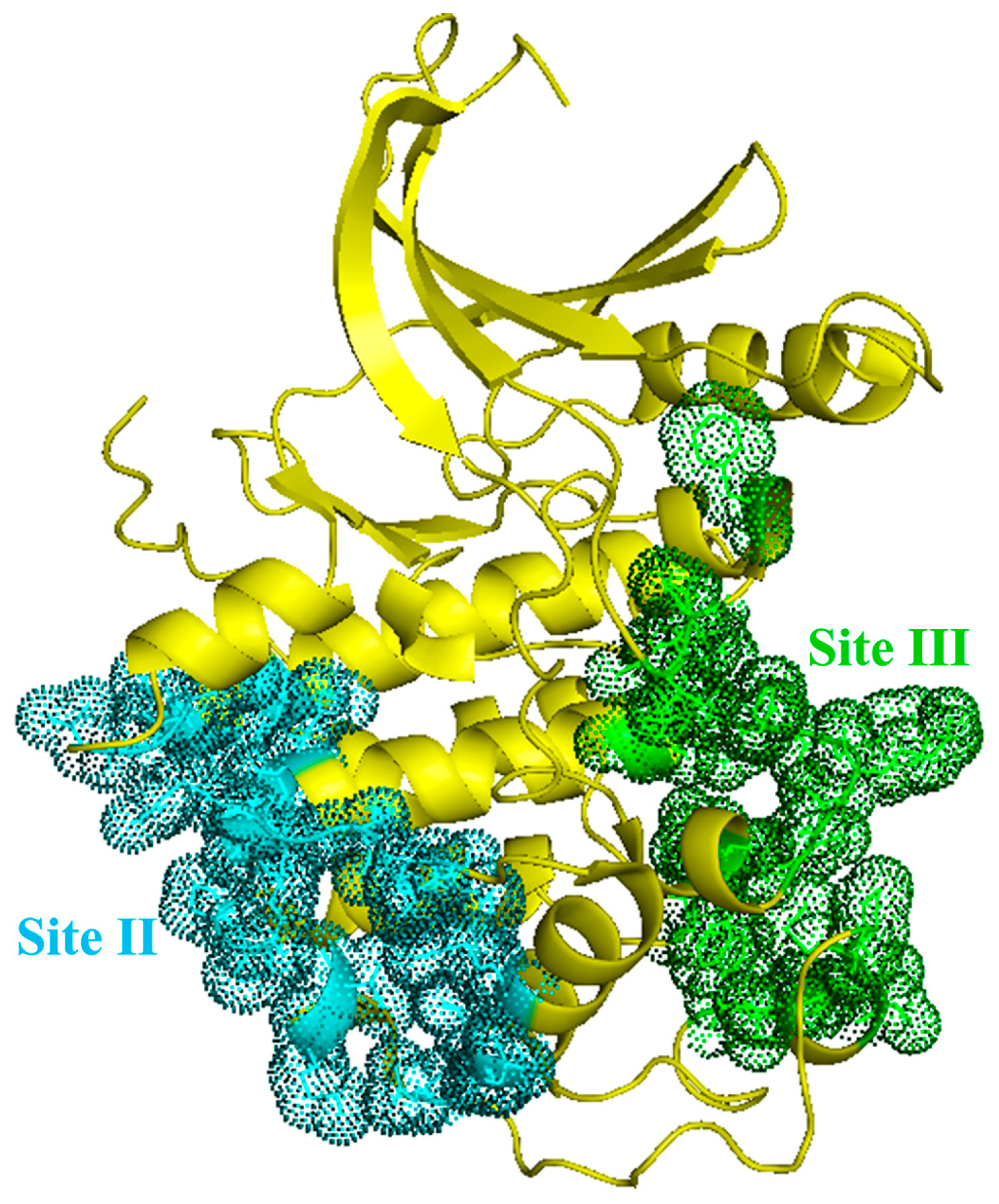{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
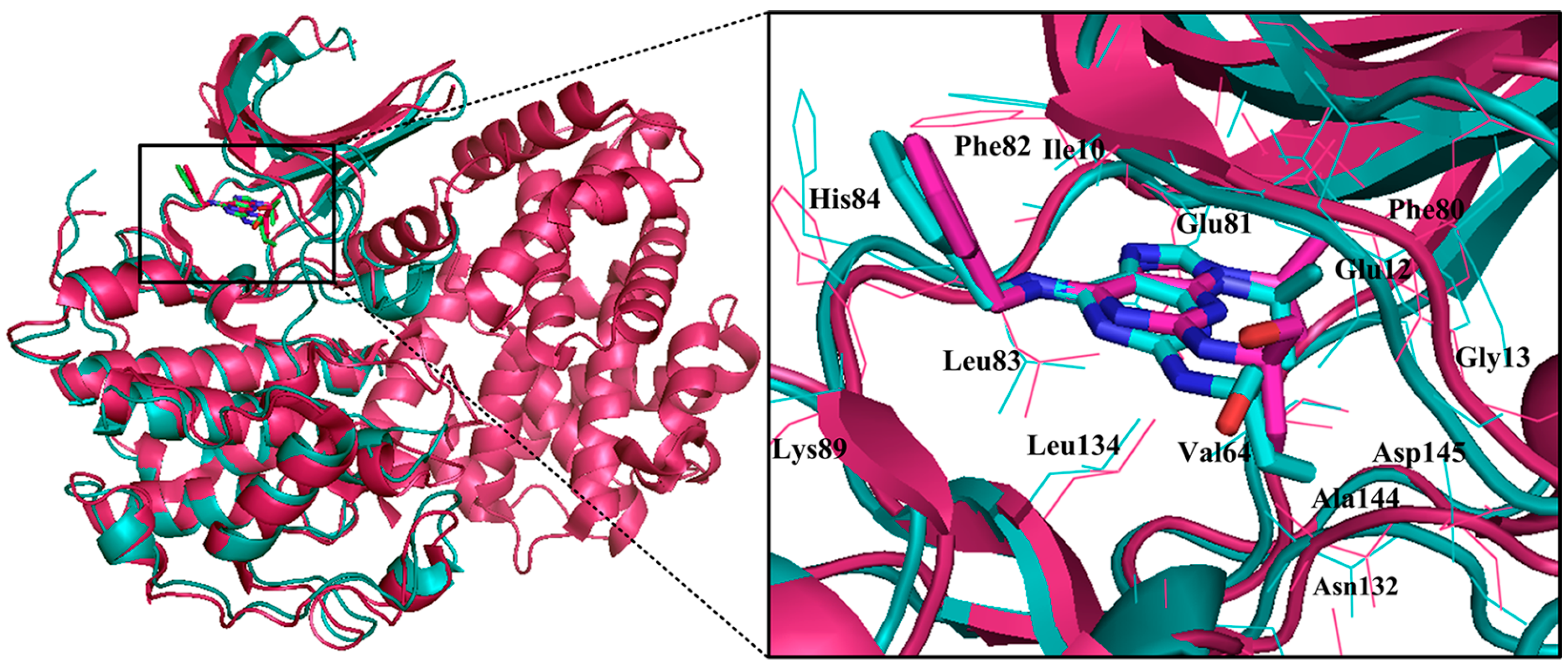{kind=link}
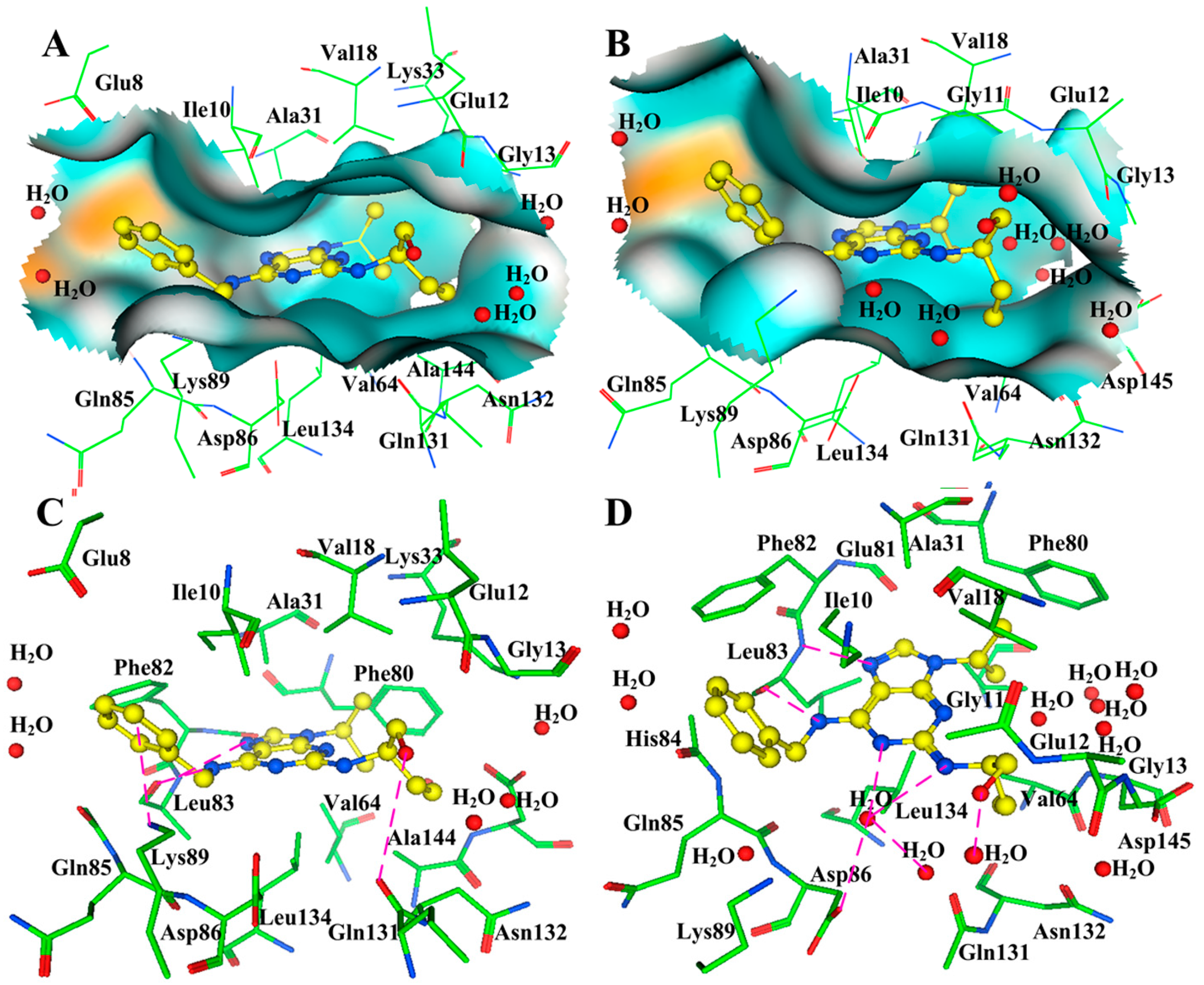{kind=link}
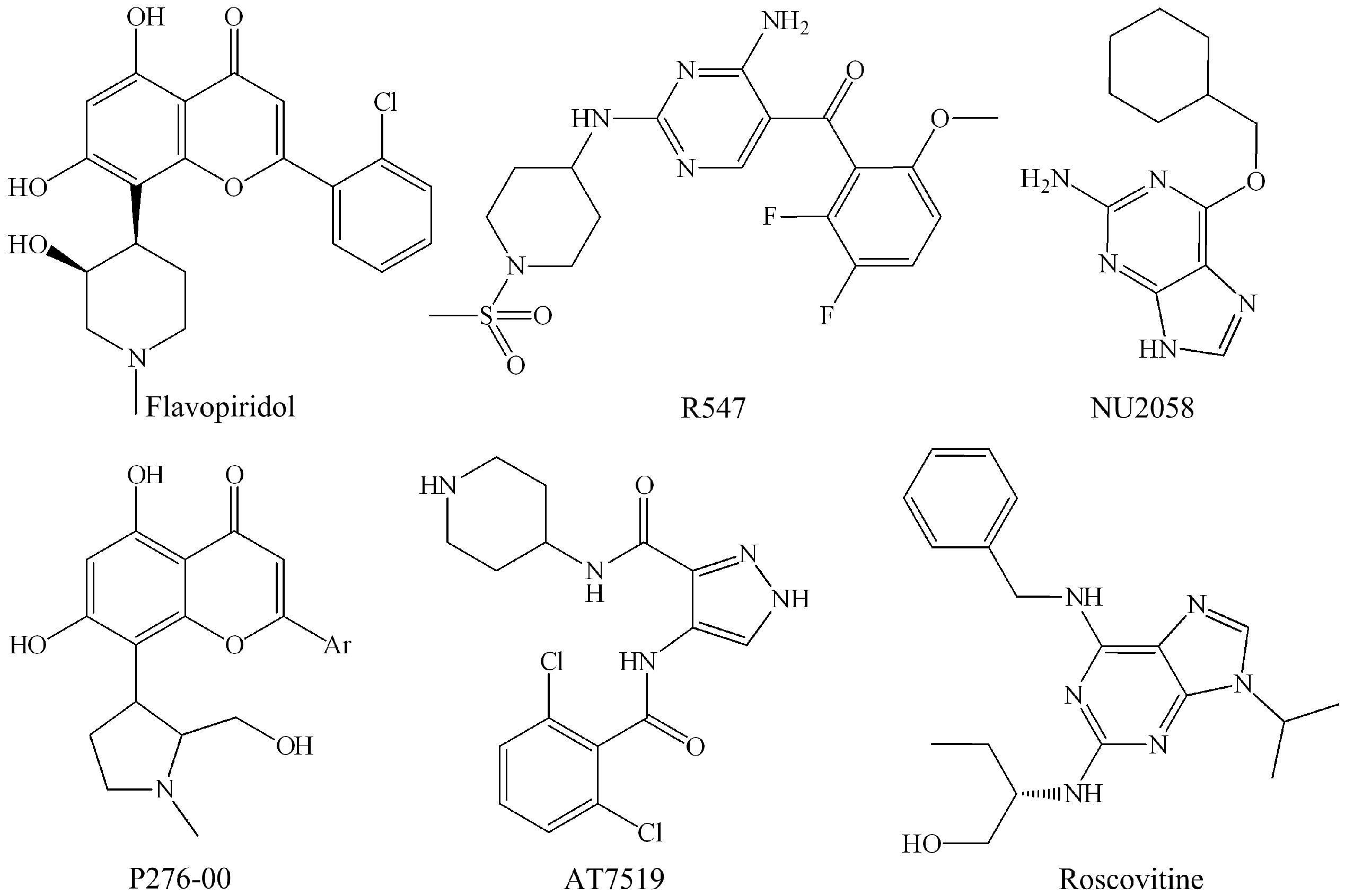
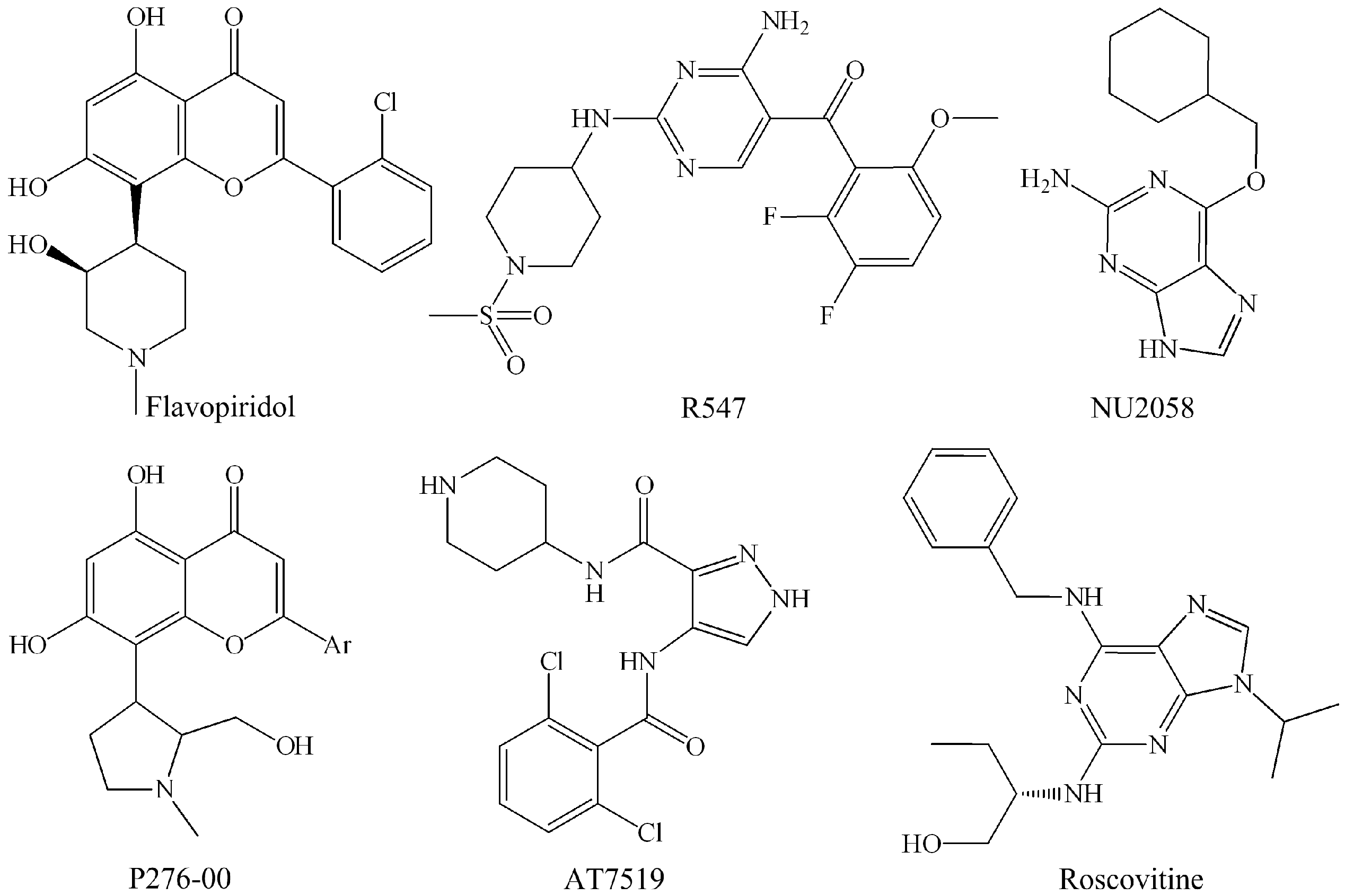
| Candidate Drug | Target (IC50) | Clinical Trials | References |
|---|---|---|---|
| Flavopiridol | CDK1 (30 nM), CDK2 (100 nM), CDK4 (20 nM), CDK6 (60 nM), CDK7 (10 nM), CDK9 (10 nM) | Phase II | Kaur et al. [37] |
| P276-00 | CDK1 (110 nM), CDK2 (10 nM), CDK4 (130 nM), CDK9 (20 nM) | Phase II | De Azevedo Jr. et al. [38]; Murthi et al. [39]; Joshi et al. [33] |
| Roscovitine | CDK1 (2.7 μM), CDK2 (0.7 μM), CDK7 (0.5 μM), CDK9 (0.8 μM) | Phase II | McClue et al. [40]; Meijer et al. [41] |
| PHA-848125 AC | CDK1 (2 nM), CDK2 (3 nM), CDK4 (5 nM), CDK5 (4 nM) | Phase II | Caporali et al. [42] |
| UCN-01 | CDK2 (42 nM), CDK4 (32 nM), CDK6 (58 nM) | Phase II | Kawakami et al. [43]; Li et al. [44]; Senderowicz [45] |
| R547 | CDK1 (0.001 μM), CDK2 (0.003 μM), CDK4 (0.001 μM) | Phase I/II | Chu et al. [46]; DePinto et al. [25] |
| AT-7519 | CDK1 (0.21 μM), CDK2 (0.047 μM), CDK4 (0.1 μM), CDK5 (0.13 μM), CDK6 (0.17 μM), CDK9 (≤0.01 μM) | Phase I/II | Mahadevan et al. [47]; Squires et al. [48] |
| Dinaciclib | CDK1 (3 nM), CDK2 (1 nM), CDK5 (1 nM), CDK9 (4 nM) | Phase I | Michael [49]; Parry et al. [50] |
| SNS-032 | CDK2 (38 nM), CDK7 (62 nM), CDK9 (4 nM) | Phase I | Tong et al. [51]; Conroy et al. [52] |
| RGB-286638 | CDK1 (2 nM), CDK2 (3 nM), CDK3 (5 nM), CDK4 (4 nM), CDK9 (1 nM) | Phase I | de Bruijn et al. [53]; van der Biessen et al. [54] |
| BAY-1000394 | CDK1, CDK2, CDK4 and CDK9 (≤11 nM) | Phase I | Siemeister et al. [55]; Siemeister et al. [56] |
| TG02 | CDK1 (9 nM), CDK2 (5 nM), CDK3 (8 nM), CDK5 (4 nM), CDK9 (3 nM) | Phase I | Poulsen et al. [57] |
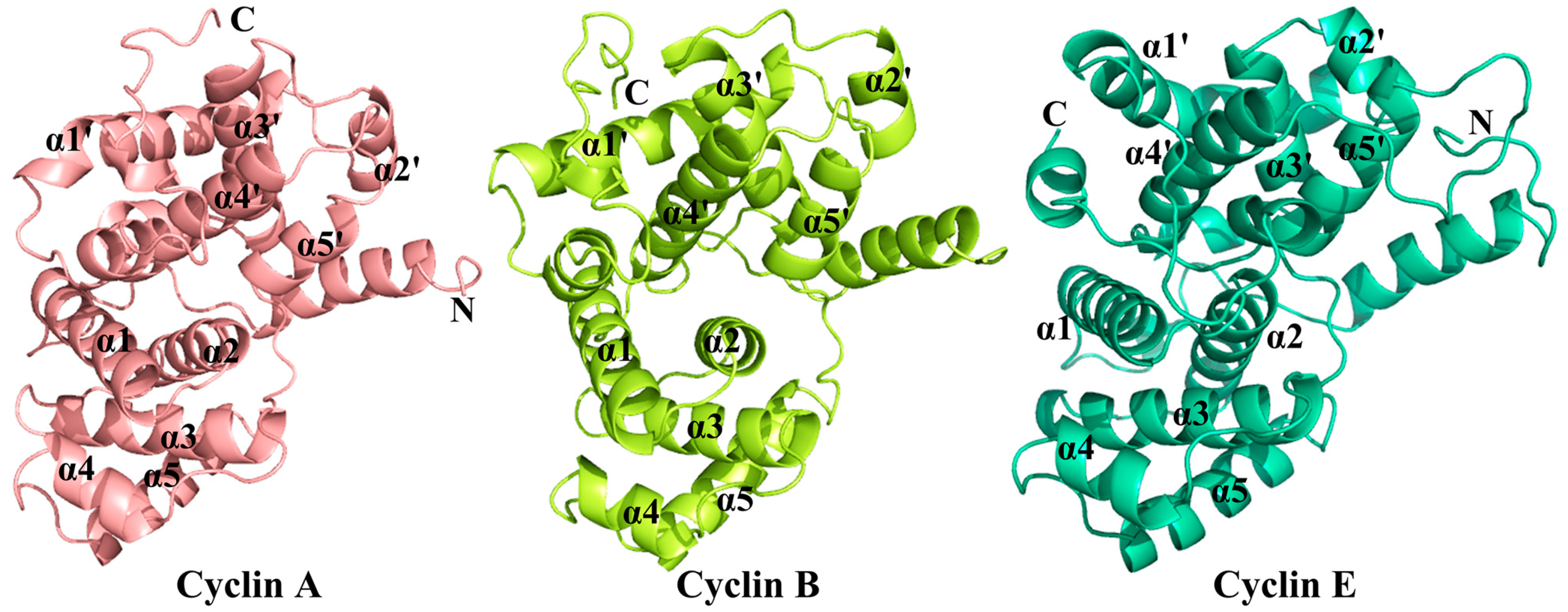
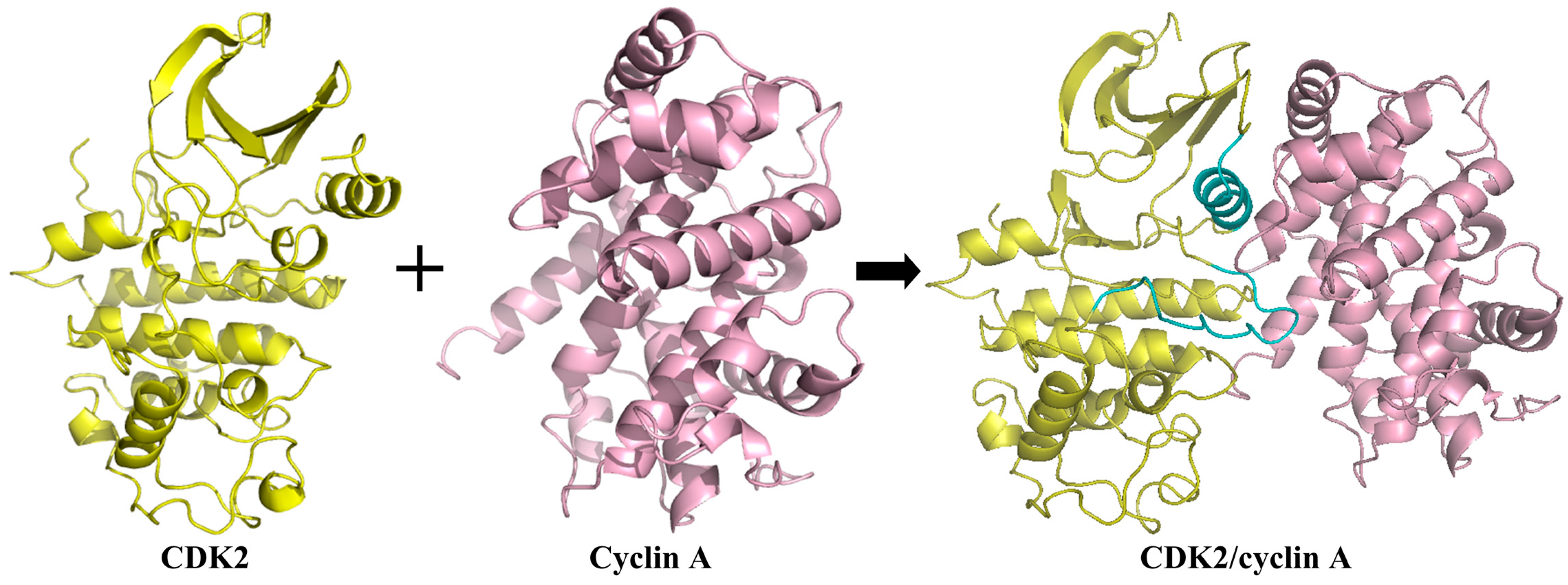
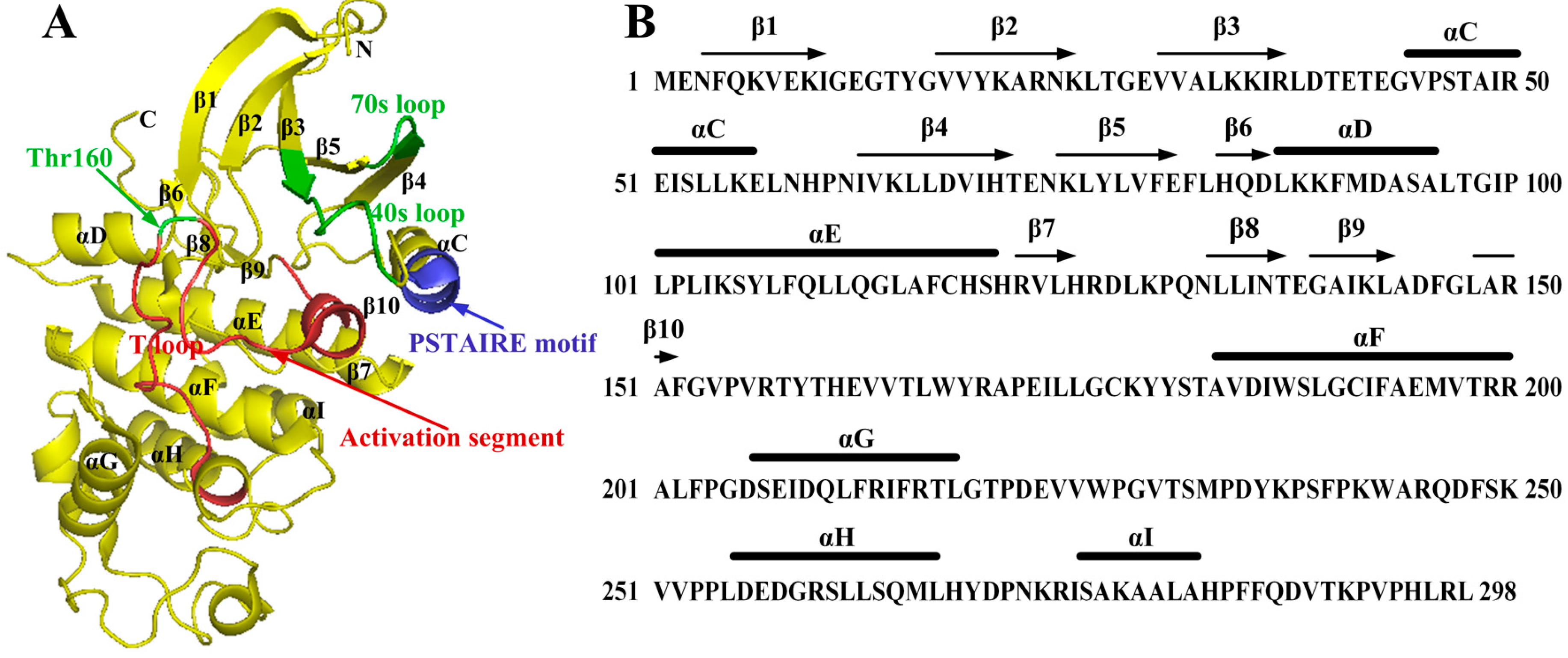
2. Binding Sites of the Monomeric Cyclin-Dependent Kinase 2 (CDK2) and the CDK2/Cyclin Complexes
2.1. Characterization of Monomeric CDK2
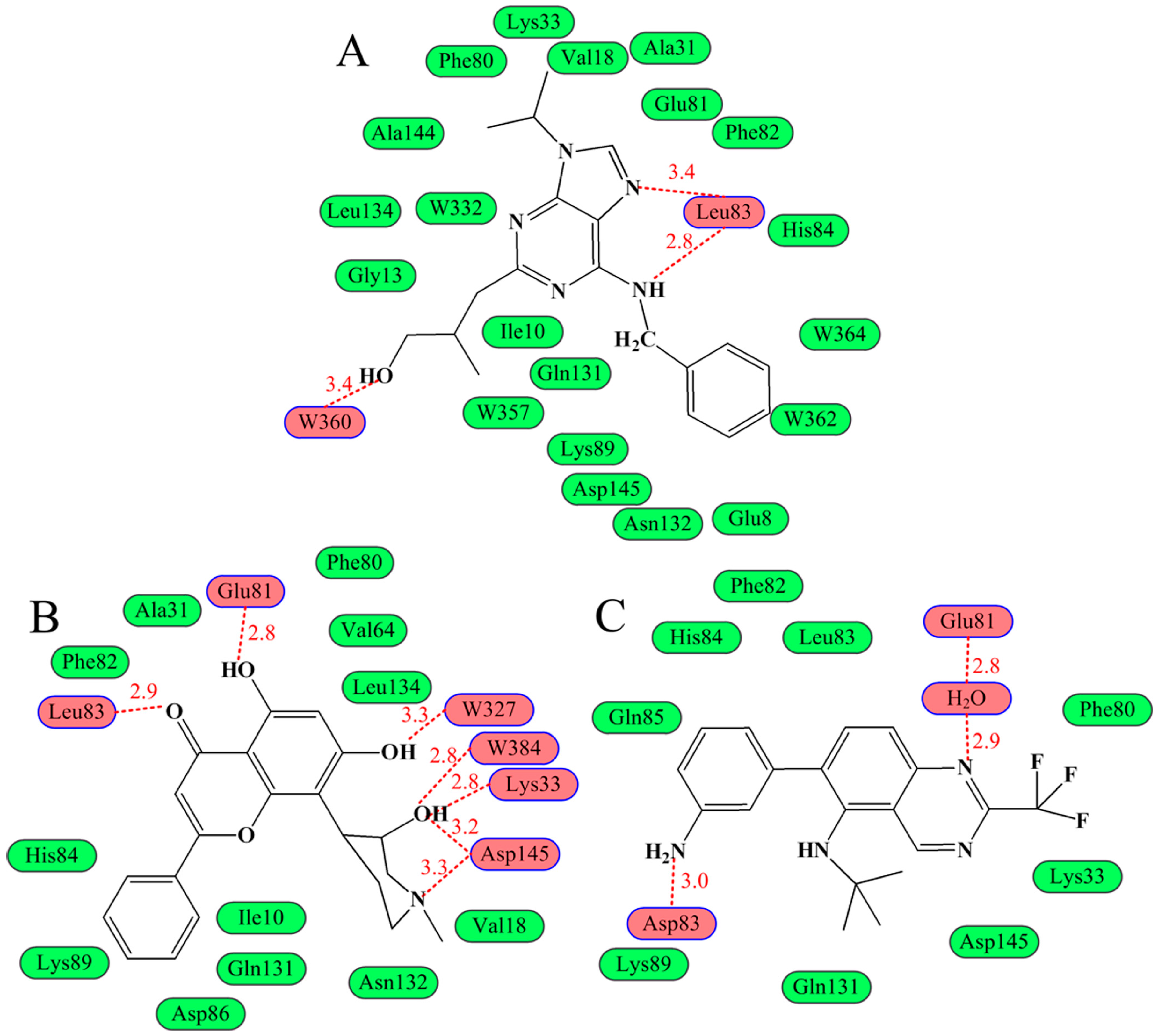

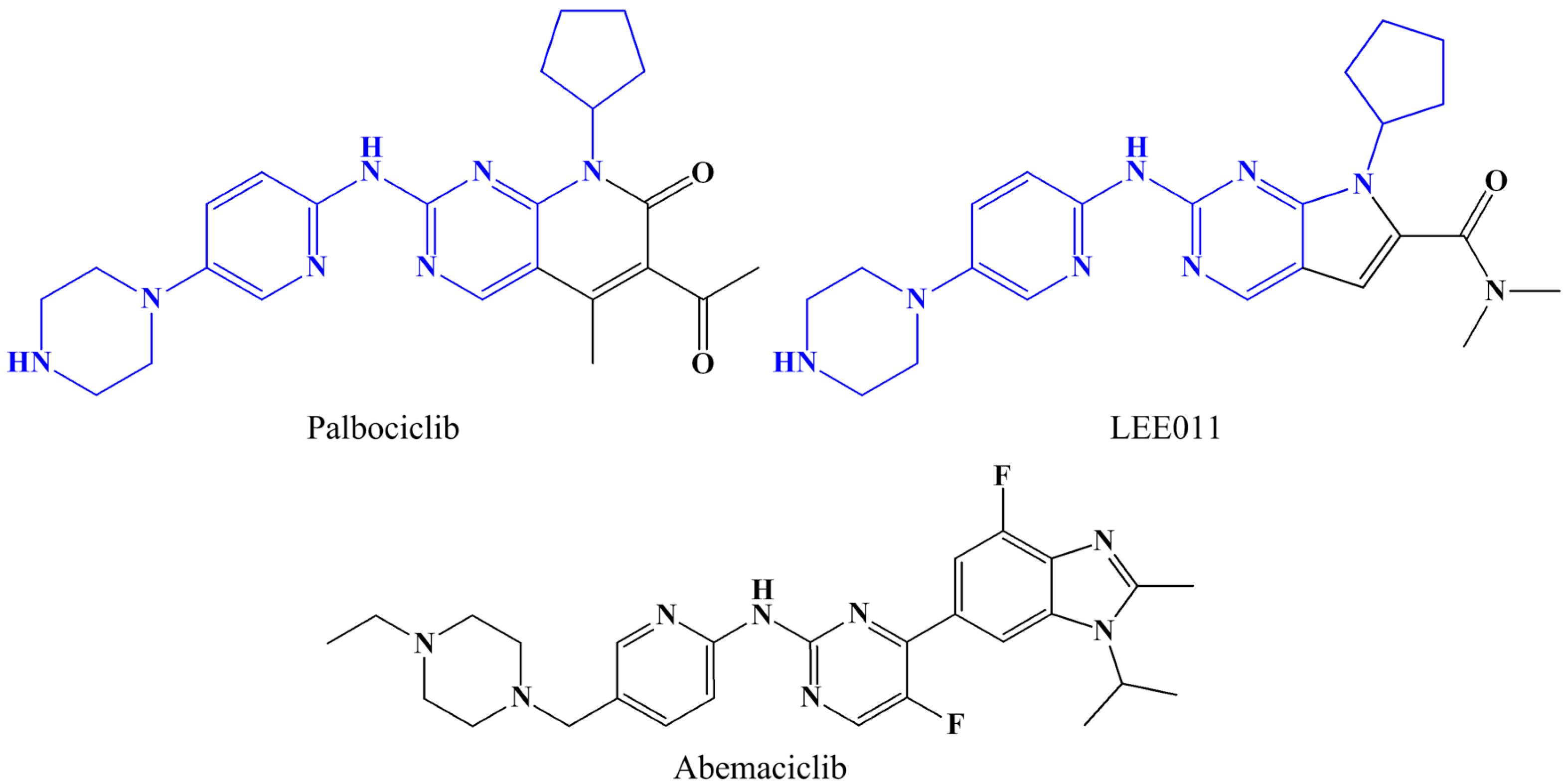

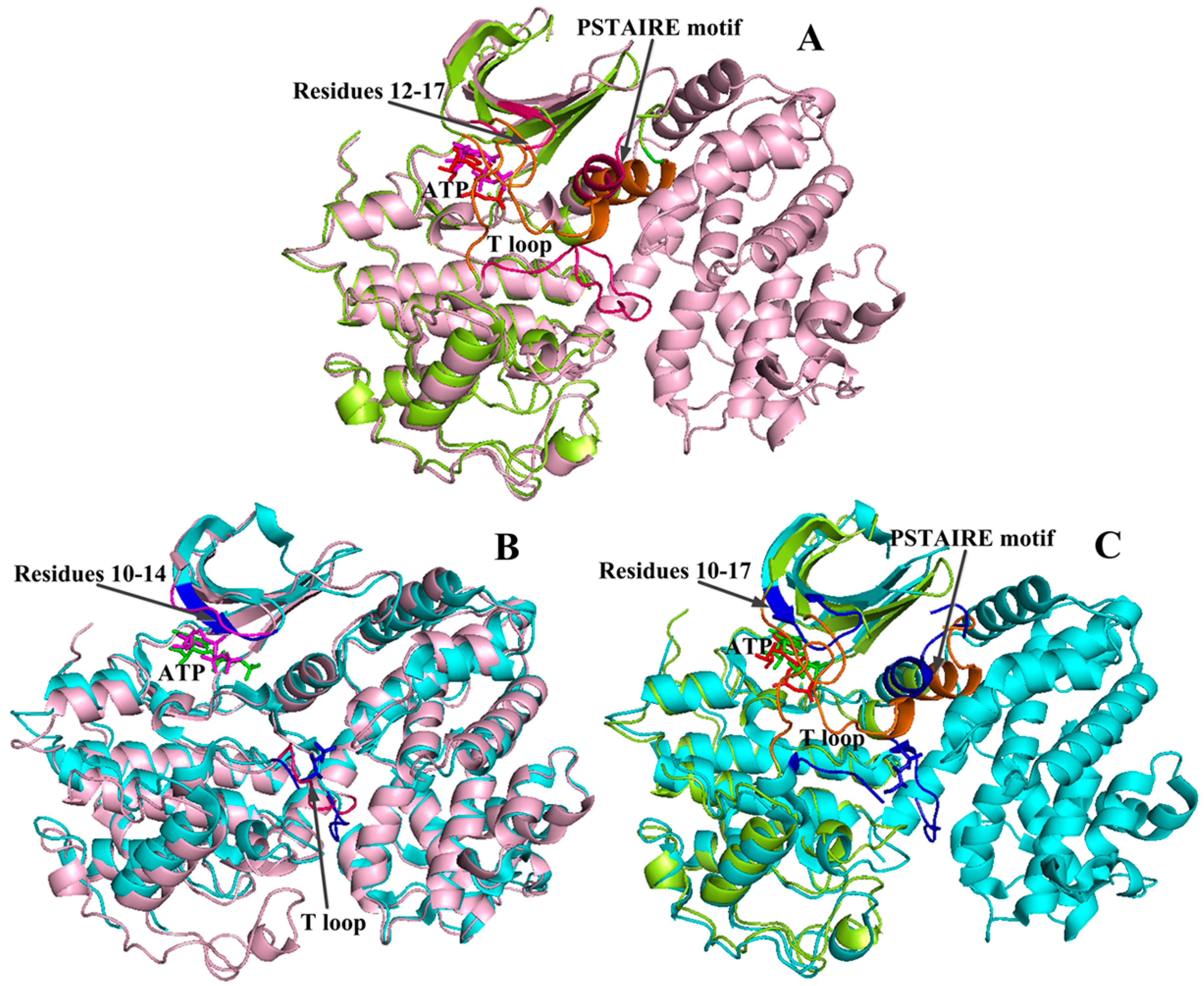
2.2. Conformational Changes of CDK2 via Cyclin Binding
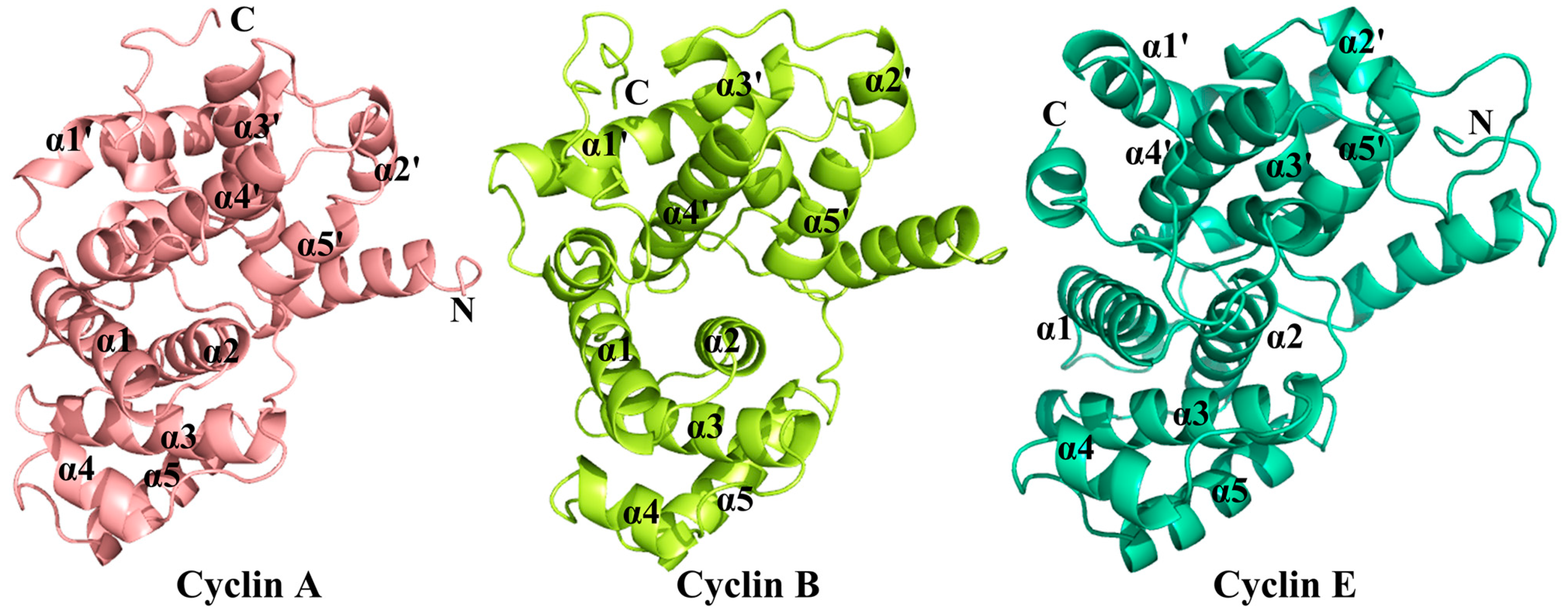

2.3. Conformational Changes of CDK2 via Phosphorylation
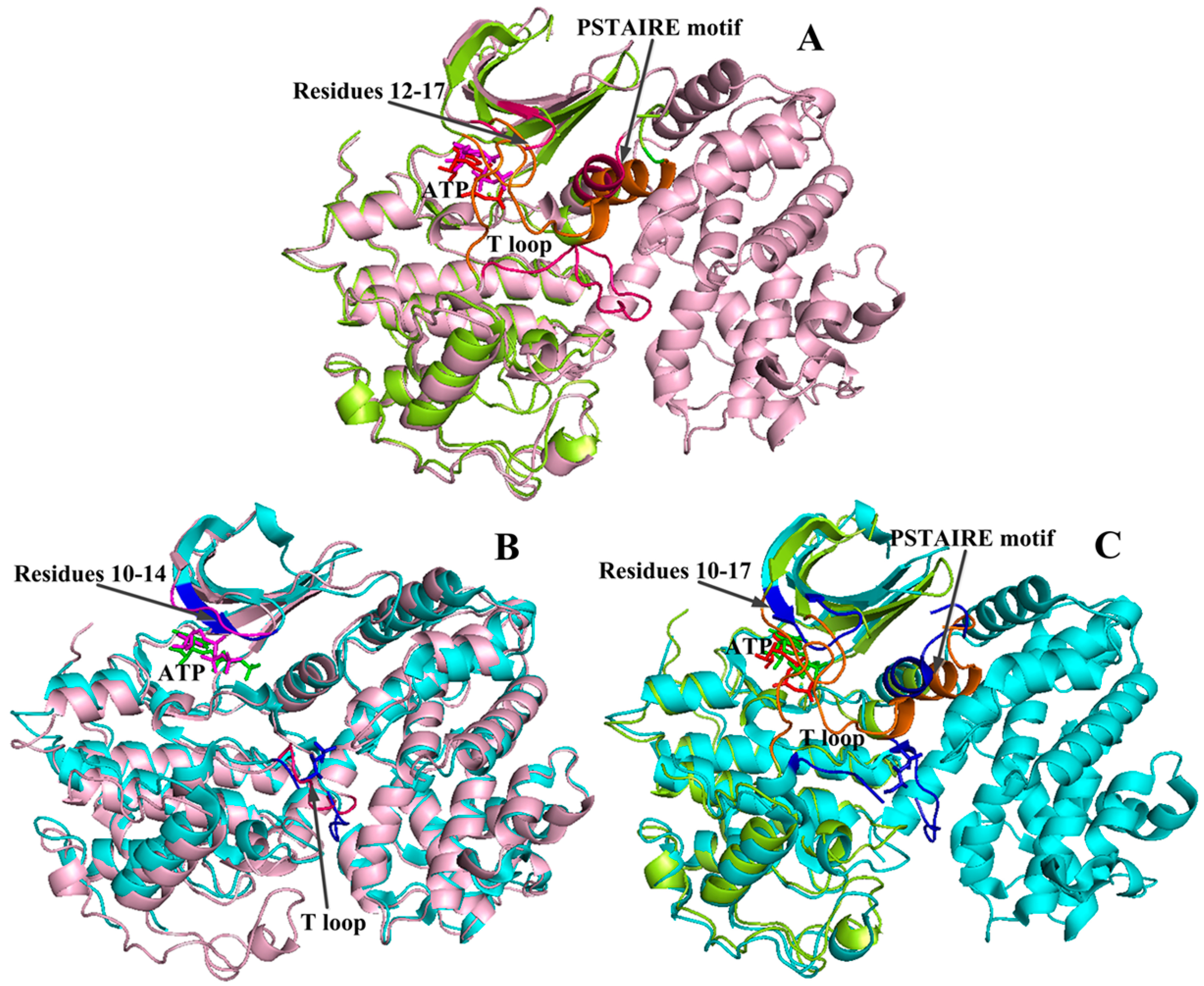
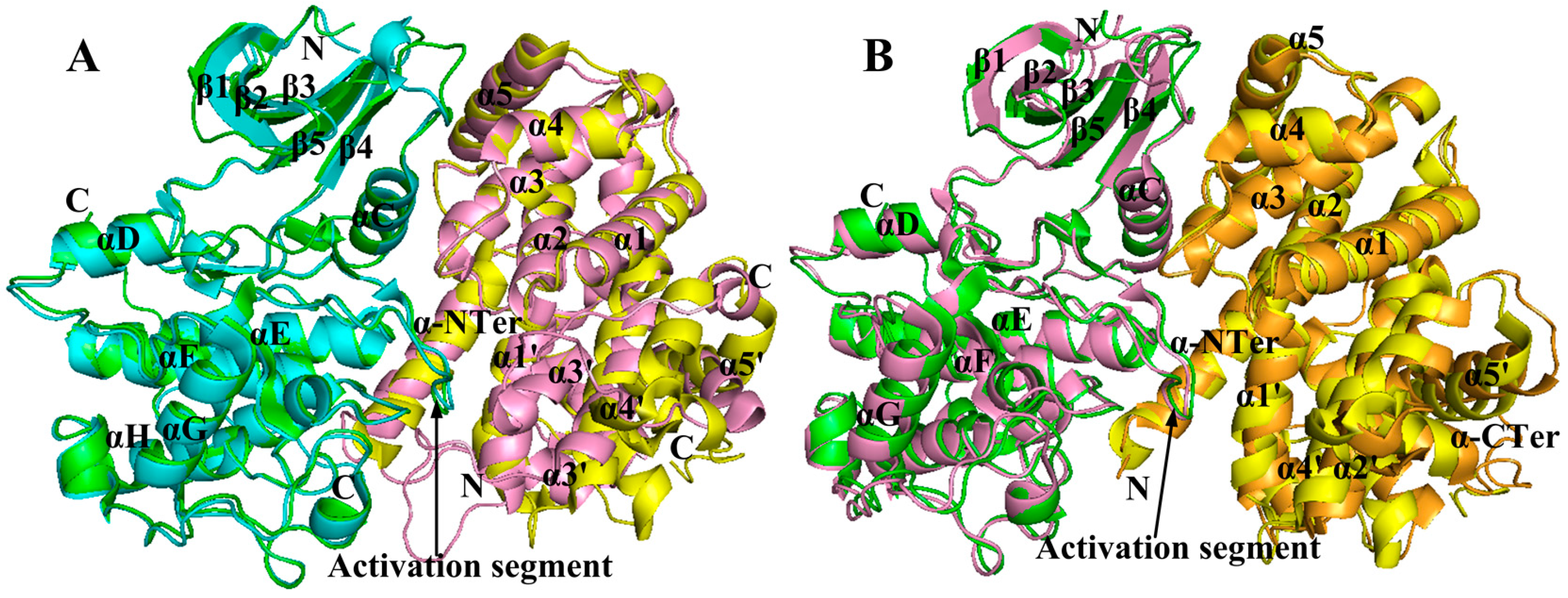
2.4. Comparison with Active CDK2/Cyclin Complexes

2.5. Changes of the Interaction via Conformational Fluctuations
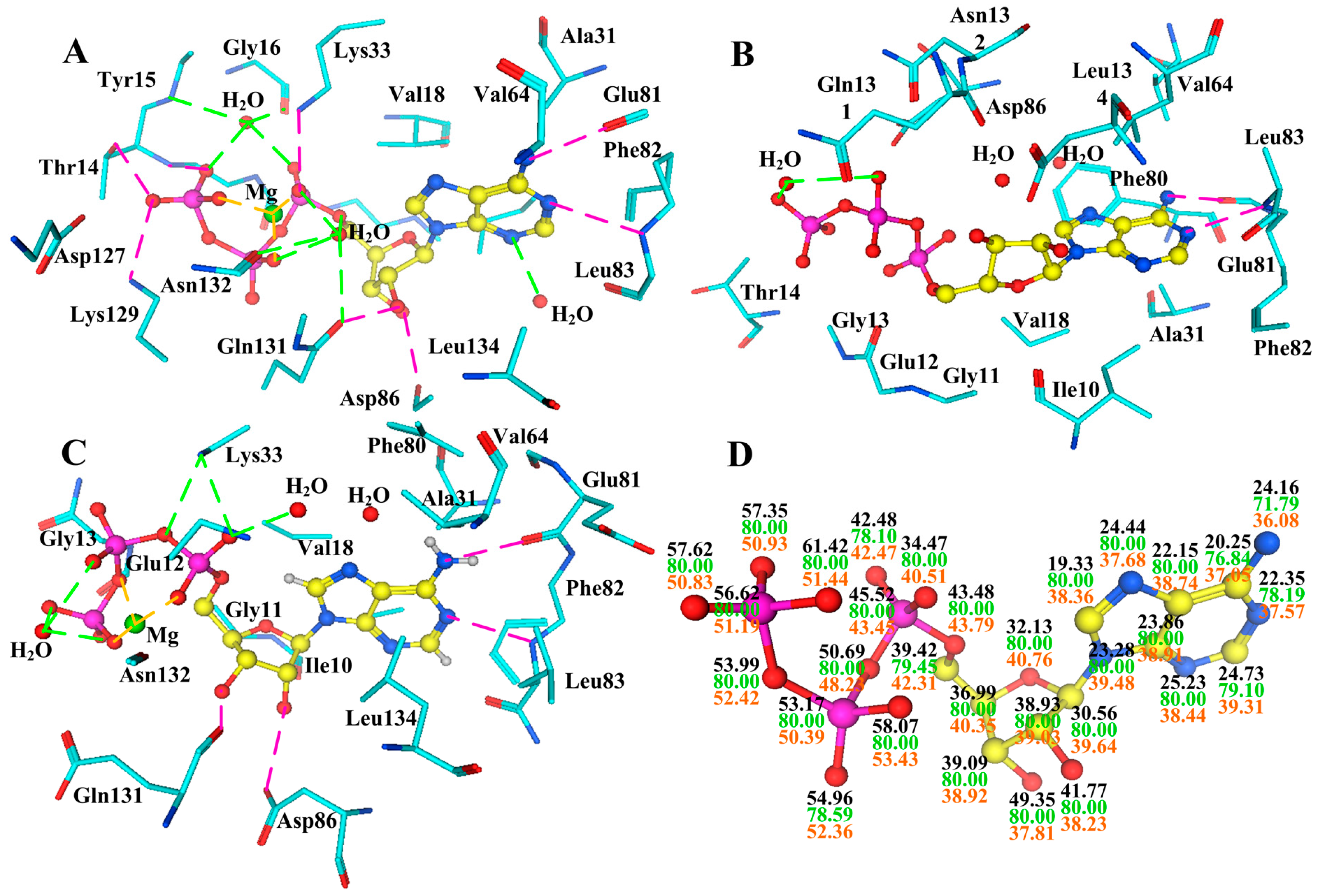


3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Gray, N.; Detivaud, L.; Doerig, C.; Meijer, L. ATP-site directed inhibitors of cyclin-dependent kinases. Curr. Med. Chem. 1999, 6, 859–875. [Google Scholar] [PubMed]
- De Vivo, M.; Bottegoni, G.; Berteotti, A.; Recanatini, M.; Gervasio, F.L.; Cavalli, A. Cyclin-dependent kinases: Bridging their structure and function through computations. Future Med. Chem. 2011, 3, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.H.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, I.R.; Golding, B.T.; Griffin, R.J. Designing inhibitors of cyclin-dependent kinases. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 325–348. [Google Scholar] [CrossRef] [PubMed]
- Lolli, G.; Johnson, L.N. CAK-Cyclin-dependent Activating Kinase: A key kinase in cell cycle control and a target for drugs? Cell Cycle 2005, 4, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massague, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, S.; Merrick, K.A.; Terret, M.E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for CDK7 in the assembly of CDK1/cyclin B and activation of CDK2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Pillai, P.; Desai, S.; Patel, R.; Sajan, M.; Farese, R.; Ostrov, D.; Acevedo-Duncan, M. A novel PKC-ι inhibitor abrogates cell proliferation and induces apoptosis in neuroblastoma. Int. J. Biochem. Cell Biol. 2011, 43, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Bica, L.; Meyerowitz, J.; Parker, S.J.; Caragounis, A.; Du, T.; Paterson, B.M.; Barnham, K.J.; Crouch, P.J.; White, A.R.; Donnelly, P.S. Cell cycle arrest in cultured neuroblastoma cells exposed to a bis(thiosemicarbazonato) metal complex. Biometals 2011, 24, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P.; Morgan, D.O. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 1994, 78, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Pavletich, N.P. Mechanisms of cyclin-dependent kinase regulation: Structures of cdks, their cyclin activators, and cip and INK4 inhibitors. J. Mol. Biol. 1999, 287, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Pines, J. Cyclins: Wheels within wheels. Cell Growth Differ. 1991, 2, 305–310. [Google Scholar] [PubMed]
- Toledo, L.M.; Lydon, N.B.; Elbaum, D. The structure-based design of ATP-site directed protein kinase inhibitors. Curr. Med. Chem. 1999, 6, 775–805. [Google Scholar] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Flores, O.; Wang, Z.; Knudsen, K.E.; Burnstein, K.L. Nuclear targeting of cyclin-dependent kinase 2 reveals essential roles of cyclin-dependent kinase 2 localization and cyclin E in vitamin D-mediated growth inhibition. Endocrinology 2010, 151, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Kawana, H.; Tamaru, J.-I.; Tanaka, T.; Hirai, A.; Saito, Y.; Kitagawa, M.; Mikata, A.; Harigaya, K.; Kuriyama, T. Role of p27Kip1 and cyclin-dependent kinase 2 in the proliferation of non-small cell lung cancer. Am. J. Pathol. 1998, 153, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Heathcote, D.A.; Kroll, S.H.; Jogalekar, A.S.; Scheiper, B.; Patel, H.; Brackow, J.; Siwicka, A.; Fuchter, M.J.; Periyasamy, M. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009, 69, 6208–6215. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Bentley, J.; Wang, L.; Newell, D.; Robson, C.; Shapiro, G.; Curtin, N. Pre-clinical evaluation of cyclin-dependent kinase 2 and 1 inhibition in anti-estrogen-sensitive and resistant breast cancer cells. Br. J. Cancer 2009, 102, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Weinberg, R.A. Tossing monkey wrenches into the clock: New ways of treating cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 4221–4223. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.; Indovina, P.; Magnotti, F.; Conti, D.; Giordano, A. Anticancer therapeutic strategies based on CDK inhibitors. Curr. Pharm. Des. 2013, 19, 5327–5332. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Pevarello, P.; Barbacid, M.; Bischoff, J.R. CDK inhibitors in cancer therapy: What is next? Trends Pharmacol. Sci. 2008, 29, 16–21. [Google Scholar] [CrossRef] [PubMed]
- DePinto, W.; Chu, X.J.; Yin, X.; Smith, M.; Packman, K.; Goelzer, P.; Lovey, A.; Chen, Y.; Qian, H.; Hamid, R.; et al. In vitro and in vivo activity of R547: A potent and selective cyclin-dependent kinase inhibitor currently in phase I clinical trials. Mol. Cancer Ther. 2006, 5, 2644–2658. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, D.E.; Melville, J.; Frame, S.; Watt, K.; Anderson, S.; Gianella-Borradori, A.; Lane, D.P.; Green, S.R. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005, 65, 5399–5407. [Google Scholar] [CrossRef] [PubMed]
- Rizzolio, F.; Tuccinardi, T.; Caligiuri, I.; Lucchetti, C.; Giordano, A. CDK inhibitors: From the bench to clinical trials. Curr. Drug Targets 2010, 11, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Shirsath, N.P.; Manohar, S.M.; Joshi, K.S. P276–00, a cyclin-dependent kinase inhibitor, modulates cell cycle and induces apoptosis in vitro and in vivo in mantle cell lymphoma cell lines. Mol. Cancer 2012, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, A.L.; Duffin, R.; Leitch, A.E.; Lucas, C.D.; Sheldrake, T.A.; Dorward, D.A.; Hirani, N.; Pinho, V.; de Sousa, L.P.; Teixeira, M.M.; et al. Induction of eosinophil apoptosis by the cyclin-dependent kinase inhibitor AT7519 promotes the resolution of eosinophil-dominant allergic inflammation. PLoS ONE 2011, 6, e25683. [Google Scholar] [CrossRef] [PubMed]
- Rigas, A.C.; Robson, C.N.; Curtin, N.J. Therapeutic potential of CDK inhibitor NU2058 in androgen-independent prostate cancer. Oncogene 2007, 26, 7611–7619. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Valius, M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011, 137, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.S.; Rathos, M.J.; Joshi, R.D.; Sivakumar, M.; Mascarenhas, M.; Kamble, S.; Lal, B.; Sharma, S. In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, P276–00. Mol. Cancer Ther. 2007, 6, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.S.; Rathos, M.J.; Mahajan, P.; Wagh, V.; Shenoy, S.; Bhatia, D.; Chile, S.; Sivakumar, M.; Maier, A.; Fiebig, H.H.; et al. P276–00, a novel cyclin-dependent inhibitor induces G1-G2 arrest, shows antitumor activity on cisplatin-resistant cells and significant in vivo efficacy in tumor models. Mol. Cancer Ther. 2007, 6, 926–934. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.H. Inhibition of cyclin-dependent kinases by purine analogues: Crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Bagella, L.; Sun, A.; Tonini, T.; Abbadessa, G.; Cottone, G.; Paggi, M.G.; de Luca, A.; Claudio, P.P.; Giordano, A. A small molecule based on the pRb2/p130 spacer domain leads to inhibition of cdk2 activity, cell cycle arrest and tumor growth reduction in vivo. Oncogene 2007, 26, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Endicott, J.A.; Noble, M.E. Structural characterization of the cyclin-dependent protein kinase family. Biochem. Soc. Trans. 2013, 41, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Stetler-Stevenson, M.; Sebers, S.; Worland, P.; Sedlacek, H.; Myers, C.; Czech, J.; Naik, R.; Sausville, E. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86–8275. J. Natl. Cancer Inst. 1992, 84, 1736–1740. [Google Scholar] [CrossRef] [PubMed]
- De Azevedo, W., Jr.; Mueller-Dieckmann, H.; Schulze-Gahmen, U.; Worland, P.; Sausville, E.; Kim, S. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Murthi, K.; Dubay, M.; McClure, C.; Brizuela, L.; Boisclair, M.; Worland, P.; Mansuri, M.; Pal, K. Structure-activity relationship studies of flavopiridol analogues. Bioorg. Med. Chem. Lett. 2000, 10, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- McClue, S.; Blake, D.; Clarke, R.; Cowan, A.; Cummings, L.; Fischer, P.; MacKenzie, M.; Melville, J.; Stewart, K.; Wang, S. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int. J. Cancer 2002, 102, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.; Blow, J.; Inagaki, N.; Inagaki, M.; Delcros, J.; Moulinoux, J. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Caporali, S.; Alvino, E.; Starace, G.; Ciomei, M.; Brasca, M.; Levati, L.; Garbin, A.; Castiglia, D.; Covaciu, C.; Bonmassar, E. The cyclin-dependent kinase inhibitor PHA-848125 suppresses the in vitro growth of human melanomas sensitive or resistant to temozolomide, and shows synergistic effects in combination with this triazene compound. Pharmacol. Res. 2010, 61, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Futami, H.; Takahara, J.; Yamaguchi, K. UCN-01, 7-hydroxyl-staurosporine, inhibits kinase activity of cyclin-dependent kinases and reduces the phosphorylation of the retinoblastoma susceptibility gene product in A549 human lung cancer cell line. Biochem. Biophys. Res. Commun. 1996, 219, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Christensen, S.; Frankel, P.; Margolin, K.; Agarwala, S.; Luu, T.; Mack, P.; Lara, P., Jr.; Gandara, D. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: A California Cancer Consortium trial. Investig. New Drugs 2012, 30, 741–748. [Google Scholar] [CrossRef]
- Senderowicz, A. The cell cycle as a target for cancer therapy: Basic and clinical findings with the small molecule inhibitors flavopiridol and UCN-01. Oncologist 2002, 7, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; DePinto, W.; Bartkovitz, D.; So, S.; Vu, B.; Packman, K.; Lukacs, C.; Ding, Q.; Jiang, N.; Wang, K. Discovery of (4-Amino-2-(1-methanesulfonylpiperidin-4-ylamino) pyrimidin-5-yl)(2, 3-difluoro-6-methoxyphenyl) methanone (R547), a potent and selective cyclin-dependent kinase inhibitor with significant in vivo antitumor activity. J. Med. Chem. 2006, 49, 6549–6560. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Plummer, R.; Squires, M.; Rensvold, D.; Kurtin, S.; Pretzinger, C.; Dragovich, T.; Adams, J.; Lock, V.; Smith, D. A phase I pharmacokinetic and pharmacodynamic study of AT7519, a cyclin-dependent kinase inhibitor in patients with refractory solid tumors. Ann. Oncol. 2011, 22, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Cooke, L.; Lock, V.; Qi, W.; Lewis, E.J.; Thompson, N.T.; Lyons, J.F.; Mahadevan, D. AT7519, a cyclin-dependent kinase inhibitor, exerts its effects by transcriptional inhibition in leukemia cell lines and patient samples. Mol. Cancer Ther. 2010, 9, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M. Chronic lymphocytic leukemia: 2013 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2013, 88, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.; Badros, A.; Popplewell, L.; Coutre, S.; Fox, J. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Conroy, A.; Stockett, D.; Walker, D.; Arkin, M.; Hoch, U.; Fox, J.; Hawtin, R. SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother. Pharmacol. 2009, 64, 723–732. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, P.; Moghaddam-Helmantel, I.; de Jonge, M.; Meyer, T.; Lam, M.; Verweij, J.; Wiemer, E.; Loos, W. Validated bioanalytical method for the quantification of RGB-286638, a novel multi-targeted protein kinase inhibitor, in human plasma and urine by liquid chromatography/tandem triple-quadrupole mass spectrometry. J. Pharm. Biomed. Anal. 2009, 50, 977–982. [Google Scholar] [CrossRef]
- Van der Biessen, D.A.; Burger, H.; de Bruijn, P.; Lamers, C.H.; Naus, N.; Loferer, H.; Wiemer, E.A.; Mathijssen, R.H.; de Jonge, M.J. Phase I study of RGB-286638, a novel, multitargeted cyclin-dependent kinase inhibitor in patients with solid tumors. Clin. Cancer Res. 2014, 20, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Siemeister, G.; Lucking, U.; Wengner, A.; Lienau, P.; Schatz, C.; Mumberg, D.; Ziegelbauer, K. Pharmacologic profile of the oral novel pan-CDK inhibitor BAY 1000394 in chemosensitive and chemorefractory tumor models. Cancer Res. 2010, 70, 3883–3883. [Google Scholar] [CrossRef]
- Siemeister, G.; Lücking, U.; Wengner, A.; Lienau, P.; Steinke, W.; Schatz, C.; Mumberg, D.; Ziegelbauer, K. BAY 1000394, a novel cyclin-dependent kinase inhibitor, with potent antitumor activity in mono-and in combination treatment upon oral application. Mol. Cancer Ther. 2012, 11, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, A.; William, A.; Blanchard, S.; Nagaraj, H.; Williams, M.; Wang, H.; Lee, A.; Sun, E.; Teo, E.-L.; Tan, E. Structure-based design of nitrogen-linked macrocyclic kinase inhibitors leading to the clinical candidate SB1317/TG02, a potent inhibitor of cyclin dependant kinases (CDKs), Janus kinase 2 (JAK2), and Fms-like tyrosine kinase-3 (FLT3). J. Mol. Model. 2013, 19, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Gu, Y.; Morgan, D.O. Activation of human cyclin-dependent kinases in vitro. Mol. Biol. Cell 1992, 3, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Connell-Crowley, L.; Solomon, M.J.; Wei, N.; Harper, J.W. Phosphorylation independent activation of human cyclin-dependent kinase 2 by cyclin A in vitro. Mol. Biol. Cell 1993, 4, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Gould, K.L.; Moreno, S.; Owen, D.J.; Sazer, S.; Nurse, P. Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J. 1991, 10, 3297–3309. [Google Scholar] [PubMed]
- Gondeau, C.; Gerbal-Chaloin, S.; Bello, P.; Aldrian-Herrada, G.; Morris, M.C.; Divita, G. Design of a novel class of peptide inhibitors of cyclin-dependent kinase/cyclin activation. J. Biol. Chem. 2005, 280, 13793–13800. [Google Scholar] [CrossRef] [PubMed]
- Krek, W.; Nigg, E.A. Cell cycle regulation of vertebrate p34cdc2 activity: Identification of Thr161 as an essential in vivo phosphorylation site. New Biol. 1992, 4, 323–329. [Google Scholar] [PubMed]
- Gu, Y.; Rosenblatt, J.; Morgan, D.O. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992, 11, 3995–4005. [Google Scholar]
- Solomon, M.J. Activation of the various cyclin/cdc2 protein kinases. Curr. Opin. Cell Biol. 1993, 5, 180–186. [Google Scholar] [CrossRef]
- Ducommun, B.; Brambilla, P.; Felix, M.A.; Franza, B.R., Jr.; Karsenti, E.; Draetta, G. cdc2 phosphorylation is required for its interaction with cyclin. EMBO J. 1991, 10, 3311–3319. [Google Scholar] [PubMed]
- De Bondt, H.L.; Rosenblatt, J.; Jancarik, J.; Jones, H.D.; Morgan, D.O.; Kim, S.H. Crystal structure of cyclin-dependent kinase 2. Nature 1993, 363, 595–602. [Google Scholar] [CrossRef]
- Cox, S.; Radzio-Andzelm, E.; Taylor, S.S. Domain movements in protein kinases. Curr. Opin. Struct. Biol. 1994, 4, 893–901. [Google Scholar]
- Brown, N.R.; Noble, M.E.; Endicott, J.A.; Johnson, L.N. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 438–443. [Google Scholar]
- Brown, N.R.; Noble, M.E.; Lawrie, A.M.; Morris, M.C.; Tunnah, P.; Divita, G.; Johnson, L.N.; Endicott, J.A. Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J. Biol. Chem. 1999, 274, 8746–8756. [Google Scholar]
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of protein kinases: Controlling activity through activation segment conformation. Mol. Cell 2004, 15, 661–675. [Google Scholar]
- Holmes, J.K.; Solomon, M.J. The role of Thr160 phosphorylation of Cdk2 in substrate recognition. Eur. J. Biochem. 2001, 268, 4647–4653. [Google Scholar]
- Davies, T.G.; Tunnah, P.; Meijer, L.; Marko, D.; Eisenbrand, G.; Endicott, J.A.; Noble, M.E. Inhibitor binding to active and inactive CDK2: The crystal structure of CDK2-cyclin A/indirubin-5-sulphonate. Structure 2001, 9, 389–397. [Google Scholar]
- Barrett, C.P.; Noble, M.E. Molecular motions of human cyclin-dependent kinase 2. J. Biol. Chem. 2005, 280, 13993–14005. [Google Scholar]
- Knockaert, M.; Greengard, P.; Meijer, L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol. Sci. 2002, 23, 417–425. [Google Scholar]
- Sims, P.A.; Wong, C.F.; McCammon, J.A. A computational model of binding thermodynamics: The design of cyclin-dependent kinase 2 inhibitors. J. Med. Chem. 2003, 46, 3314–3325. [Google Scholar]
- Huang, D.; Zhou, T.; Lafleur, K.; Nevado, C.; Caflisch, A. Kinase selectivity potential for inhibitors targeting the ATP binding site: A network analysis. Bioinformatics 2010, 26, 198–204. [Google Scholar]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar]
- Johnson, L.N.; de Moliner, E.; Brown, N.R.; Song, H.; Barford, D.; Endicott, J.A.; Noble, M.E. Structural studies with inhibitors of the cell cycle regulatory kinase cyclin-dependent protein kinase 2. Pharmacol. Ther. 2002, 93, 113–124. [Google Scholar]
- Xiang, J.; Yang, H.; Che, C.; Zou, H.; Wei, Y.; Quan, J.; Zhang, H.; Yang, Z.; Lin, S. Identifying tumor cell growth inhibitors by combinatorial chemistry and zebrafish assays. PLoS ONE 2009, 4, e4361. [Google Scholar]
- Li, Y.; Gao, W.; Li, F.; Wang, J.; Zhang, J.; Yang, Y.; Zhang, S.; Yang, L. An in silico exploration of the interaction mechanism of pyrazolo[1,5-a]pyrimidine type CDK2 inhibitors. Mol. Biosyst. 2013, 9, 2266–2281. [Google Scholar]
- Kim, K.S.; Kimball, S.D.; Misra, R.N.; Rawlins, D.B.; Hunt, J.T.; Xiao, H.Y.; Lu, S.; Qian, L.; Han, W.C.; Shan, W.; et al. Discovery of aminothiazole inhibitors of cyclin-dependent kinase 2: Synthesis, X-ray crystallographic analysis, and biological activities. J. Med. Chem. 2002, 45, 3905–3927. [Google Scholar]
- Sielecki, T.M.; Johnson, T.L.; Liu, J.; Muckelbauer, J.K.; Grafstrom, R.H.; Cox, S.; Boylan, J.; Burton, C.R.; Chen, H.; Smallwood, A.; et al. Quinazolines as cyclin dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1157–1160. [Google Scholar]
- Shafiq, M.I.; Steinbrecher, T.; Schmid, R. Fascaplysin as a specific inhibitor for CDK4: Insights from molecular modelling. PLoS ONE 2012, 7, e42612. [Google Scholar] [PubMed]
- Mariaule, G.; Belmont, P. Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where Are We Now? A short survey. Molecules 2014, 19, 14366–14382. [Google Scholar] [CrossRef] [PubMed]
- Cadoo, K.A.; Gucalp, A.; Traina, T.A. Palbociclib: An evidence-based review of its potential in the treatment of breast cancer. Breast Cancer 2014, 6, 123–133. [Google Scholar] [PubMed]
- Witkiewicz, A.K.; Knudsen, E.S. Retinoblastoma tumor suppressor pathway in breast cancer: Prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014, 16, 207. [Google Scholar] [CrossRef] [PubMed]
- McInnes, C.; Wang, S.; Anderson, S.; O’Boyle, J.; Jackson, W.; Kontopidis, G.; Meades, C.; Mezna, M.; Thomas, M.; Wood, G. Structural determinants of CDK4 inhibition and design of selective ATP competitive inhibitors. Chem. Biol. 2004, 11, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Schulze-Gahmen, U. Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J. Med. Chem. 2006, 49, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; van Duyne, R.; Zhang, N.; Kashanchi, F.; Zeng, C. A novel binding pocket of cyclin-dependent kinase 2. Proteins 2009, 74, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Barr, R.K.; Ketterman, A.J. Peptide inhibitors of protein kinases-discovery, characterisation and use. Biochim. Biophys. Acta 2005, 1754, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Lowe, E.D.; Dubinina, E.; Skamnaki, V.; Cook, A.; Brown, N.R.; Johnson, L.N. The structure of cyclin E1/CDK2: Implications for CDK2 activation and CDK2-independent roles. EMBO J. 2005, 24, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.R.; Lowe, E.D.; Petri, E.; Skamnaki, V.; Antrobus, R.; Johnson, L.N. Cyclin B and cyclin A confer different substrate recognition properties on CDK2. Cell Cycle 2007, 6, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Kaldis, P.; Sutton, A.; Solomon, M.J. The Cdk-activating kinase (CAK) from budding yeast. Cell 1996, 86, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Betzi, S.; Alam, R.; Martin, M.; Lubbers, D.J.; Han, H.; Jakkaraj, S.R.; Georg, G.I.; Schonbrunn, E. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem. Biol. 2011, 6, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Alam, R.; Betzi, S.; Ingles, D.J.; Zhu, J.Y.; Schonbrunn, E. A novel approach to the discovery of small-molecule ligands of CDK2. Chembiochem 2012, 13, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Anighoro, A.; Chripkova, M.; Carrassa, L.; Broggini, M. Structure-based discovery of the first allosteric inhibitors of cyclin-dependent kinase 2. Cell Cycle 2014, 13, 2296–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pines, J. Four-dimensional control of the cell cycle. Nat. Cell Biol. 1999, 1, E73–E79. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996, 3, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Latham, V.M.; Zhang, X.; Shapiro, G.I. Combined depletion of cell cycle and transcriptional cyclin-dependent kinase activities induces apoptosis in cancer cells. Cancer Res. 2006, 66, 9270–9280. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhang, J.; Gao, W.; Zhang, L.; Pan, Y.; Zhang, S.; Wang, Y. Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2. Int. J. Mol. Sci. 2015, 16, 9314-9340. https://doi.org/10.3390/ijms16059314
Li Y, Zhang J, Gao W, Zhang L, Pan Y, Zhang S, Wang Y. Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2. International Journal of Molecular Sciences. 2015; 16(5):9314-9340. https://doi.org/10.3390/ijms16059314
Chicago/Turabian StyleLi, Yan, Jingxiao Zhang, Weimin Gao, Lilei Zhang, Yanqiu Pan, Shuwei Zhang, and Yonghua Wang. 2015. "Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2" International Journal of Molecular Sciences 16, no. 5: 9314-9340. https://doi.org/10.3390/ijms16059314
APA StyleLi, Y., Zhang, J., Gao, W., Zhang, L., Pan, Y., Zhang, S., & Wang, Y. (2015). Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2. International Journal of Molecular Sciences, 16(5), 9314-9340. https://doi.org/10.3390/ijms16059314