
Gene Overexpression and RNA Silencing Tools for the Genetic Manipulation of the S-(+)-Abscisic Acid Producing Ascomycete Botrytis cinerea

Abstract
:1. Introduction
2. Results
2.1. Construction of Three Binary Vectors Suitable for the ATMT of B. cinerea Strain Bc-6
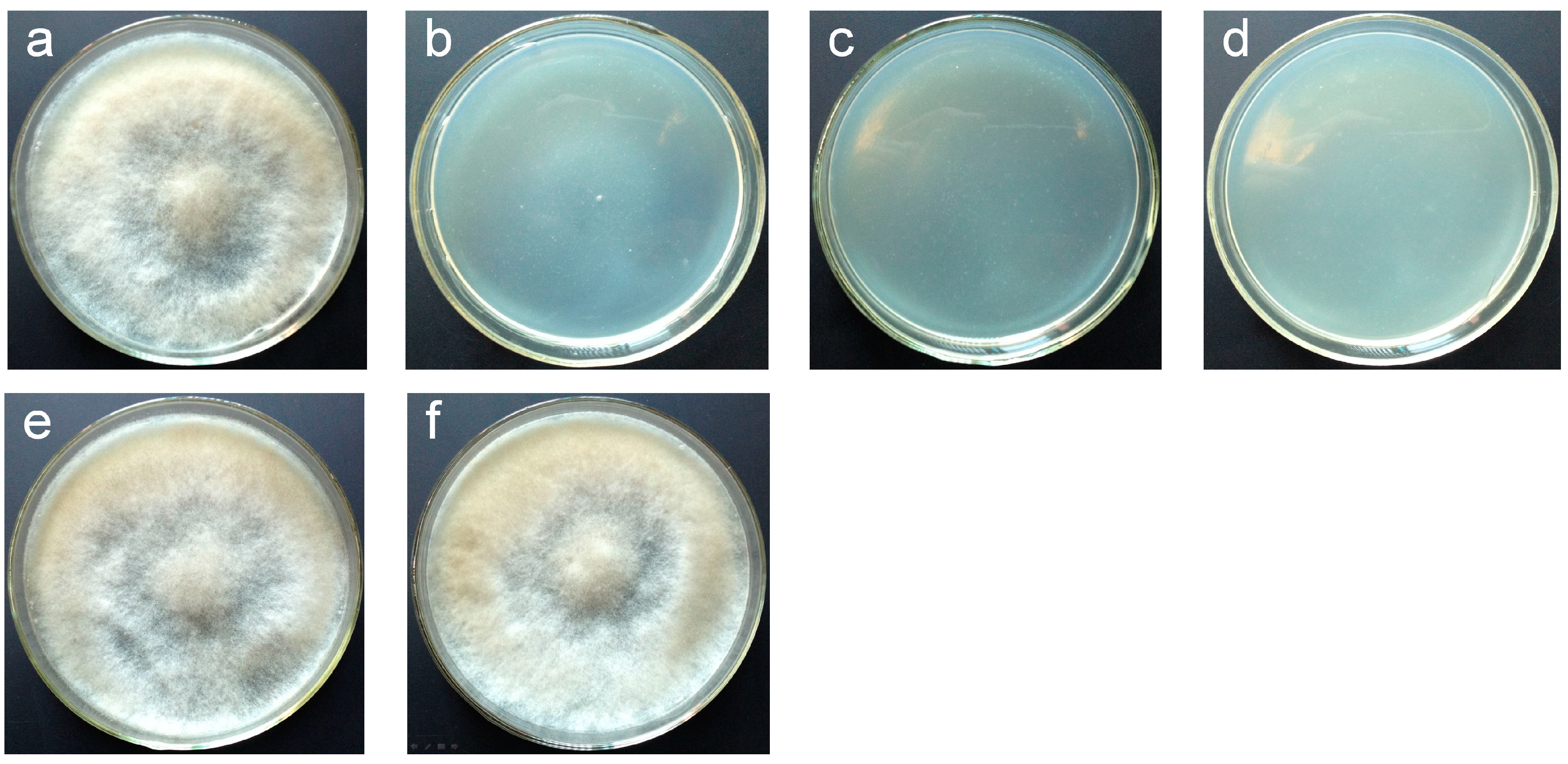
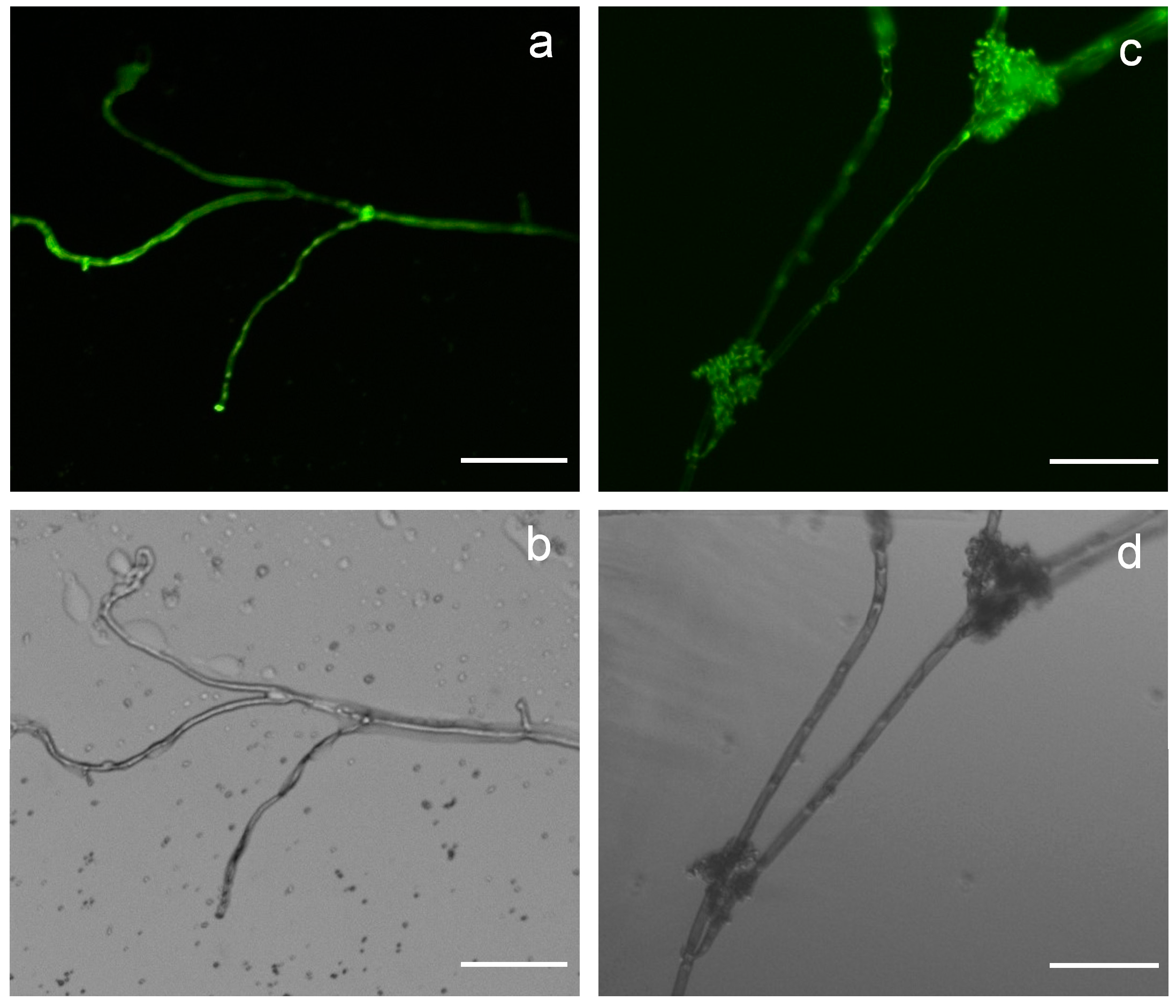
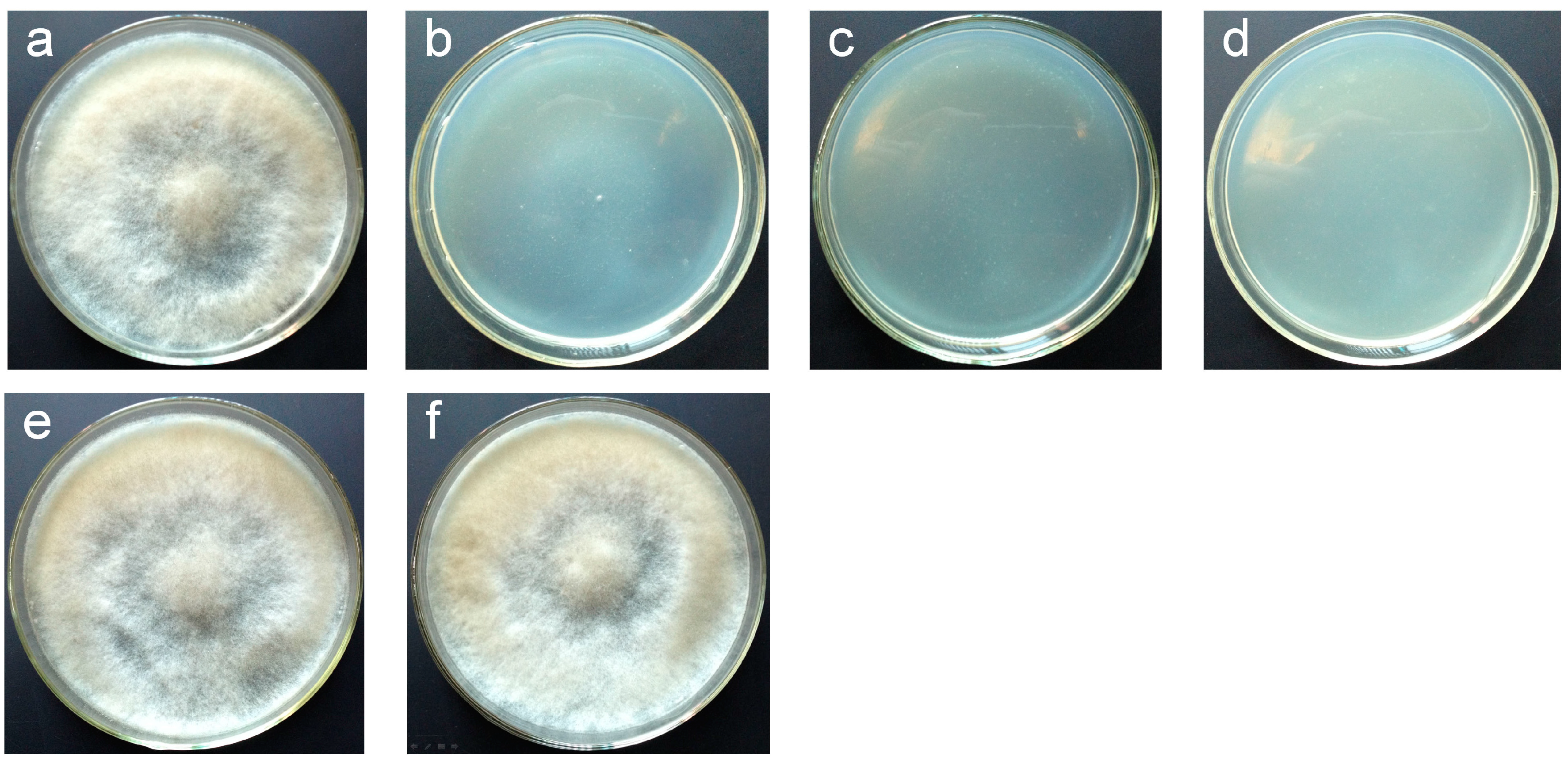
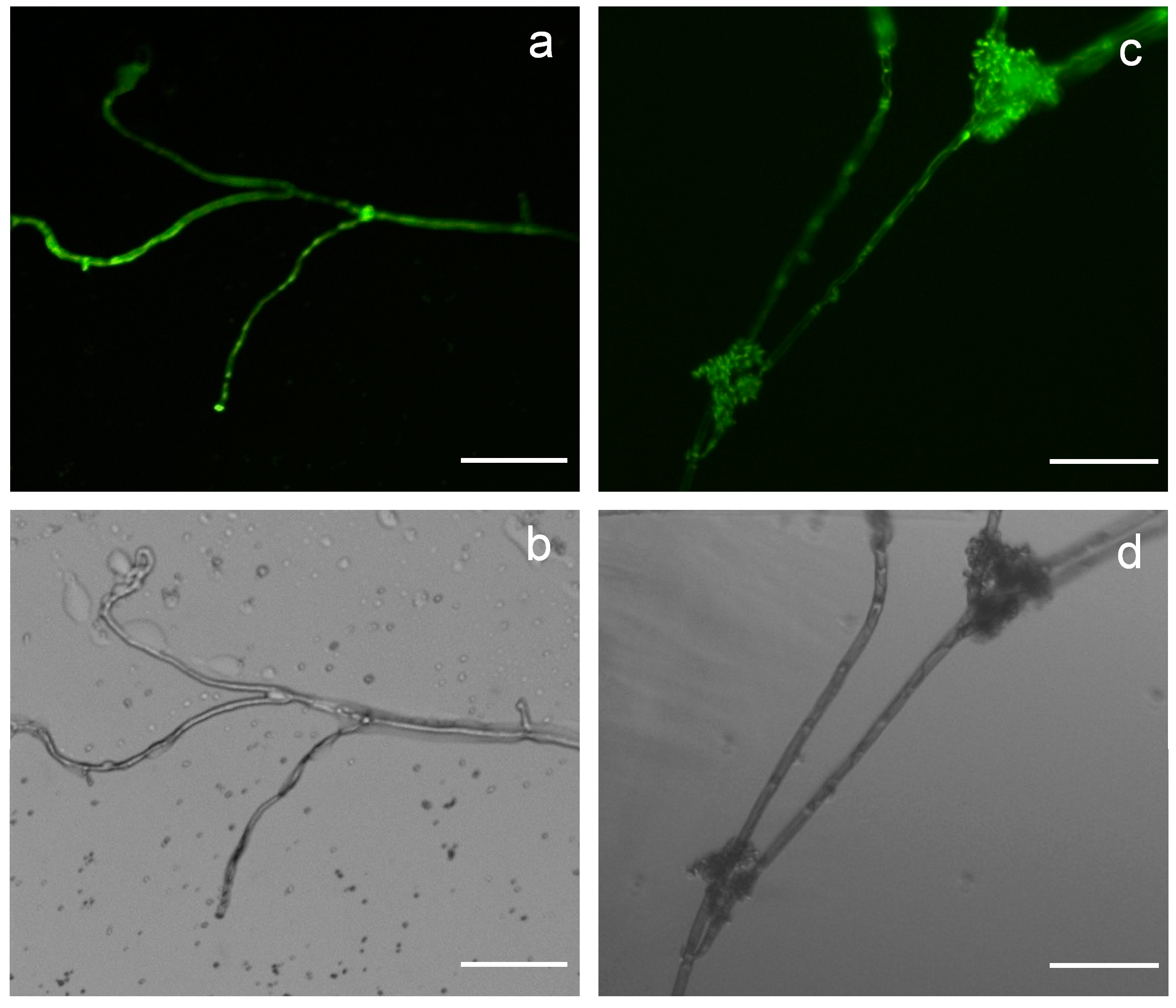
2.2. Vectors pCBh1 and pCBg1 Expressed eGFP in B. cinerea Bc-6 with High Efficiency

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | 5'–3' DNA Sequence |
|---|---|
| pUC19-AnoliC | AGCTCGGTACCCGGGCTGCAGCTGTGGAGCCGCAT |
| pUC19-PstI-AnoliC | CTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCGGATCGATTGTGATGTGATG |
| pUC19-BamHI-AntrpC | GGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGTAGATGCCGACCGGGATC |
| pUC19-AntrpC | GCCAAGCTTGCATGCTAGGCAACCATGCATGGTTAC |
| p1300-AntrpC | CATGATTACGAATTCTAGGCAACCATGCATGGTTACTATTG |
| p1300-AnoliC | TGCCAAGCTTGCATGCTGCAGCTGTGGAGCCGCATTCCG |
| AnoliC-eGFP | CATCACAATCGATCCATGGTGAGCAAGGGCGAGGAGCTG |
| AntrpC-eGFP | CGGTCGGCATCTACTCTAGATCCGGTGGATCCCGGGC |
| bar-5 | ATGAGCCCAGAACGACGCCCG |
| bar-3 | ATCTACTTCAGATCTCGGTGACGGGCAGGACCGGACGGGGCGGTAGCGGCAGG |
| p1300-EcoRI | TATGACCATGATTACTAGGCAACCATGCATGGTTACTATTG |
| p1300-HidIII | ACGACGGCCAGTGCCCTGCAGCTGTGGAGCCGCATTCCG |
| 5-AnoliC-CUT | CATCACAATCGATCCTCTAGAGGTACCGCTGGAGGATACAGG |
| 3-AntrpC-CUT | CGGTCGGCATCTACTGAGCTCGGATCCGCCGTTCCCTGGCTG |
| PAntrpC-3 | GGTAGAATAGGTAAGTCAGATTGAATCTG |
| CaMV35S-5 | GGGATCTCGAGTTTCTCCATAATAATG |
| BstXI-PAntrpC-5 | GGCTAGAGCAGCTTGCCAACATGGTGGGTCGACAGAAGATG |
| SacII-leftborder-3 | GAGCCGATTTTGAAACCGCGGTGATCACAGGCAGCAACGC |
| Silent-eGFP-Se-5 | CATCACAATCGATCCATGGTGAGCAAGGGCGAGG |
| Silent-eGFP-Se-3 | CCTGTATCCTCCAGCCTTGTACAGCTCGTCCATGCCG |
| Silent-eGFP-AS-5 | CAGCCAGGGAACGGCCTTGTACAGCTCGTCCATGCCG |
| Silent-eGFP-AS-3 | CGGTCGGCATCTACTATGGTGAGCAAGGGCGAGG |
| RT-gfp-5 | CACTACCTGAGCACCCAGTC |
| RT-gfp-3 | CACGAACTCCAGCAGGACC |
| RT-ABA4-5 | AAGACTTGGACGAGTGGGAGTT |
| RT-ABA4-3 | CCGTTGTTAGCCATTACTTTCAG |
| RT-tubA-5 | GCGTTCGTGCATTGGTATGT |
| RT-tubA-3 | CACGGGCCTCAGAGAATTCA |
| AnoliC-ABA4 | CATCACAATCGATCCATGTCCTCTCAACCATTCACGAAC |
| AntrpC- ABA4 | CGGTCGGCATCTACTCTAACATCTCCATCCGCCATCAATGC |
| PAnoliC-579 | GGCTTCGTACGGGAGGTTCGGCGTAG |
| TAntrpC-132 | TCTGCTTCGCCGGAGCCTGAAGGGCG |
| Silent-ABA4-Se-5 | CATCACAATCGATCCATGTCCTCTCAACCATTCACGAAC |
| Silent-ABA4-Se-3 | CCTGTATCCTCCAGCACATCTCCATCCGCCATCAATGCT |
| Silent-ABA4-AS-5 | CAGCCAGGGAACGGCACATCTCCATCCGCCATCAATGCT |
| Silent-ABA4-AS-3 | CGGTCGGCATCTACTATGTCCTCTCAACCATTCACGAAC |
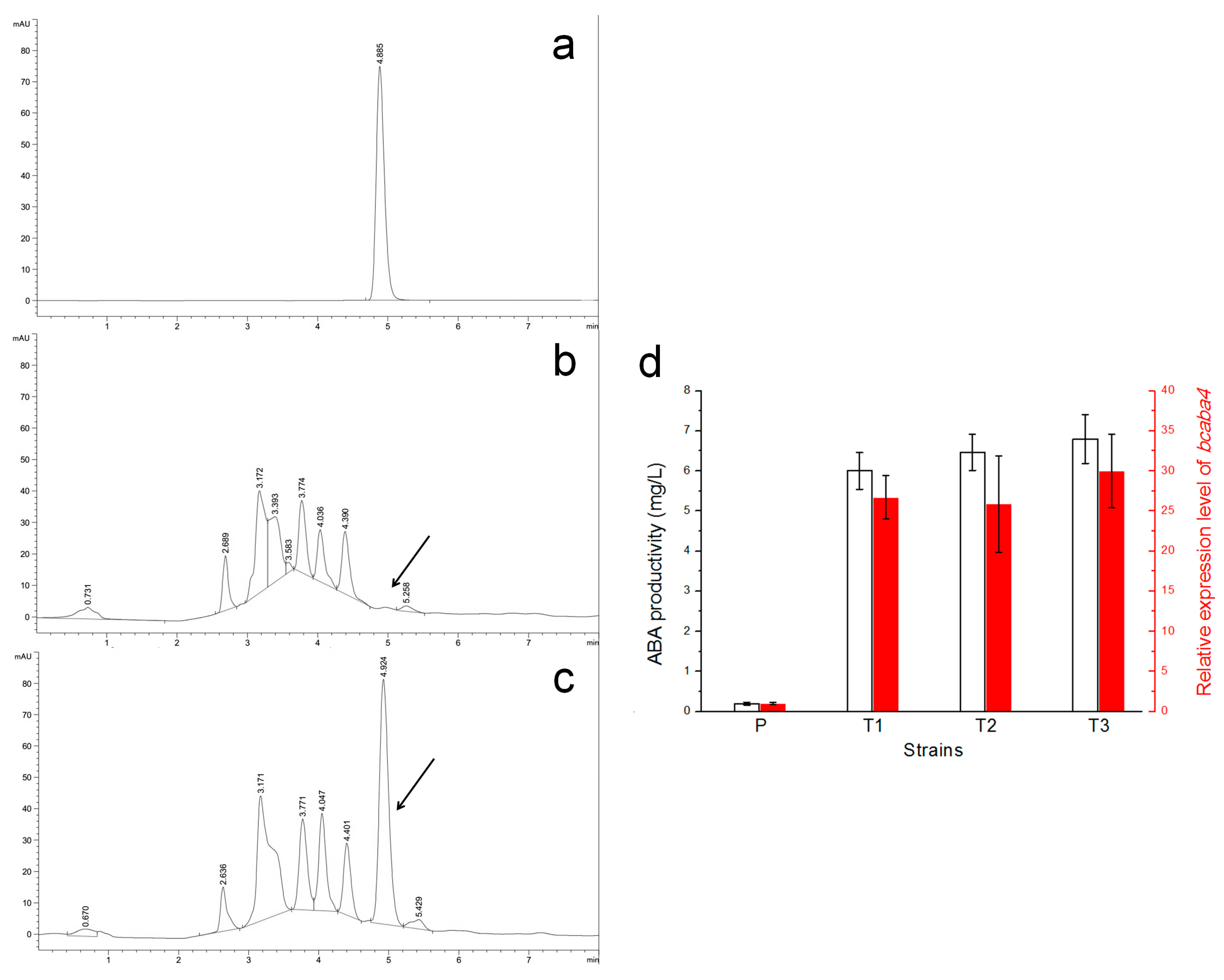
2.3. Vectors pCBh1 and pCBg1 Expressed Bcaba4 in B. cinerea Bc-6 with High Efficiency
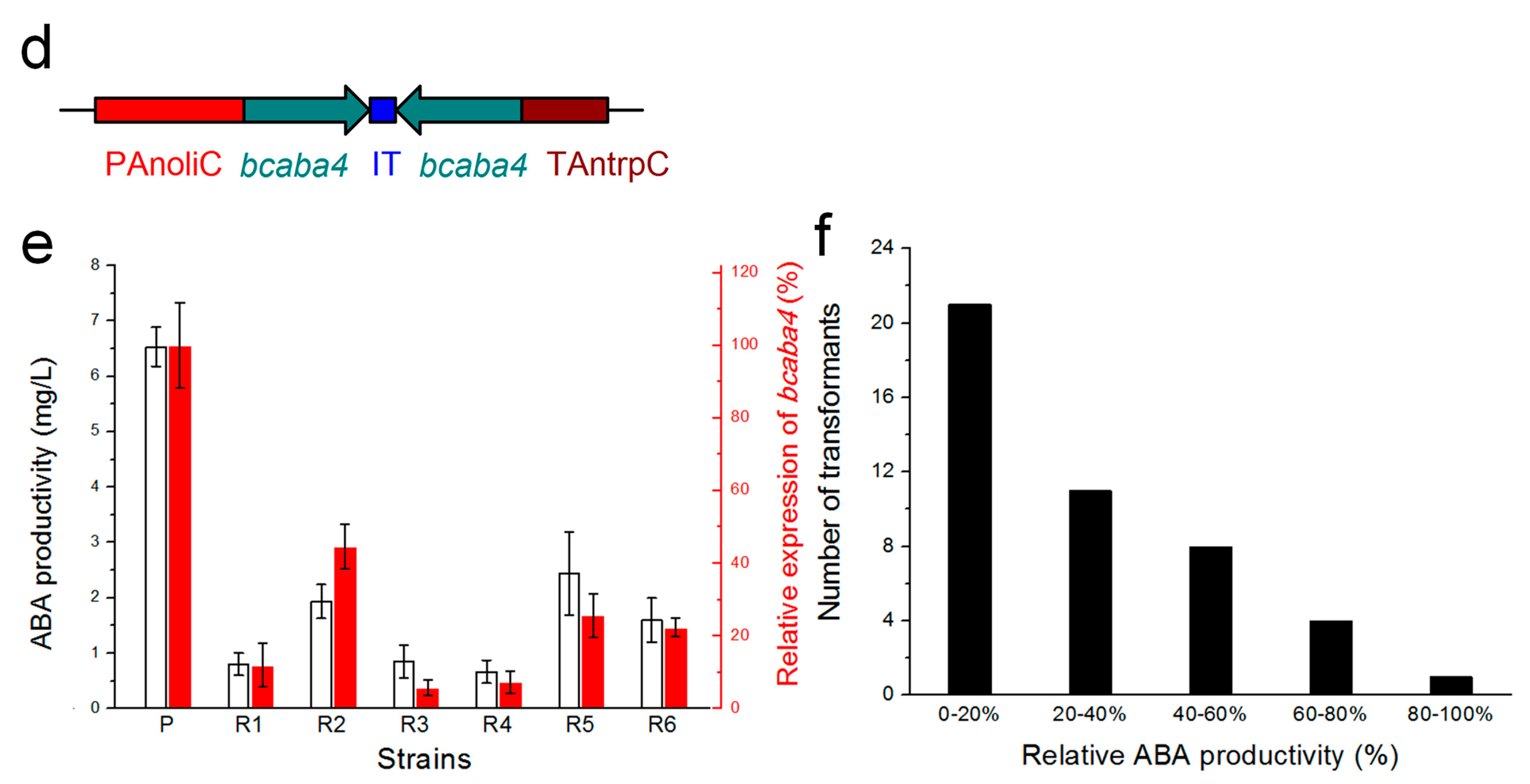
2.4. Efficient Silencing of the eGFP Gene by a pCBSilent1-Based Vector
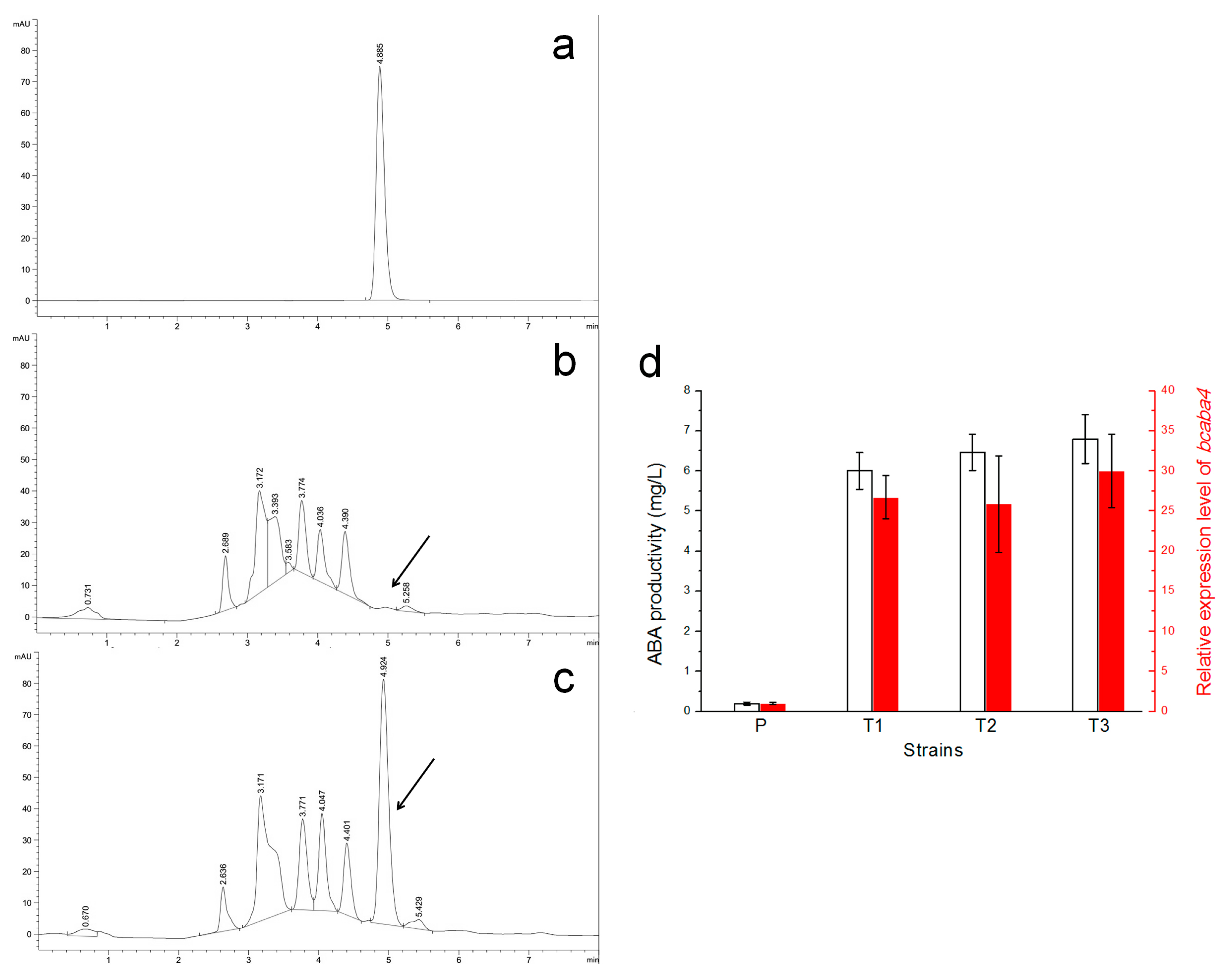

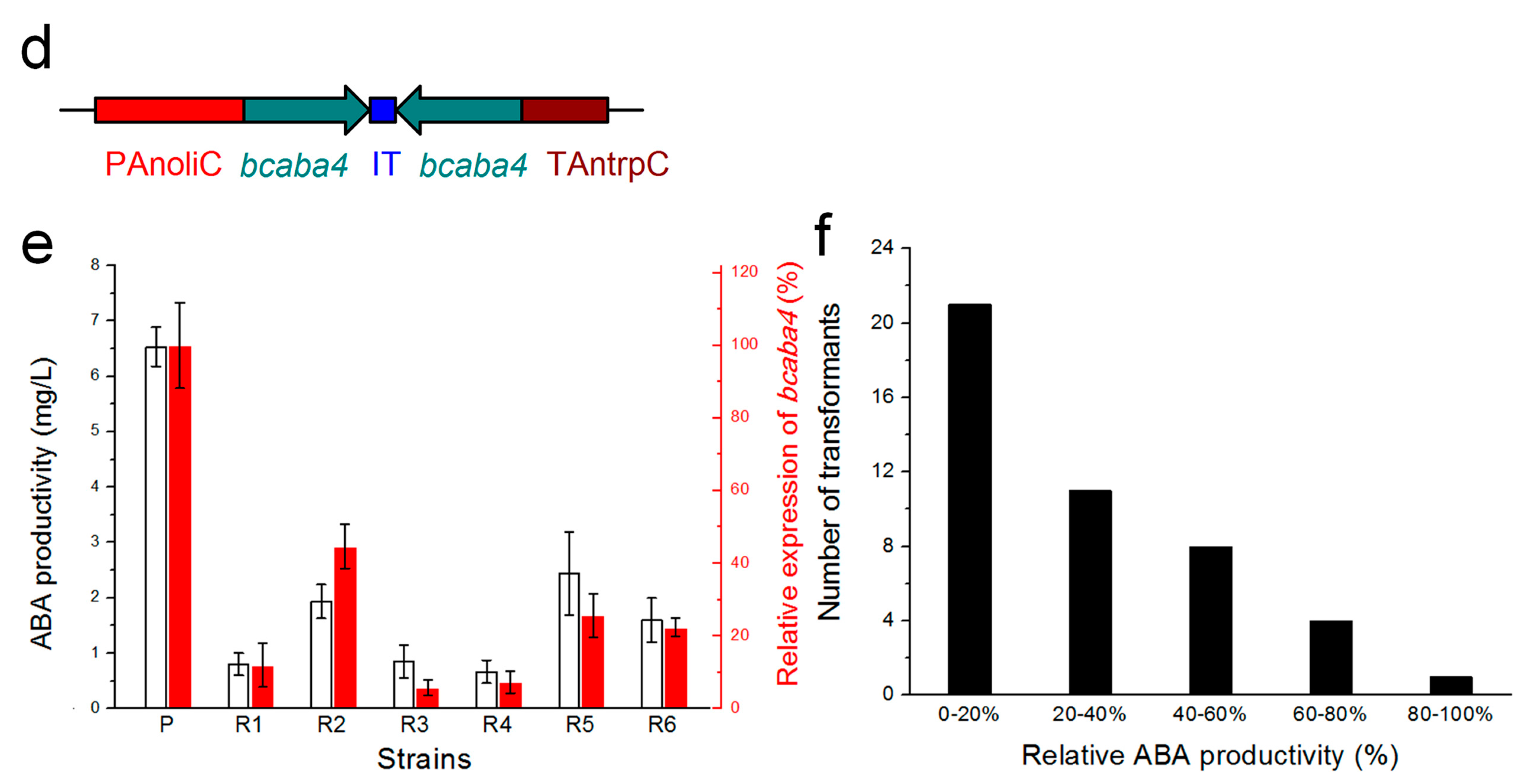
2.5. Efficient Silencing of the Bcaba4 Gene in an ABA-Producing B. cinerea Mutant
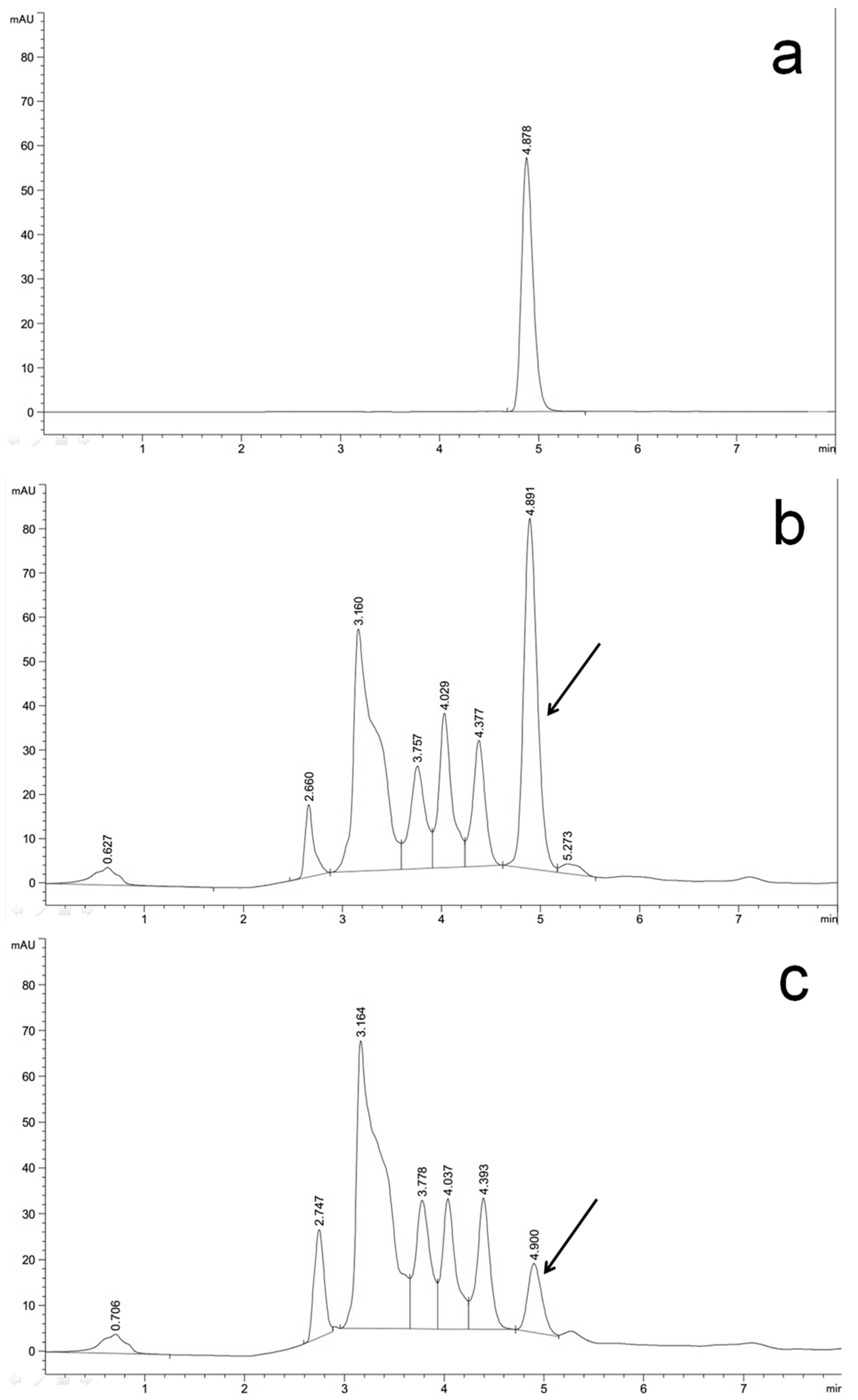
3. Discussion

4. Experimental Section
4.1. Strains, Media, and ATMT
4.2. Construction of the Gene Overexpression Binary Vector pCBh1
4.3. Construction of the Gene Overexpression Binary Vector pCBg1
4.4. Construction of the RNA Silencing Binary Vector pCBSilent1
4.5. Overexpression and Gene Silencing of eGFP in B. cinerea Strains
4.6. Imaging of GFP Fluorescence
4.7. Overexpression and Gene Silencing of Bcaba4 Gene in B. cinerea Strains
4.8. Extracellular ABA Determination
4.9. qRT-PCR Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Elad, Y.; Williamson, B.; Tudzynski, P.; Delen, N. Botrytis: Biology, Pathology and Control; Kluwer: Dordrecht, The Netherlands, 2004; pp. 1–8. [Google Scholar]
- Williamson, B.; Tudzynski, B.; Tudzynski, P.; van Kan, J.A. Botrytis cinerea: The cause of grey mould disease. Mol. Plant Pathol. 2007, 8, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Tudzynski, P.; Kokkelink, L. Botrytis cinerea: Molecular aspects of a necrotrophic life style. In The Mycota V, Plant Relationships, 2nd ed.; Deising, H., Ed.; Springer: Berlin, Germany, 2009; pp. 29–50. [Google Scholar]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Ilvesoksa, J.; Laakso, S.; Rosenqvist, H. Interaction of abscisic acid and indole-3-acetic acid-producing fungi with Salix leaves. J. Plant Growth Regul. 1993, 12, 149–156. [Google Scholar] [CrossRef]
- Qadir, A.; Hewett, E.W.; Long, P.G. Ethylene production by Botrytis cinerea. Postharvest Biol. Technol. 1997, 11, 85–91. [Google Scholar] [CrossRef]
- Talieva, M.N.; Filimonova, M.V. On parasitic specialization of the Botrytis species in the light of new experimental-data. Zh. Obshch. Biol. 1992, 53, 225–231. [Google Scholar]
- Marumo, S.; Katayama, M.; Komori, E.; Ozaki, Y.; Natsume, M.; Kondo, S. Microbial production of abscisic acid by Botrytis cinerea. Agric. Biol. Chem. 1982, 46, 1967–1968. [Google Scholar] [CrossRef]
- Gong, T.; Shu, D.; Zhao, M.; Zhong, J.; Deng, H.Y.; Tan, H. Isolation of genes related to abscisic acid production in Botrytis cinerea Bc-3-H8 by cDNA-AFLP. J. Basic Microbiol. 2014, 54, 204–214. [Google Scholar] [CrossRef]
- Matsumoto, S.; Ito, M.; Marumo, S. Production of Natural Type Abscisic Acid. JPS63296697A, 2 12 1988. [Google Scholar]
- Tan, H.; Gong, G.; Li, Z.D.; Peng, S.L.; Lei, B.L.; Liu, D.J.; Ding, L.S. High-yield ABA producing strain obtained by UV irradiation of protoplasts. Chin. J. Appl. Environ. Biol. 1998, 4, 281–285. [Google Scholar]
- Hamada, W.; Reignault, P.; Bompeix, G.; Boccara, M. Transformation of B. cinerea with the hygromycin B resistance gene. HPH. Curr. Genet. 1994, 26, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Van Kan, J.A.L.; van’t Klooster, J.W.; Wagemakers, C.A.M.; van der Vlugt-Bergmans, C.J.B; Dees, D.C.T. Cutinase A of Botrytis cinerea is expressed, but not essential, during penetration of gerbera and tomato. Mol. Plant Microbe Interact. 1997, 10, 30–38. [Google Scholar] [CrossRef]
- Noda, J.; Brito, N.; Espino, J.J.; González, C. Methodological improvements in the expression of foreign genes and in gene replacement in the phytopathogenic fungus Botrytis cinerea. Mol. Plant Pathol. 2007, 8, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Viaud, M.; Brunet-Simon, A.; Brygoo, Y.; Pradier, J.M.; Levis, C. Cyclophilin A and calcineurin functions investigated by gene inactivation, cyclosporin A inhibition and cDNA arrays approaches in the phytopathogenic fungus Botrytis cinerea. Mol. Microbiol. 2003, 50, 1451–1465. [Google Scholar] [CrossRef] [PubMed]
- Choquer, M.; Robin, G.; le Pecheur, P.; Giraud, C.; Levis, C.; Viaud, M. Ku70 or Ku80 deficiencies in the fungus Botrytis cinerea facilitate targeting of genes that are hard to knock out in a wild-type context. FEMS Microbiol. Lett. 2008, 289, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; van Kan, J.A.L.; Bailey, A.M.; Foster, G.D. RNA-mediated gene silencing of superoxide dismutase (bcsod1) in Botrytis cinerea. Phytopathology 2008, 98, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Giesbert, S.; Schumacher, J.; Kupas, V.; Espino, J.; Segmüller, N.; Haeuser-Hahn, I.; Schreier, P.H.; Tudzynski, P. Identification of pathogenesis-associated genes by T-DNA mediated insertional mutagenesis in Botrytis cinerea: A type 2A phosphoprotein phosphatase and an SPT3 transcription factor have significant impact on virulence. Mol. Plant Microbe Interact. 2012, 25, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; van Kan, J.A.L.; Bailey, A.M.; Foster, G.D. Inadvertent gene silencing of argininosuccinate synthase (bcass1) in Botrytis cinerea by the pLOB1 vector system. Mol. Plant Pathol. 2010, 11, 613–624. [Google Scholar] [PubMed]
- Schumacher, J.; Viaud, M.; Simon, A.; Tudzynski, B. The Gα subunit BCG1, the phospholipase C (BcPLC1) and the calcineurin phosphatase co-ordinately regulate gene expression in the grey mould fungus Botrytis cinerea. Mol. Microbiol. 2008, 67, 1027–1050. [Google Scholar] [CrossRef] [PubMed]
- Espino, J.; González, M.; González, C.; Brito, N. Efficiency of different strategies for gene silencing in Botrytis cinerea. Appl. Microbiol. Biotechnol. 2014, 98, 9413–9424. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.G.; Bruel, C.A. Sulphur and nitrogen regulation of the protease-encoding ACP1 gene in the fungus Botrytis cinerea: Correlation with a phospholipase D activity. Microbiology 2008, 154, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Antal, Z.; Rascle, C.; Cimerman, A.; Viaud, M.; Billon-Grand, G.; Choquer, M.; Bruel, C. The homeobox BcHOX8 gene in Botrytis cinerea regulates vegetative growth and morphology. PLoS ONE 2012, 7, e48134. [Google Scholar] [CrossRef] [PubMed]
- Reis, H.; Pfiffi, S.; Hahn, M. Molecular and functional characterization of a secreted lipase from Botrytis cinerea. Mol. Plant Pathol. 2005, 6, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Siewers, V.; Kokkelink, L.; Smedsgaard, J.; Tudzynski, P. Identification of an abscisic acid gene cluster in the grey mold Botrytis cinerea. Appl. Environ. Microbiol. 2006, 72, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Siewers, V.; Smedsgaard, J.; Tudzynski, P. The P450 monooxygenase BcABA1 is essential for abscisic acid biosynthesis in Botrytis cinerea. Appl. Environ. Microbiol. 2004, 70, 3868–3876. [Google Scholar] [CrossRef] [PubMed]
- De Groot, M.J.A.; Bundock, P.; Hooykaas, P.J.J.; Beijersbergen, A.G.M. Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat. Biotechnol. 1998, 16, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Dobinson, K.F.; Grant, S.J.; Kang, S. Cloning and targeted disruption, via Agrobacterium tumefaciens-mediated transformation, of a trypsin protease gene from the vascular wilt fungus Verticillium dahliae. Curr. Genet. 2004, 45, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Mullins, E.D.; Chen, X.; Romaine, P.; Raina, R.; Geiser, D.M.; Kang, S. Agrobacterium-mediated transformation of Fusarium oxysporum: An efficient tool for insertional mutagenesis and gene transfer. Phytopathology 2001, 91, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Rho, H.S.; Kang, S.; Lee, Y.H. Agrobacterium tumefaciens mediated transformation of the plant pathogenic fungus, Magnaporthe grisea. Mol. Cells 2001, 12, 407–411. [Google Scholar] [PubMed]
- Zhang, Y.J.; Zhao, J.J.; Xie, M.; Peng, D.L. Agrobacterium tumefaciens-mediated transformation in the entomopathogenic fungus Lecanicillium lecanii and development of benzimidazole fungicide resistant strains. J. Microbiol. Methods 2014, 105, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.; Jobic, C.; Fèvre, M.; Bruel, C. Agrobacterium-mediated transformation of Botrytis cinerea, simple purification of monokaryotic transformants and rapid conidia-based identification of the transfer-DNA host genomic DNA flanking sequences. Curr. Genet. 2003, 44, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Ding, Z.; Yang, X.; Wang, K.; Zhu, T. Gene disruption and characterization of a class V chitin synthase in Botrytis cinerea. Can. J. Microbiol. 2009, 55, 1267–1274. [Google Scholar] [CrossRef]
- Cui, Z.; Wang, Y.; Lei, N.; Wang, K.; Zhu, T. Botrytis cinerea chitin synthase BcChsVI is required for normal growth and pathogenicity. Curr. Genet. 2013, 59, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.Y.; Ma, X.R.; Li, Z.D.; Tan, H. Cloning and characterization of farnesyl pyphosphate synthase gene from the ABA-producing fungi Botrytis cinerea. DNA Seq. 2008, 19, 313–318. [Google Scholar] [PubMed]
- Zhang, L.; Thiewes, H.; van Kan, J.A.L. The D-galacturonic acid catabolic pathway in Botrytis cinerea. Fungal Genet. Biol. 2011, 48, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Nakayashiki, H.; Hanada, S.; Nguyen, B.Q.; Kadotani, N.; Tosa, Y.; Mayama, S. RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet. Biol. 2005, 42, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Benoit, R.M.; Wilhelm, R.N.; Scherer-Becker, D.; Ostermeier, C. An improved method for fast, robust, and seamless integration of DNA fragments into multiple plasmids. Protein Expr. Purif. 2006, 45, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Sleight, S.C.; Bartley, B.A.; Lieviant, J.A.; Sauro, H.M. In-Fusion BioBrick assembly and re-engineering. Nucleic Acids Res. 2010, 38, 2624–2636. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Cai, G.; Hall, E.O.; Freeman, G.J. In-Fusion assembly: Seamless engineering of multidomain fusion proteins, modular vectors, and mutations. Biotechniques 2007, 43, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.; Bruel, C.; Rascle, C.; Girard, V.; Billon-Grand, G.; Poussereau, N. pH controls both transcription and post-translational processing of the protease BcACP1 in the phytopathogenic fungus Botrytis cinerea. Microbiology 2009, 155, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- De Lucas, J.R.; Gregory, S.; Turner, G. Analysis of the regulation of the Aspergillus nidulans acuD gene, encoding isocitrate lyase, by construction of a hybrid promoter. Mol. Gen. Genet. 1994, 243, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Heneghan, M.N.; van Kan, J.A.L.; Bailey, A.M.; Foster, G.D. The pOT and pLOB vector systems: Improving ease of transgene expression in Botrytis cinerea. J. Gen. Appl. Microbiol. 2008, 54, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Kück, U.; Hoff, B. New tools for the genetic manipulation of filamentous fungi. Appl. Microbiol. Biotechnol. 2010, 86, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Hellens, R.; Mullineaux, P.; Klee, H. A guide to Agrobacterium binary Ti vectors. Trends Plant Sci. 2000, 5, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning. A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Pall, M.L.; Brunelli, J.P. A series of six compact fungal transformation vectors containing polylinkers with multiple unique restriction sites. Fungal Genet. Newlett. 1993, 40, 59–62. [Google Scholar]
- Dulermo, T.; Rascle, C.; Billon-Grand, G.; Gout, E.; Bligny, R.; Cotton, P. Novel insights into mannitol metabolism in the fungal plant pathogen Botrytis cinerea. Biochem. J. 2010, 427, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative Ct method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Z.-T.; Zhang, Z.; Luo, D.; Zhou, J.-Y.; Zhong, J.; Yang, J.; Xiao, L.; Shu, D.; Tan, H. Gene Overexpression and RNA Silencing Tools for the Genetic Manipulation of the S-(+)-Abscisic Acid Producing Ascomycete Botrytis cinerea. Int. J. Mol. Sci. 2015, 16, 10301-10323. https://doi.org/10.3390/ijms160510301
Ding Z-T, Zhang Z, Luo D, Zhou J-Y, Zhong J, Yang J, Xiao L, Shu D, Tan H. Gene Overexpression and RNA Silencing Tools for the Genetic Manipulation of the S-(+)-Abscisic Acid Producing Ascomycete Botrytis cinerea. International Journal of Molecular Sciences. 2015; 16(5):10301-10323. https://doi.org/10.3390/ijms160510301
Chicago/Turabian StyleDing, Zhong-Tao, Zhi Zhang, Di Luo, Jin-Yan Zhou, Juan Zhong, Jie Yang, Liang Xiao, Dan Shu, and Hong Tan. 2015. "Gene Overexpression and RNA Silencing Tools for the Genetic Manipulation of the S-(+)-Abscisic Acid Producing Ascomycete Botrytis cinerea" International Journal of Molecular Sciences 16, no. 5: 10301-10323. https://doi.org/10.3390/ijms160510301