
Organocatalytic Upgrading of Furfural and 5-Hydroxymethyl Furfural to C10 and C12 Furoins with Quantitative Yield and Atom-Efficiency

Abstract
: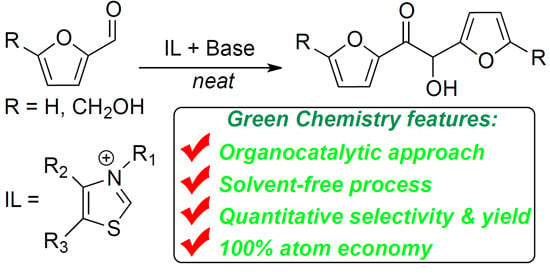
1. Introduction
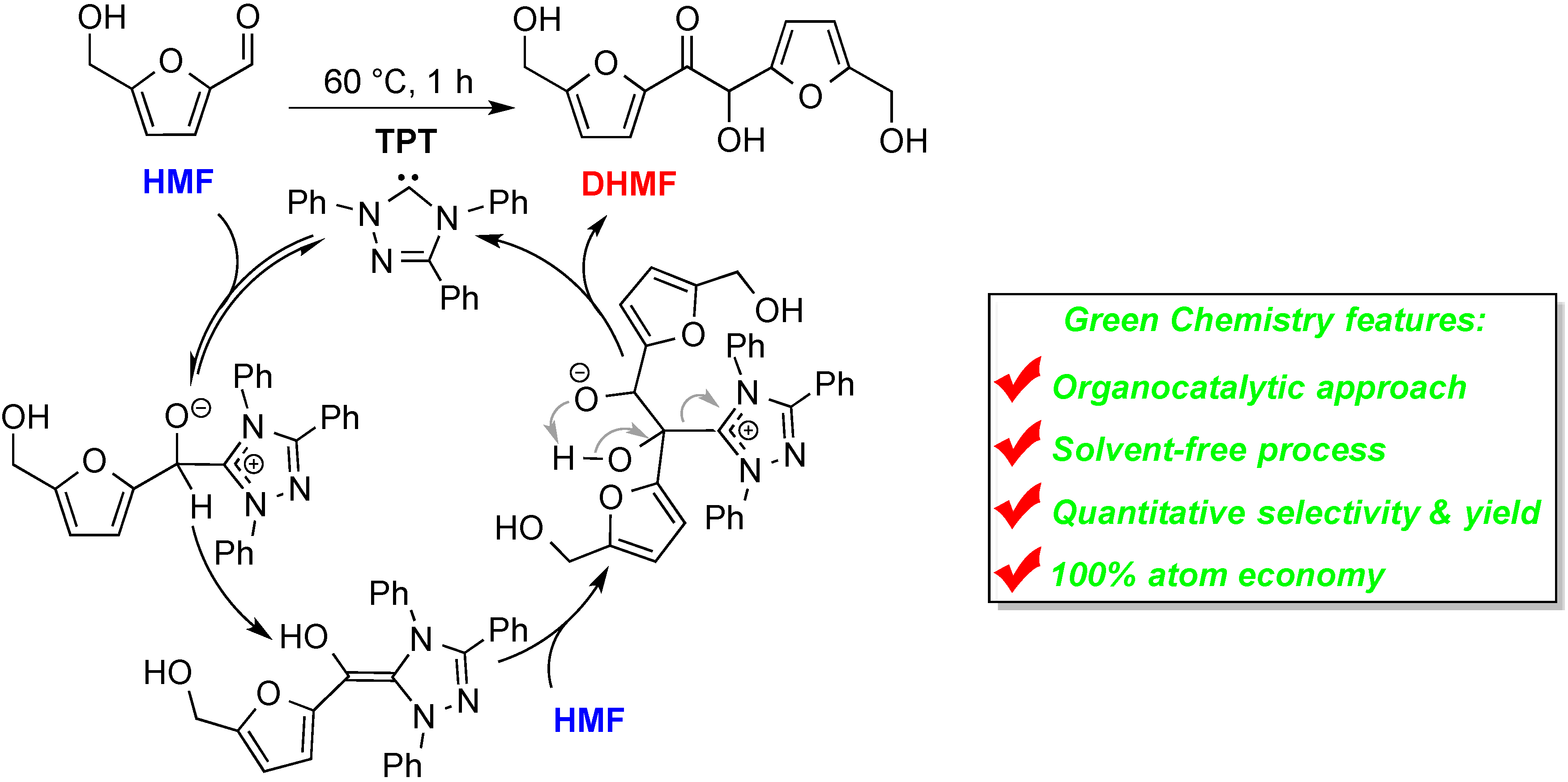
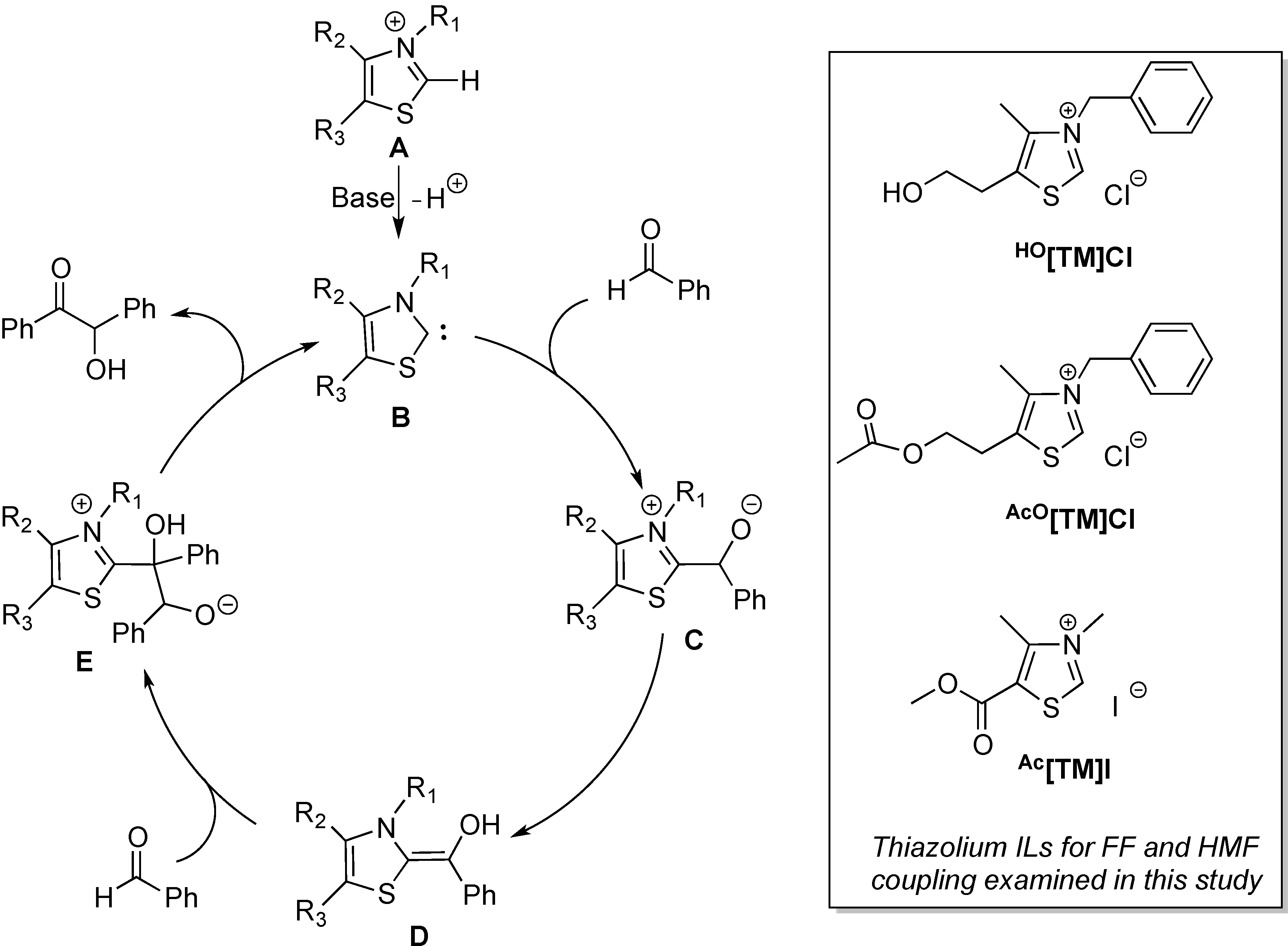
2. Results and Discussion
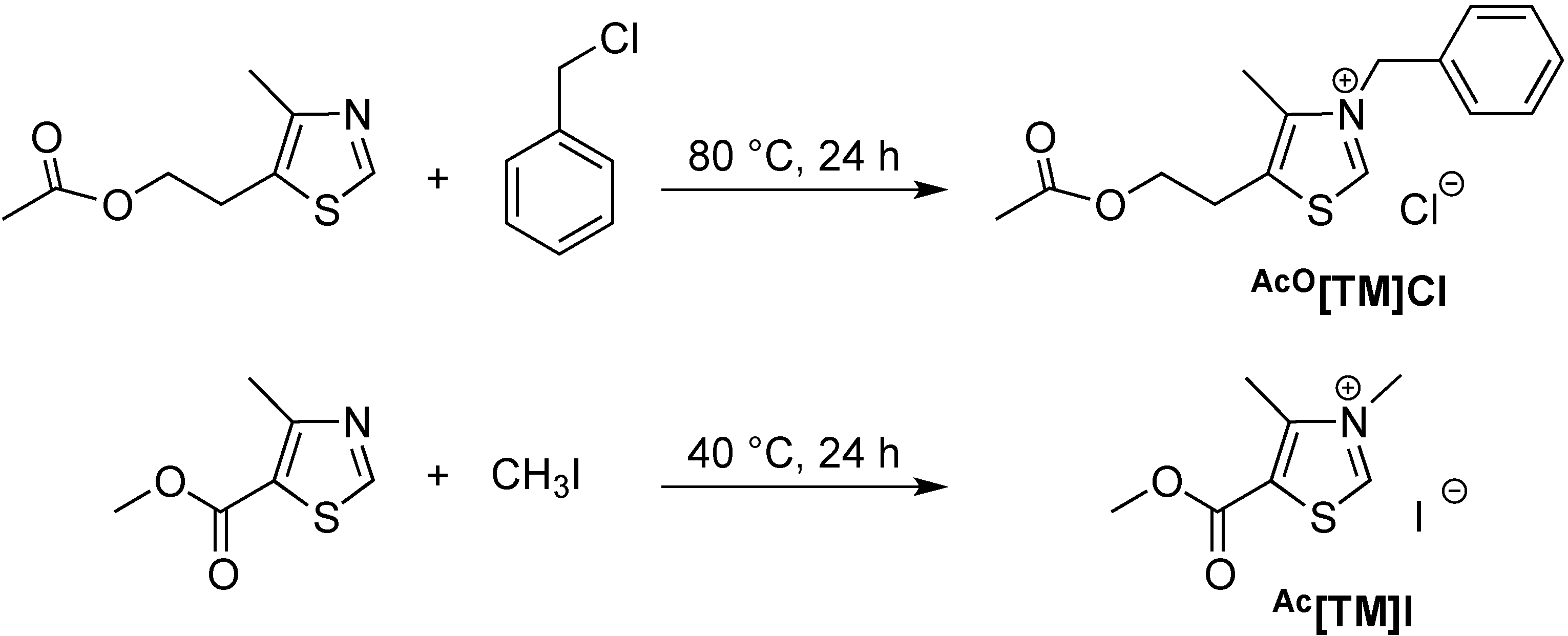
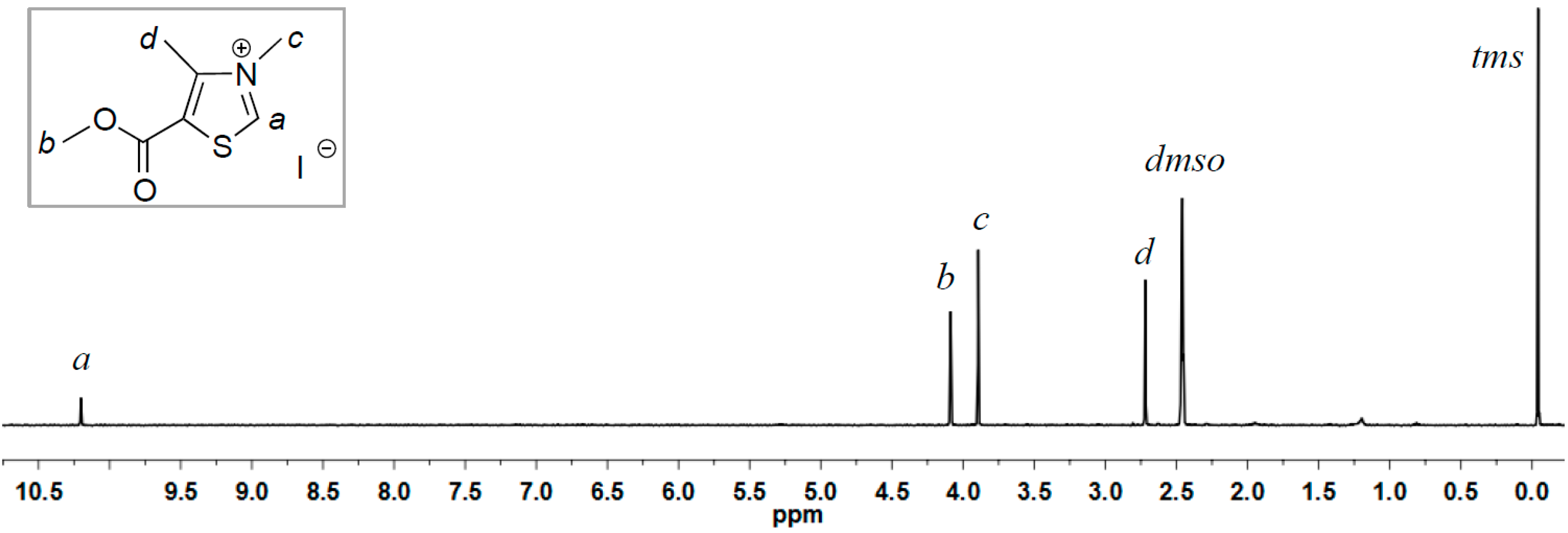
2.1. Synthesis of Two New Thiazolium ILs AcO[TM]Cl and Ac[TM]I

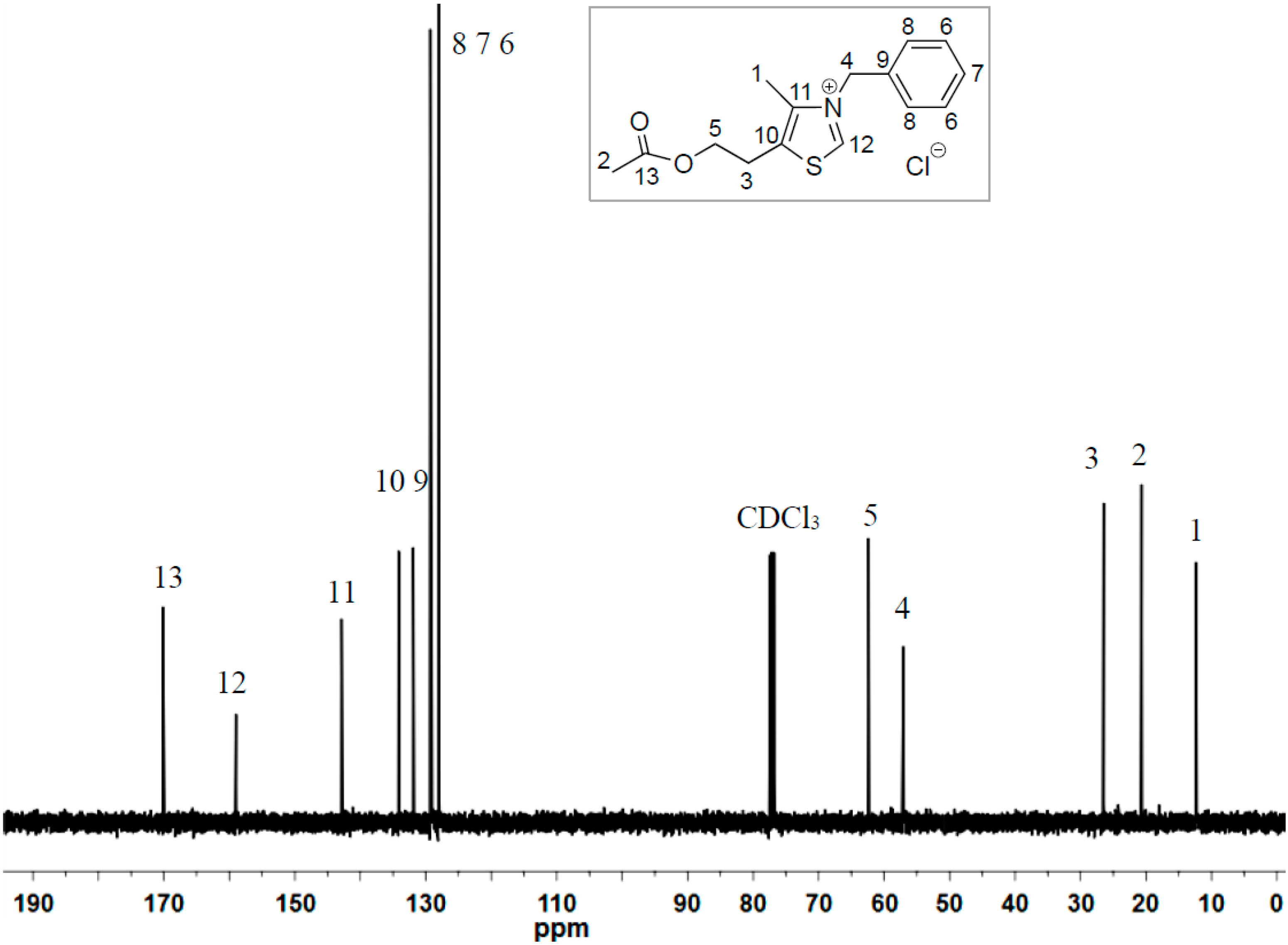
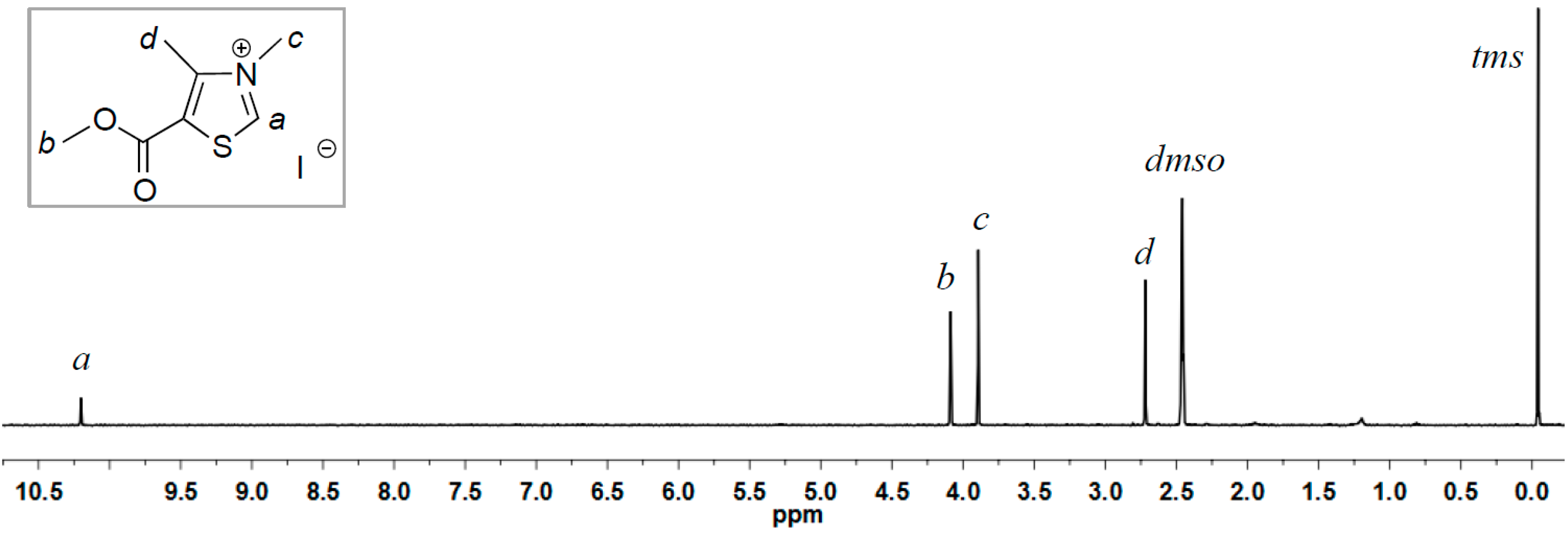
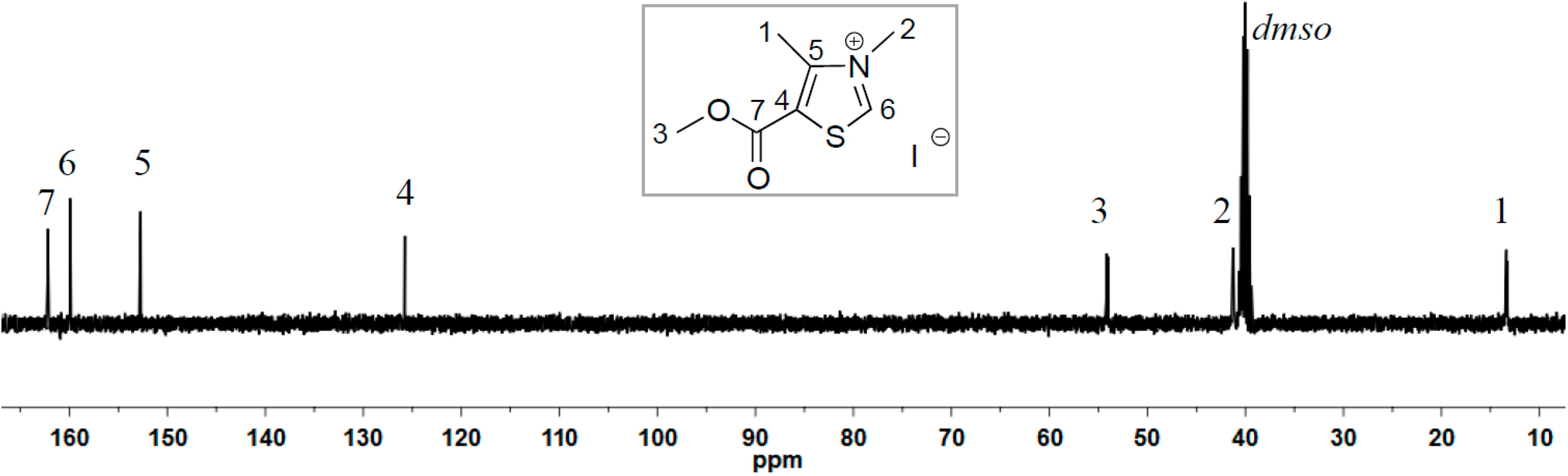
2.2. Coupling Reaction of FF to Furoin by HO[TM]Cl, AcO[TM]Cl, and Ac[TM]I

{kind=link}
{kind=link}
{kind=link}
{kind=link}
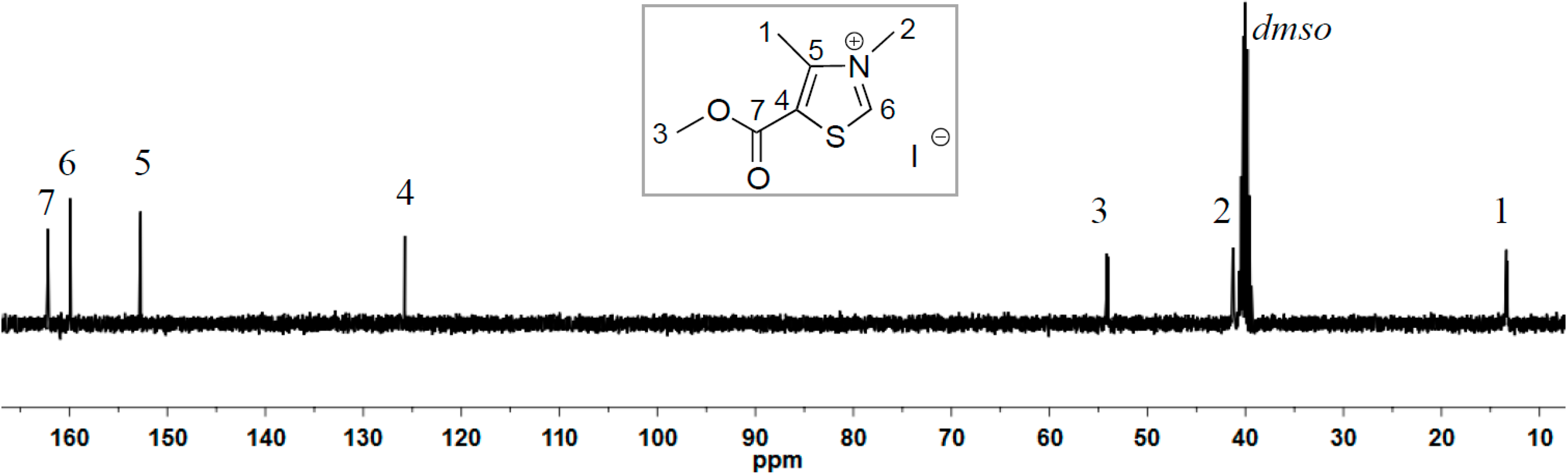{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Pre-Catalyst (mol %) | Base (mol %) | Temperature (°C) | Time (h) | Furoin Yield (%) |
|---|---|---|---|---|---|
| 1 | AcO[TM]Cl, 1 | Et3N, 2 | 80 | 3 | 99 |
| 2 | AcO[TM]Cl, 0.5 | Et3N, 1 | 80 | 3 | 99 |
| 3 | AcO[TM]Cl, 0.1 | Et3N, 0.2 | 80 | 3 | >99 |
| 4 | AcO[TM]Cl, 0.1 | Et3N, 0.1 | 60 | 1 | 81 |
| 5 | AcO[TM]Cl, 0.1 | Et3N, 0.2 | 60 | 1 | 86 |
| 6 | AcO[TM]Cl, 0.1 | Et3N, 0.4 | 60 | 1 | >99 |
| 7 | AcO[TM]Cl, 0.1 | KOtBu, 0.2 | 60 | 1 | 60 |
| 8 | AcO[TM]Cl, 0.1 | DBU, 0.2 | 60 | 1 | 56 |
| 9 | HO[TM]Cl, 0.5 | Et3N, 1 | 80 | 3 | 99 |
| 10 | HO[TM]Cl, 0.1 | Et3N, 0.4 | 60 | 1 | 84 |
| 11 | Ac[TM]I, 0.1 | Et3N, 0.2 | 80 | 3 | 0 |
| 12 | Ac[TM]I, 0.5 | Et3N, 1 | 80 | 3 | 16 |
| 13 | Ac[TM]I, 5 | Et3N, 10 | 80 | 3 | 49 |
| 14 | Ac[TM]I, 10 | Et3N, 20 | 80 | 3 | 84 |
2.3. Coupling Reaction of HMF to DHMF by HO[TM]Cl, AcO[TM]Cl, and Ac[TM]I
| Entry | Pre-Catalyst (mol %) | Base (mol %) | Temperature (°C) | Time (h) | Furoin Yield (%) |
|---|---|---|---|---|---|
| 1 | AcO[TM]Cl, 1 | Et3N, 2 | 80 | 3 | 34 |
| 2 | AcO[TM]Cl, 5 | Et3N,10 | 80 | 3 | 50 |
| 3 | AcO[TM]Cl, 10 | Et3N, 20 | 80 | 3 | 94 |
| 4 | AcO[TM]Cl, 10 | Et3N, 20 | 100 | 3 | 96 |
| 5 | AcO[TM]Cl, 10 | Et3N, 20 | 120 | 3 | 97 |
| 6 | AcO[TM]Cl, 10 | KOtBu, 20 | 80 | 3 | 89 |
| 7 | AcO[TM]Cl, 10 | KOtBu, 20 | 100 | 3 | 93 |
| 8 | AcO[TM]Cl, 10 | DBU, 20 | 80 | 3 | 86 |
| 9 | HO[TM]Cl, 10 | Et3N, 20 | 80 | 3 | 93 |
| 10 | HO[TM]Cl, 10 | Et3N, 20 | 120 | 3 | 97 |
| 11 | Ac[TM]I, 0.5 | Et3N, 1 | 80 | 3 | 37 |
| 12 | Ac[TM]I, 10 | Et3N, 20 | 80 | 3 | 86 |
| 13 | Ac[TM]I, 10 | KOtBu, 20 | 80 | 3 | 59 |
| 14 | Ac[TM]I, 10 | DBU, 20 | 80 | 3 | 71 |
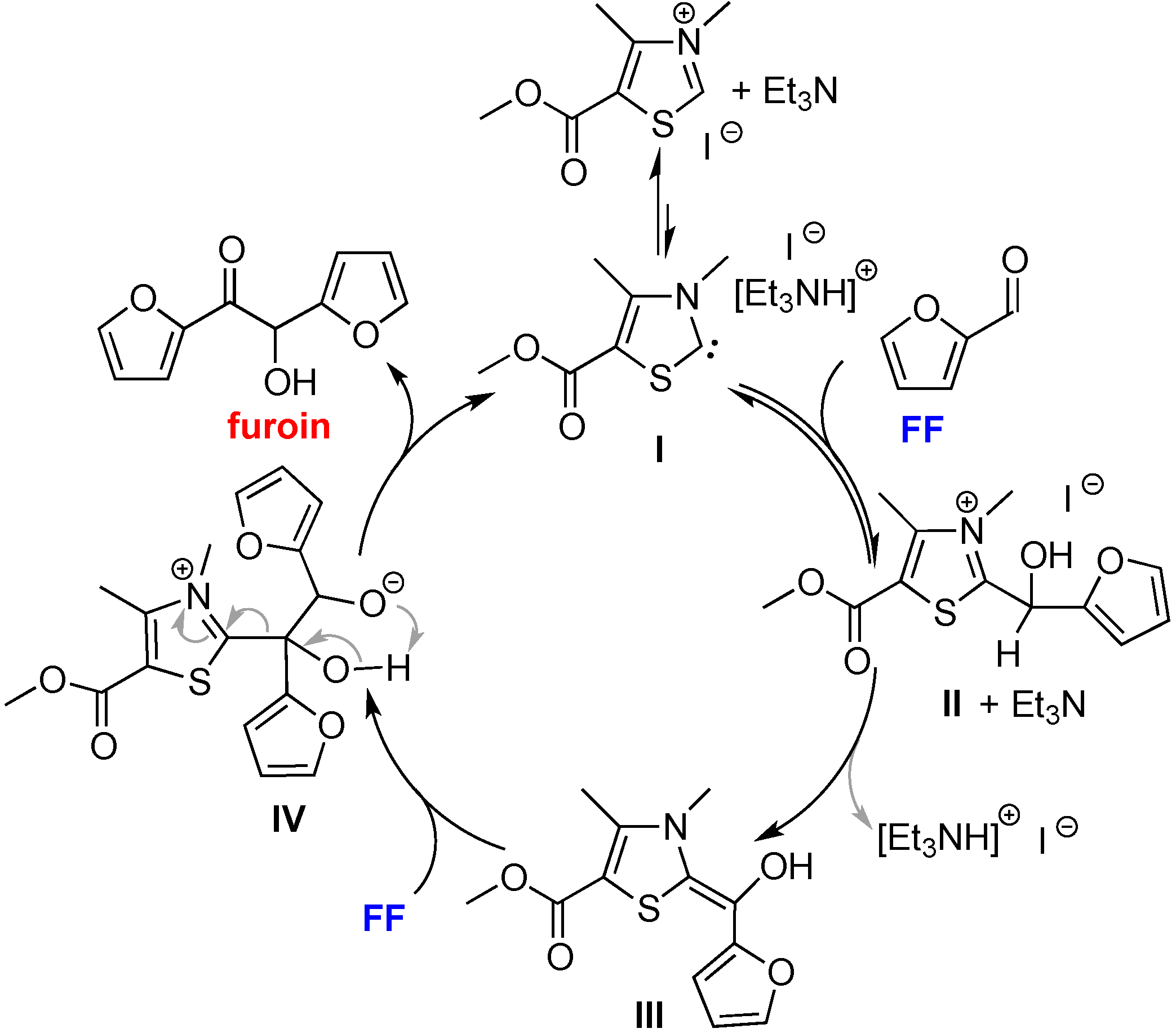
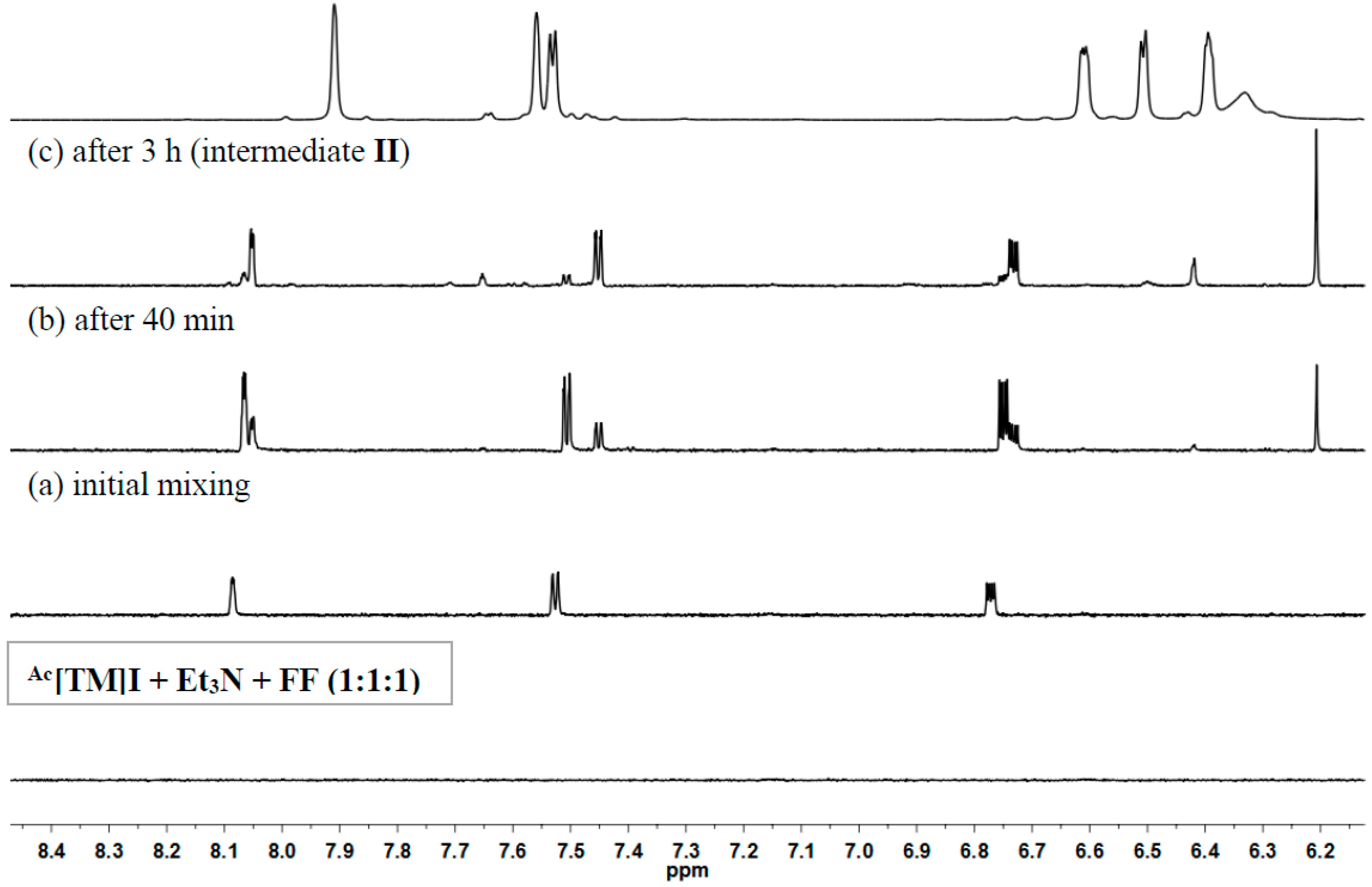
2.4. Proposed Mechanism for FF and HMF Coupling Reaction
3. Experimental Section
3.1. Materials, Reagents, and Methods
3.2. Synthesis of Ionic Liquid AcO[TM]Cl
3.3. Synthesis of Ionic Liquid Ac[TM]I
3.4. Typical Procedure for Coupling Reaction of FF into Furoin
3.5. Typical Procedure for Coupling Reaction of HMF into DHMF
3.6. Generation of Intermediate II from Reaction of Ac[TM]I + Et3N +FF
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, T.; Nolte, M.W.; Shanks, B.H. Catalytic dehydration of C6 carbohydrates for the production of hydroxymethylfurfural (HMF) as a versatile platform chemical. Green Chem. 2014, 16, 548–572. [Google Scholar] [CrossRef]
- Saha, B.; Abu-Omar, M.M. Advances in 5-hydroxymethylfurfural production from biomass in biphasic solvents. Green Chem. 2014, 16, 24–38. [Google Scholar] [CrossRef]
- Teong, S.P.; Yi, G.; Zhang, Y. Hydroxymethylfurfural production from bioresources: Past, present and future. Green Chem. 2014, 16, 2015–2026. [Google Scholar] [CrossRef]
- Van Putten, R.-J.; van der Waal, J.C.; de Jong, E.; Rasrendra, C.B.; Heeres, H.J.; de Vries, J.G. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar]
- Lange, J.-P.; van der Heide, E.; van Buijtenen, J.; Richard Price, R. Furfural—A promising platform for lignocellulosic biofuels. ChemSusChem 2012, 5, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Rosatella, A.A.; Simeonov, S.P.; Frade, R.F.M.; Afonso, C.A.M. 5-Hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754–793. [Google Scholar] [CrossRef]
- Zakrzewska, M.E.; Bogel-Łukasik, E.; Bogel-Łukasik, R. Ionic liquid-mediated formation of 5-hydroxymethylfurfural—A promising biomass-derived building block. Chem. Rev. 2011, 111, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, T.; Fu, W.; Woodley, J.M.; Riisager, A. Synthesis of 5-(hydroxymethyl)furfural in ionic liquids: Paving the way to renewable chemicals. ChemSusChem 2011, 4, 451–458. [Google Scholar] [CrossRef] [PubMed]
- James, O.O.; Maity, S.; Usman, L.A.; Ajanaku, K.O.; Ajani, O.O.; Siyanbola, T.O.; Sahu, S.; Chaubey, R. Towards the conversion of carbohydrate biomass feedstocks to biofuels via hydroxylmethylfurfural. Energy Environ. Sci. 2010, 3, 1833–1850. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US department of energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass; U.S. Department of Energy: Washington, DC, USA, 2004. [Google Scholar]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Catalytic conversion of biomass to biofuels. Green Chem. 2010, 12, 1493–1513. [Google Scholar] [CrossRef]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Huber, G.W. Synergies between bio- and oil refineries for the production of fuels from biomass. Angew. Chem. Int. Ed. 2007, 46, 7184–7201. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Wegenhart, B.L.; Liu, S.; Thom, M.; Stanley, D.; Abu-Omar, M.M. A Solvent-free method for making dioxolane and dioxane from the biorenewables glycerol and furfural catalyzed by oxorhenium(V) oxazoline. ACS Catal. 2012, 2, 2524–2530. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Sacia, E.R.; Bell, A.T. Etherification and reductive etherification of 5-(hydroxymethyl)furfural: 5-(Alkoxymethyl)furfurals and 2,5-bis(alkoxymethyl)furans as potential bio-diesel candidates. Green Chem. 2012, 14, 1626–1634. [Google Scholar] [CrossRef]
- Corma, A.; de la Torre, O.; Renz, M. Production of high quality diesel from cellulose and hemicellulose by the Sylvan process: Catalysts and process variables. Energy Environ. Sci. 2012, 5, 6328–6344. [Google Scholar] [CrossRef]
- Corma, A.; de la Torre, O.; Renz, M.; Villandier, N. Production of high-quality diesel from biomass waste products. Angew. Chem. Int. Ed. 2011, 50, 2375–2378. [Google Scholar] [CrossRef]
- Huber, G.W.; Chheda, J.N.; Barrett, C.J.; Dumesic, J.A. Production of liquid alkanes by aqueous-phase processing of biomass-derived carbohydrates. Science 2005, 308, 1446–1450. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.D.; Waldie, F.D.; Wu, R.; Schlaf, M.; Silks, L.A., III; Gordon, J.C. The hydrodeoxygenation of bioderived furans into alkanes. Nat. Chem. 2013, 5, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-B.; Yang, Z.; Dai, J.-J.; Guo, Q.-X.; Fu, Y. Production of high quality fuels from lignocellulose-derived chemicals: A convenient C–C bond formation of furfural, 5-methylfurfural and aromatic aldehyde. RSC Adv. 2012, 2, 11211–11214. [Google Scholar] [CrossRef]
- Liu, D.; Chen, E.Y.-X. Organocatalysis in biorefining for biomass conversion and upgrading. Green Chem. 2014, 16, 964–981. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; MacMillan, D.W.C. Organocatalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 20618–20619. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Houk, K.N.; List, B. Asymmetric organocatalysis. Acc. Chem. Res. 2004, 37, 487–631. [Google Scholar] [CrossRef]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-Heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- Wende, R.C.; Schreiner, P. Evolution of asymmetric organocatalysis: Multi- and retrocatalysis. Green Chem. 2012, 14, 1821–1849. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-heterocyclic carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, E.Y.-X. Diesel and alkane fuels from biomass by organocatalysis and metal-acid tandem catalysis. ChemSusChem 2013, 6, 2236–2239. [Google Scholar] [CrossRef] [PubMed]
- Caes, B.R.; Palte, M.J.; Raines, R.T. Organocatalytic conversion of cellulose into a platform chemical. Chem. Sci. 2013, 4, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, T.; Rodriguez-Rodriguez, S.; Fristrup, P.; Riisager, A. Metal-free dehydration of glucose to 5-(hydroxymethyl)furfural in ionic liquids with boric acid as a promoter. Chem. Eur. J. 2011, 17, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, Y.; Chen, E.Y.-X. Organocatalytic upgrading of the key biorefining building block by a catalytic ionic liquid and N-heterocyclic carbenes. Green Chem. 2012, 14, 2738–2746. [Google Scholar] [CrossRef]
- Enders, D.; Breuer, K.; Kallfass, U.; Balensiefer, T. Preparation and application of 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene, a new stable carbine. Synthesis 2003, 8, 1292–1295. [Google Scholar]
- Enders, D.; Breuer, K.; Raabe, G.; Runsink, J.; Teles, J.H.; Melder, J.-P.; Ebel, K.; Brode, S. Preparation, structure, and reactivity of 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene, a new stable carbine. Angew. Chem. Int. Ed. 1995, 34, 1021–1023. [Google Scholar] [CrossRef]
- Remsing, R.C.; Swatloski, R.P.; Rogers, R.D.; Moyna, G. Mechanism of cellulose dissolution in the ionic Liquid 1-n-butyl-3-methylimidazolium chloride: A 13C and 35/37Cl NMR relaxation study on model systems. Chem. Commun. 2006, 12, 1271–1273. [Google Scholar]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of cellulose with ionic liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, M.E.; Bogel-Łukasik, E.; Bogel-Łukasik, R. Solubility of carbohydrates in ionic liquids. Energy Fuels 2010, 24, 737–745. [Google Scholar] [CrossRef]
- El Seoud, O.A.; Koschella, A.; Fidale, L.C.; Dorn, S.; Heinze, T. Applications of ionic liquids in carbohydrate chemistry: A window of opportunities. Biomacromolecules 2007, 8, 2629–2647. [Google Scholar] [CrossRef] [PubMed]
- Wasserscheid, P.; Welton, T. Ionic Liquids in Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Pârvulescu, V.I.; Hardacre, C. Catalysis in ionic liquids. Chem. Rev. 2007, 107, 2615–2665. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.; Seddon, K. Ionic liquids. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley and Sons: New York, NY, USA, 2007; Volume 26, pp. 836–920. [Google Scholar]
- Sun, N.; Rodriguez, H.; Rahman, M.; Rogers, R.D. Where are ionic liquid strategies most suited in the pursuit of chemicals and energy from lignocellulosic biomass? Chem. Commun. 2011, 47, 1405–1421. [Google Scholar] [CrossRef]
- Pinkert, A.; Marsh, K.N.; Pang, S.; Staiger, M.P. Ionic liquids and their interaction with cellulose. Chem. Rev. 2009, 109, 6712–6728. [Google Scholar] [CrossRef]
- Moore, J.L.; Rovis, T. Carbene catalysts. Top. Curr. Chem. 2009, 291, 77–144. [Google Scholar]
- Kluger, R.; Tittmann, K. Thiamin diphosphate catalysis: Enzymic and nonenzymic covalent intermediates. Chem. Rev. 2008, 108, 1797–1833. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R. On the mechanism of thiamine action. IV. Evidence from studies on model systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Breslow, R. Rapid deuterium exchange in thiazolium salts. J. Am. Chem. Soc. 1957, 79, 1762–1763. [Google Scholar] [CrossRef]
- Kabro, A.; Escudero-Adán, E.C.; Grushin, V.V.; van Leeuwen, P.W. Biomass conversion to high value chemicals: From furfural to chiral hydrofuroins in two steps. Org. Lett. 2012, 14, 4014–4017. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem. Int. Ed. 2002, 41, 1743–1745. [Google Scholar] [CrossRef]
- Stetter, H.; Raemsch, R.Y.; Kuhlmann, H. The preparative use of thiazolium salt-catalyzed acyloin and benzoin formation, I. Preparation of simple acyloins and benzoins. Synthesis 1976, 733, 733–735. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Wölfle, M.; Teles, J.H.; Frey, W. Bisphenols from furfurals by organocatalysis and gold catalysis. Synlett 2007, 11, 1747–1752. [Google Scholar] [CrossRef]
- Lee, C.K.; Kim, M.S.; Gong, J.S.; Lee, I.-S.H. Benzoin condensation reaction of 5-membered heterocyclic compounds. J. Heterocycl. Chem. 1992, 29, 149–153. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zang, H.; Chen, E.Y.X. Organocatalytic Upgrading of Furfural and 5-Hydroxymethyl Furfural to C10 and C12 Furoins with Quantitative Yield and Atom-Efficiency. Int. J. Mol. Sci. 2015, 16, 7143-7158. https://doi.org/10.3390/ijms16047143
Zang H, Chen EYX. Organocatalytic Upgrading of Furfural and 5-Hydroxymethyl Furfural to C10 and C12 Furoins with Quantitative Yield and Atom-Efficiency. International Journal of Molecular Sciences. 2015; 16(4):7143-7158. https://doi.org/10.3390/ijms16047143
Chicago/Turabian StyleZang, Hongjun, and Eugene Y. X. Chen. 2015. "Organocatalytic Upgrading of Furfural and 5-Hydroxymethyl Furfural to C10 and C12 Furoins with Quantitative Yield and Atom-Efficiency" International Journal of Molecular Sciences 16, no. 4: 7143-7158. https://doi.org/10.3390/ijms16047143
APA StyleZang, H., & Chen, E. Y. X. (2015). Organocatalytic Upgrading of Furfural and 5-Hydroxymethyl Furfural to C10 and C12 Furoins with Quantitative Yield and Atom-Efficiency. International Journal of Molecular Sciences, 16(4), 7143-7158. https://doi.org/10.3390/ijms16047143