1. Introduction
In human endometrium the epithelial glandular cells are surrounded by a rich mesenchymal component that contains fibroblast-like stromal cells. Morphological changes in both epithelial and stromal cells during menstrual cycles have been well documented [
1,
2,
3]. In the proliferative phase, estrogen exerts mitogenic effects on both cell groups, leading to a quick expansion of glandular epithelial and stromal cells [
4]. The increased levels of progesterone in the secretory phase suppress cell growth but promote differentiation [
5]. At the end of the secretory phase, endometrial gland maturation is accompanied by predecidualization changes of stromal cells around blood vessels. During the decidualization process stromal cells become terminally differentiated upon implantation. In the absence of implantation, withdrawal of ovarian hormones leads to shedding of the endometrium, which marks the completion of a menstrual cycle [
6,
7]. Thus, steroid hormones serve as a driving force behind the synchronized actions of stromal and epithelial glandular cells. However, most previous studies concentrated on epithelial cells with relatively little attention paid to the hormonal response by stromal cells. The molecular events controlled by ovarian hormones in stromal cells, such as those occurring on the gene transcription level, remain largely unknown.
In addition to their function of supporting normal reproductivity, stromal cells and their responses to ovarian steroids are involved in the pathogenesis of endometrial hyperplasia and endometriosis. It has been observed that synthetic progestins such as levonorgestrel and oral medroxyprogesterone acetate significantly affect the expression of estrogen and progesterone receptors in glandular and stromal cells [
8]. This indicates the presence of autoregulatory feedback loops that may amplify hormonal responses in these cells. Interestingly, endometrial stromal cells isolated from women with endometriosis exhibited aberrant responses to ovarian hormones in the migratory and invasive behaviors, suggesting the involvement of these cells in the pathogenesis of endometriosis that is characterized by ectopic endometrial overgrowth [
9].
Endometrial stromal sarcoma is a rare but life-threatening uterine malignancy with a poor prognosis [
10]. Current radiation and chemotherapy regimens are relatively ineffective for treating stromal sarcomas [
11]. Malignant mixed mullerian tumor, also referred to as carcinosarcoma, represents a highly aggressive uterine malignancy containing both carcinomatous and sarcomatous components [
12]. While most carcinosarcomas are believed to arise from monoclonal endometrial stem cells, some cases may be true collision tumors derived from epithelial and stromal cells independently [
12]. It is noteworthy that elevated estrogen levels associated with obesity and nulliparity represent the most recognized risk factor for stromal sarcoma and mixed mullerian tumors [
13]. Exposure to exogenous estrogenic compounds, including the use of oral contraceptives, postmenopausal hormone replacement therapy [
14], and use of tamoxifen for breast cancer treatment [
15], have all been linked to an increased risk for stromal sarcomas. These observations underscore the significance of carcinogenic effects by estrogens in human endometrial stromal cells.
An increasing body of evidence supports the involvement of mesenchymal-epithelial interactions in cancer development [
16]. In prostate cancers, steroid-induced production of EGF and TGF-α by stromal cells is considered to be a major factor for continued cancer expansion [
17]. This paracrine action may contribute to a seemingly androgen-independent growth of epithelial cells, and account for the failure of predicting the response to endocrine therapies merely based on the androgen receptor levels of cancer cells [
17]. More interestingly, frequent loss of heterozygosity (LOH) has been found in normal-appearing stromal cells micro-dissected from mammary ductal carcinomas. This finding suggests that abnormal stromal-epithelial interactions may play a significant part in the progression of mammary neoplasia, and alterations in stromal cells may precede the epithelial transformation [
18].
In vitro studies have shown that uterine stromal cells are able to promote the growth [
19] and invasiveness [
20] of endometrial cancer cells when they were co-cultured in a three-dimensional model. In this system, matrix metalloproteinases (MMPs) produced by normal stromal cells were found to translocate to the surface of endometrial adenocarcinoma cells. Interestingly, MMP-2 secretion and translocation in the co-cultures were greatly enhanced by estrogen [
20]. It was also reported that stromal cells produce vascular endothelial growth factor and soluble vascular endothelial growth factor receptor, and the expression of these factors are regulated by estrogen and selective estrogen receptor modulators [
21]. Taken together, these findings suggested that endometrial stromal cells represent an important regulation target of ovarian hormones, and aberrant hormonal regulation may contribute to the development of endometrial malignancies.
In this study, we used primary stromal cell culture as a study model to investigate the morphological as well as transcriptome changes induced by estrogen, progesterone, and tamoxifen. Changes of gene expression in response to these hormones were detected, validated, and compared. Numerous genes governing diversified cell functions were found to be targets for hormonal regulation. A large number of novel genes with unknown functions were also identified. These findings provided useful information for studying the pathophysiological functions of stromal cells in the normal endometrium and the development of uterine hyperplasia and neoplasia.
3. Discussion
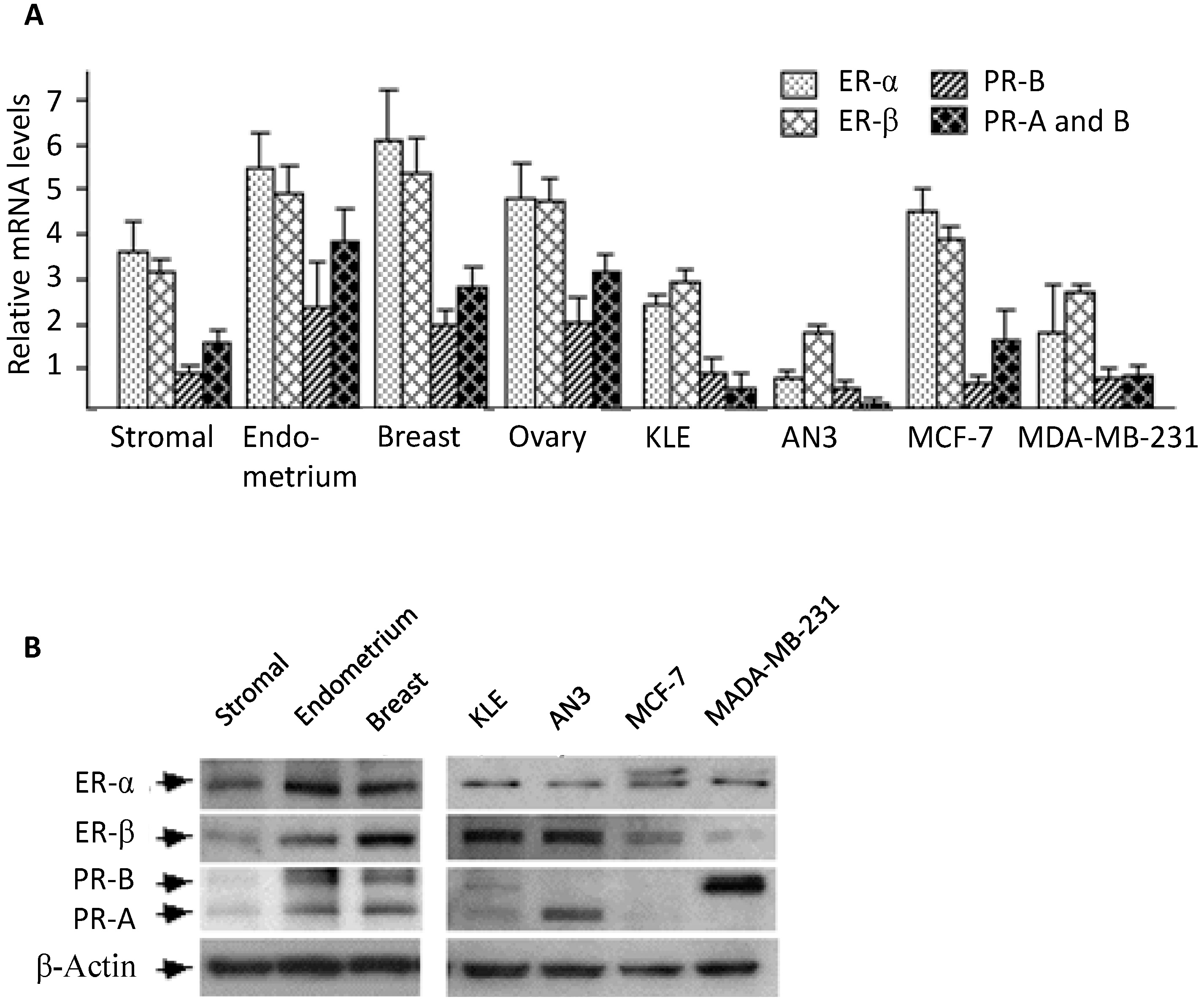
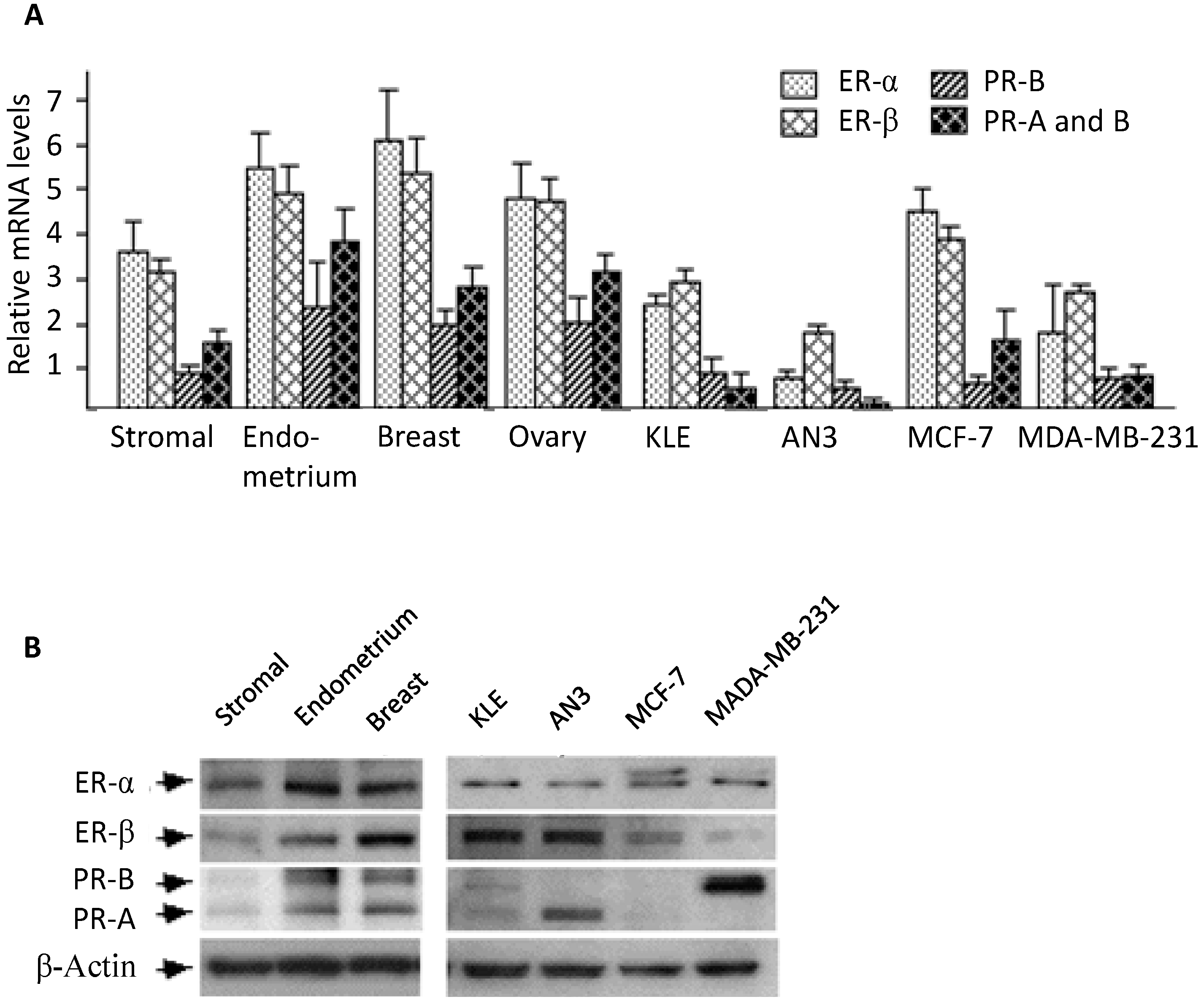
The expression of estrogen and progesterone receptors in human endometrial stromal cells has been reported by several groups [
22,
23,
24,
25]. These previous observations, however, were mostly based on immunohistochemistry studies [
24,
25], and the receptors’ expression levels relative to other tissues were not compared. In this study, we performed real-time PCR and Western blotting analysis to compare mRNA and protein levels between endometrial stromal cells and several reproductive tissues. In general, stromal cells appeared to express substantial amounts of estrogen and progesterone receptors, a result consistent with most previous reports [
22].
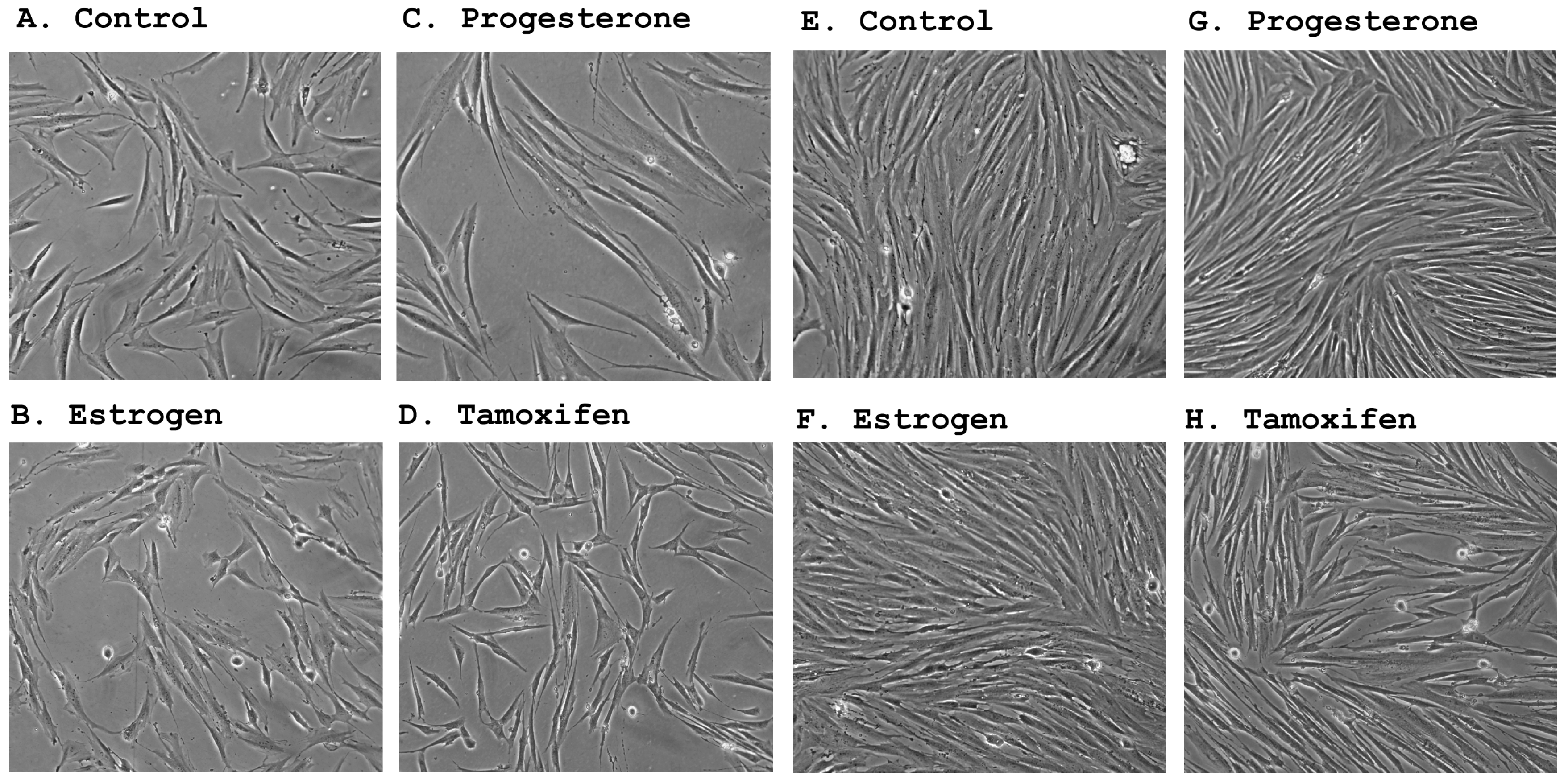
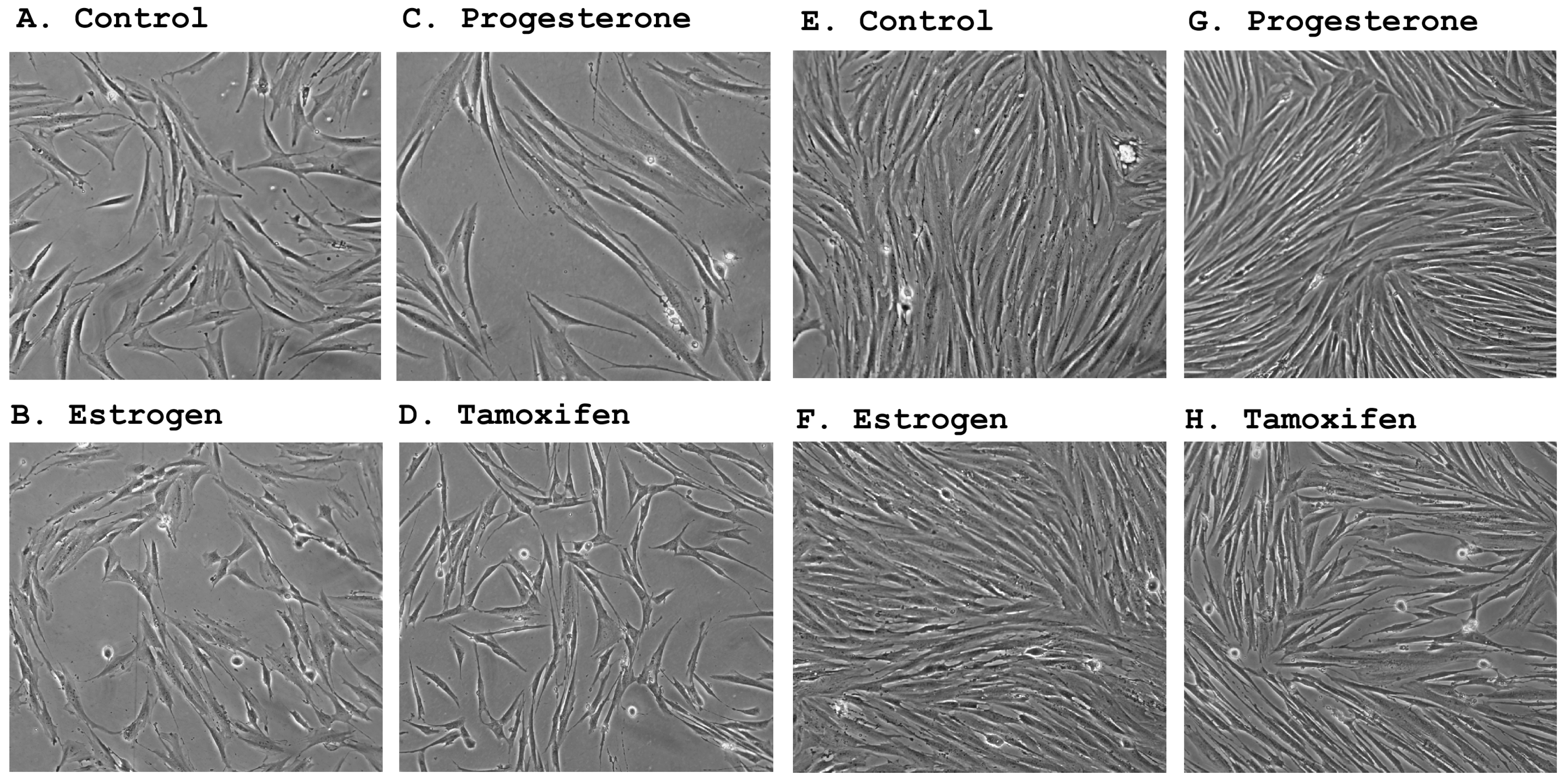
In accordance with their estrogen and progesterone receptor expression status, primary stromal cultures underwent significant morphological changes following treatment by steroid hormones. The counteraction of estrogen and progesterone appeared to be reminiscent of their opposing functions observed during the menstrual cycle where estrogen is responsible for cell proliferation [
26,
27] and progesterone for cell differentiation [
28,
29,
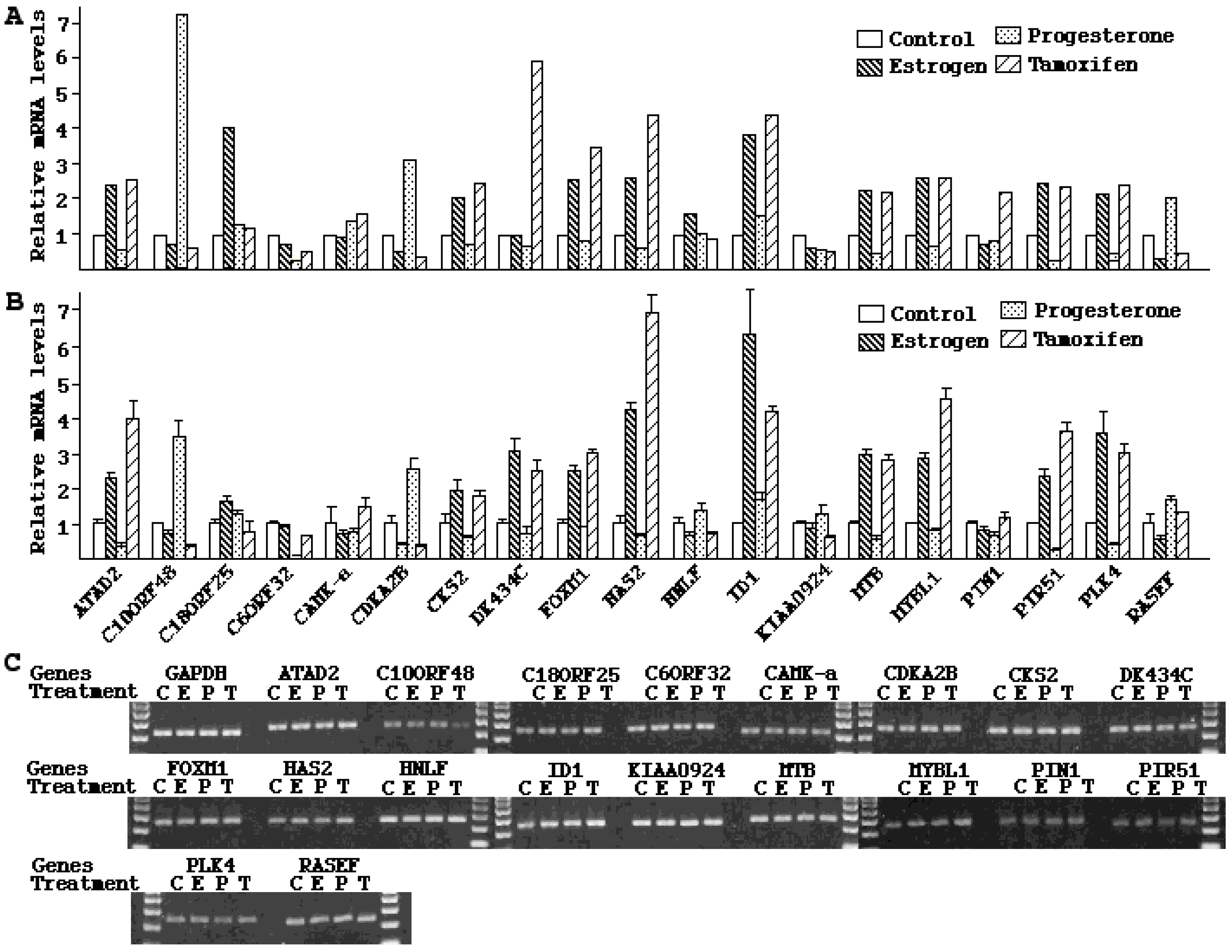
30]. Accompanying the morphological changes were dramatic and divergent alterations in mRNA expression by the two hormones. The opposite effects of estrogen and progesterone are best illustrated by the results from a number of cyclins. As shown in
Table 4 under cell cycle and apoptosis genes,
cyclin A2,
B1 and
B2 were all up-regulated by estrogen for more than 2-fold, but down-regulated by progesterone. These mitogens are known to promote cell proliferation and stimulate malignant transformation.
Cyclin A2 activation is considered a prerequisite for G1/S and G2/M transition [
31]. Elevated expression of
cyclin A2 and their catalytic partners
CDK1 and
CDK2 have been detected in male germ cell tumors, and their levels are quantitatively associated with tumor invasiveness [
32]. Over-expression of a non-degradable form of
cyclin A2 (δ 152) affected the cell cycle and promoted aneuploidy as well as transformation of rat fibroblasts [
33]. On the other hand, decreased levels of
cyclin A2 and
B1 were associated with P53-dependent G2 arrest in human squamous carcinoma cells [
34]. Therefore, estrogen-induced up-regulation of cyclins may be partially responsible for the carcinogenic effects observed on human endometrium and stroma. Their down-regulation by progesterone may contribute to this hormone’s cytostatic effect in the uterine cells. Since both stromal sarcomas and endometrial cancers respond to progestational treatment [
35,
36,
37], further exploration on the regulation and function of these genes may potentially lead to the improvement of hormonal prevention and/or treatment of uterine malignancies.
DNA synthesis represents a critical step in cell proliferation. A series of enzymes required for DNA replication such as DNA helicase, topoisomerase, and polymerase, were significantly affected by estrogen and progesterone. The DNA helicase complex functions as a replication licensing system that ensures precise chromosomal DNA replication before cell division [
38]. DNA topoisomerase II is required for chromosome segregation following DNA replication [
39]. DNA polymerase α and epsilon are the two major eukaryotic processing polymerases that are recruited to replication origins during the late G1 phase [
40]. Drug inhibition of DNA polymerase α and epsilon led to suppression of cancer cell growth [
41]. Lee
et al. reported that a human trophoblast cell line transfected with SV40 T antigen displayed increased proliferation and invasiveness, but was unable to form colonies on soft agar or tumors in nude mice. Interestingly, in these premalignant cells, DNA polymerase epsilon was identified as one of the genes up-regulated compared to the untransfected control. In this study, DNA helicase-2, DNA polymerase α and epsilon, and DNA topoisomerase II were all up-regulated by estrogen, but down-regulated by progesterone (
Table 3, Enzymes), suggesting that these enzymes may play important role(s) in the estrogen-dependent neoplastic diseases of the uterus [
42].
The estrogenic and carcinogenic effects of tamoxifen in stromal and endometrial cells have been well documented [
15,
27]. Since tamoxifen has been used to prevent breast cancer recurrence in high-risk women, investigating its effects on stromal cells bears strong clinical significance. Characterization of gene expression signatures represents the first step to better understand tamoxifen-mediated tumorigenic effects in the endometrium. Our data indicate that gene expression patterns induced by tamoxifen share some similarity with those of estrogen. In fact, all the genes discussed above, including those of
cyclin A2,
B1 and
B2, DNA polymerase α and epsilon, and DNA topoisomerase II were found to be up-regulated by both estrogen and tamoxifen (
Table 4, enzymes, cell cycle and apoptosis genes). It was reported that the subtle structural differences of estrogen and tamoxifen cause distinct conformational changes in the ER upon binding by these hormones [
43,
44], leading to the recruitment of alternative co-activators into the transcription initiation complex [
45,
46]. The few genes differentially regulated by estrogen and tamoxifen may provide an ideal model to study the distinct transactivation mechanisms by estrogen and tamoxifen.
Estrogen and progesterone concentrations undergo dynamic changes under different physio-pathological conditions. In addition, stromal cells were exposed simultaneously to both estrogen and progesterone, the combined effects by the two hormones added another dimension to the complexity of hormonal regulation. In this study, one hormone a time at a single concentration was examined for each hormone. In addition, stromal-epithelial interactions were not considered when purified stromal cells were used. Results from experiments applying these much-simplified conditions could not accurately reflect the in vivo situation. Thus, the observations needs to be further verified under physiological conditions. Nevertheless, the microarray analysis led to the identification of numerous candidate target genes, which provided useful information for future mechanistic and functional studies concerning the hormonal regulation in the endometrial stromal cell population.
4. Material and Methods
4.1. Tissue, Cell Lines, and Reagents
Normal breast, ovarian and endometrium tissues were collected from patients with benign conditions. Endometrial stromal cells were isolated by collagenase/DNase I digestion following established protocols [
47,
48]. Endometrial adenocarcinoma cell lines KLE and AN3, and breast cancer cell lines MCF-7 and MDA-MB-231 were obtained from American Type Culture Collection (Rockville, MD, USA). Stromal KLE and cells were grown in F12 medium. AN3, MCF-7, and MDA-MB-231 cells were maintained in MEM medium. All the media were supplemented with 10% FCS (BioWhittaker, Walkersville, MD, USA), 100 µg/mL streptomycin, 100 units/mL penicillin, and 2 mM glutamine. Cell cultures were maintained at 37 °C in an atmosphere containing 5% CO
2 and 100% humidity.
β-estradiol, progesterone, tamoxifen, and monoclonal antibody for ER-β (SAB4500814) and goat anti-rabbit secondary antibody (A4062) were purchased from Sigma (St. Louis, MO, USA). Antibodies against ER-α (MC-20), PR-A or PR-B (C-20) were products of Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-β-actin antibody (TA306308) was purchased from Oncogene (Boston, MA, USA).
4.2. Cell Treatment, RNA Isolation, and Quantitative Real-Time PCR
Stromal cell cultures were grown in 10 cm diameter dishes. Cells were de-induced in medium containing 10% charcoal-stripped serum for two days before treated with 1 × 10
−7 M of β-estradiol, 2 × 10
−7 M of tamoxifen, or 2 × 10
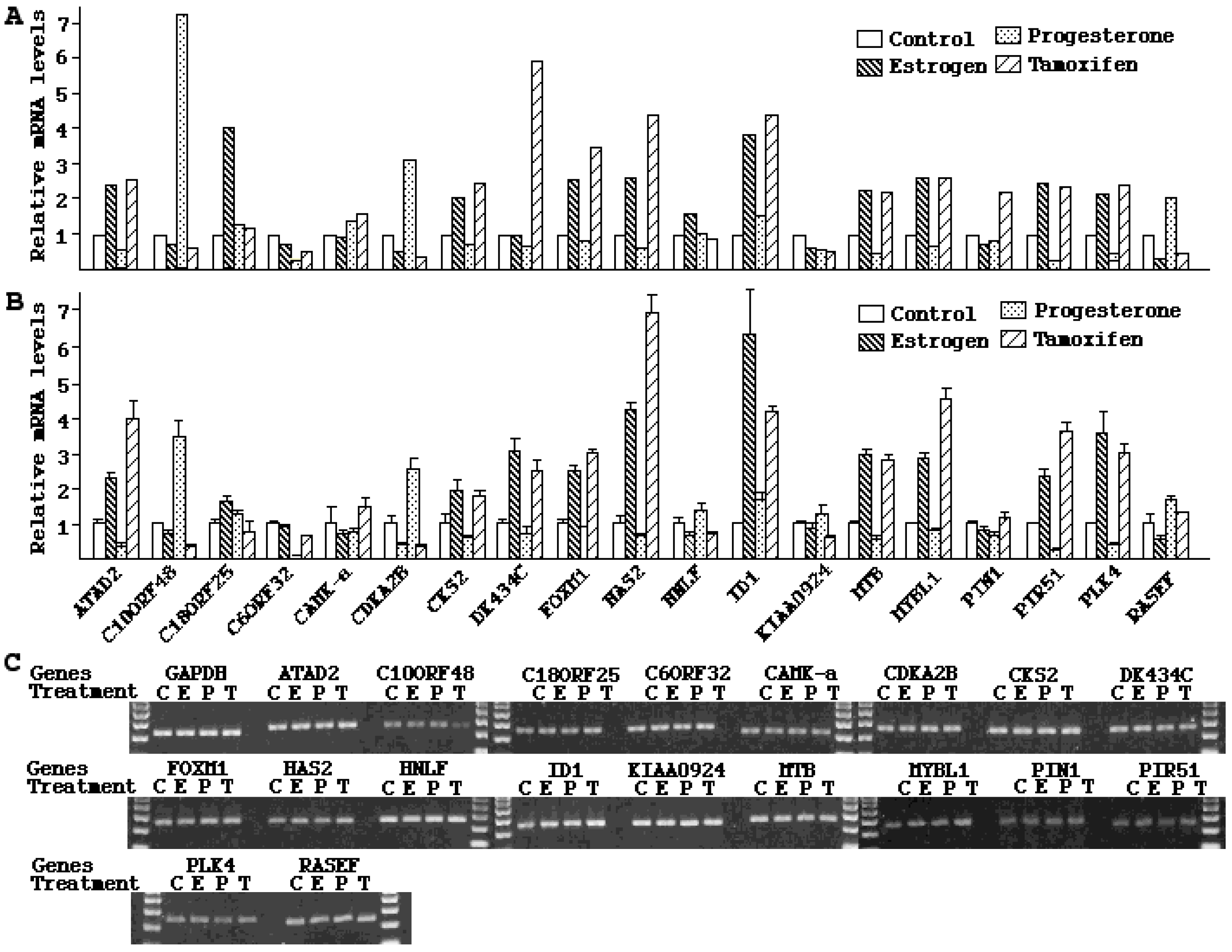
−7 M of progesterone for 48 h. Total RNA was isolated using Trizol reagents (Invitrogen, Carlsbad, CA, USA). The RNA samples were treated with DNA Free™ (Ambion, Austib, TX, USA) to eliminate contaminated genomic DNA. cDNA was synthesized from 1 µg of RNA with random primers using SuperScript kit (Invitrogen, Carlsbad, CA, USA). The 20 µL of reverse transcription products were diluted to 100 and 2 µL was used for each real-time PCR. PCR reactions were carried out in 25 µL containing 140 ng of primers and 12.5 µL SYBR Green Master Mix (Stratagene, Cedar Creek, TX, USA). The designations and sequences of PCR primers are described in
Table 6. Real-time PCR was performed under the following conditions: initial denature, 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 56 °C for 40 s, and extension at 72 °C for 30 s. The threshold cycle number (
Ct) values were standardized against
GAPDH controls, converted to fold (2
Ct) relative to
GAPDH, and compared between experimental and control groups. All data groups were analyzed by ANOVA to determine if there was significance (
p < 0.05) among the groups. For all experimental groups that satisfied the initial ANOVA criterion, individual comparisons were performed with the use of
post hoc Bonferroni
t tests based on assumptions of two-tailed distribution and two samples with equal variance. Statistical significance (
p ≤ 0.05) is indicated by asterisks in the figures.
Table 6.
Primers and sizes of amplicons in real-time PCR.
Table 6.
Primers and sizes of amplicons in real-time PCR.
| Gene Name | Description | 5' Primer | 3' Primer | Size (bp) |
|---|
| ER-α | Estrogen receptor α | aattcagataatcgacgccag | gtgtttcaacattctccctcctc | 344 |
| ER-β | Estrogen receptor β | tgcggaacctcaaaagagtc | cttcacacgaccagactcca | 206 |
| PR-AB | Progesterone receptor A and B | atgagccggtccgggtgcaag | gccacccagagcccgaggttt | 243 |
| PR-B | Progesterone receptor B | gactgagagcttcacagtat | tctcctaactcggggagttct | 187 |
| ATAD2 | ATPase family, AAA domain containing 2 | gattatcttccgcaggacca | gttgcattggatcaacatcg | 255 |
| C10ORF48 | chromosome 10 open reading frame 48 | gggtcaatagtgcagccagt | tgcgcttactgttactgcaaa | 247 |
| C18ORF25 | chromosome 18 open reading frame 25 | gtaggggccagactgaatga | agtgtccccagctttttcaa | 250 |
| C6ORF32 | chromosome 6 open reading frame 32 | aggagaaaatgccactgtcg | tcctctgggtcttcctcctt | 250 |
| CAMK-a | calcium/calmodulin-dependent protein kinase II | acgagaagctgagcccctac | ttgggggagttagacaccag | 221 |
| CDKN2B | cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) | tcgtttgcttttcagggttt | cctcctccactttgtcctca | 248 |
| CKS2 | CDC28 protein kinase regulatory subunit 2 | ggagtggaggagacttggtg | cagctcatgcacaggtatgg | 236 |
| DK434C | DKFZP434C245 protein | taagctgtgggacaagagca | ttgagtcactggaggctgtg | 248 |
| FOXM1 | forkhead box M1 | cgtggattgaggaccacttt | gattcggtcgtttctgctgt | 249 |
| HAS2 | hyaluronan synthase 2 | agagcactgggacgaagtgt | atgcactgaacacacccaaa | 245 |
| HNLF | putative NFkB activating protein HNLF | agaagcgctgtttcatcgag | gccatcctggtagaattgga | 253 |
| ID1 | inhibitor of DNA binding 1, dominant negative helix-loop-helix protein | cccattctgtttcagccagt | agccgttcatgtcgtagagc | 245 |
| KIAA0924 | KIAA0924 protein | atcgctcattttgaggttgc | gcagaggacagggcagtaaa | 246 |
| MTB | more than blood homolog | tgcgggaggttctgagttac | ggaccatcgggtaaggatct | 261 |
| MYBL1 | v-myb myeloblastosis viral oncogene homolog (avian)-like 1 | gtccgaaacgttggtctgtt | gaccttccgacgcattgtag | 248 |
| PIN1 | protein (peptidyl-prolyl cis/trans isomerase) NIMA-interacting 1 | tgccaccgtcacacagtatt | gagtctgcctccagcacct | 253 |
| PIR51 | RAD51 associated protein 1 | ttctggaaggcagtgatggt | gagcagagtccaccgaagtc | 243 |
| PLK4 | polo-like kinase 4 (Drosophila) | gccaaggaccttattcacca | ttatttgggagtggctgacc | 251 |
| RASEF | RAS and EF hand domain containing | atcaaccttgtggagccaag | ctgaggtcactgagggcttc | 245 |
4.3. Western Blot Analysis
Cell extracts (20 µg) were resolved in SDS polyacrylamide gels (Ready Gel, 4%–15% gradient, Bio-Rad Laboratories, Hercules, CA, USA) and electrically transferred onto an Immun-Blot polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked for 2 h in PBS buffer containing 0.1% Tween-20 and 10% nonfat dried milk. Specific antibodies against ER-α, ER-β, PR-A and PR-B, or β-actin were applied following the manufacturer’s recommendations. Primary antibody binding was performed at 4 °C overnight with constant rotation. The secondary antibody binding was carried out at room temperature for 1 h at 1:5000 dilutions. Immunobloting signals were detected using the Chemiluminescence Plus Western Blotting Detection System (Amersham Corp., Arlington Heights, IL, USA). The blots were re-probed with β-actin antibody and the results provided controls for protein loading.
4.4. Microarray Hybridization
Affymetrix GeneChip™ Human Genome U133 Plus 2.0 microarrays were used for mRNA profiling. Microarray analysis was performed at Mayo Microarray Core facilities by technologists following standard procedures. Briefly, RNA samples were subject to Agilent analysis for quality controls. cDNA was prepared from 10 μg of RNA, quantified by spectrometry, and used as a template for the synthesis of biotinylated cRNA using RNA transcript labeling reagent (Affymetrix, Santa Clara, CA, USA). The quality of the cRNA probes was verified by gel electrophoresis and pilot hybridization with the Test-3 array. Hybridization solution containing fragmented cRNA probes and control cRNA (BioB, BioC, and BioD) was supplemented with herring sperm DNA and bovine serum albumin. The probe solution was heated at 99 °C for 5 min followed by incubation at 45 °C for 5 min before use. Hybridization was carried out at 45 °C for 16 h with constant rotation at 60 rpm. The arrays were washed and stained with streptavidin-phycoerythrin (Molecular Probes, Eugene, OR, USA). After washes, arrays were scanned using the GeneChip system confocal scanner (Hewlett Packard, Palo Alto, CA, USA).
4.5. Microarray Data Analysis
Gene expression profiles were analyzed at the Mayo General Clinical Research Center Genomics, Proteomics, and Metabolic Core Facility using established protocols [
49,
50]. The GeneChip 5.0 (Affymetrix) program was used to scan and quantitatively document the hybridization signals. Compilation of candidate genes and calculation of changes were performed on SpotFire and Microsoft Excel programs. To minimize the false-positive conclusion, only genes with hybridization signal reached an absolute level that was significantly higher than that of the background (
p < 0.05) entered analyses. Ingenuity Pathway Analysis (Ingenuity Systems), and the Entrez Search Engine website, from the NCBI (
http://www.ncbi.nlm.nih.gov/) were applied to selected genes for analysis on their biological interactions.



{kind=link}
{kind=link}
{kind=link}