
A Heme Oxygenase-1 Transducer Model of Degenerative and Developmental Brain Disorders

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Transduction and Pathogenesis of Chronic Human Brain Disease
2. Convergent Neuropathology in Chronic CNS Disorders
2.1. Alzheimer Disease
2.2. Parkinson Disease
2.3. Schizophrenia
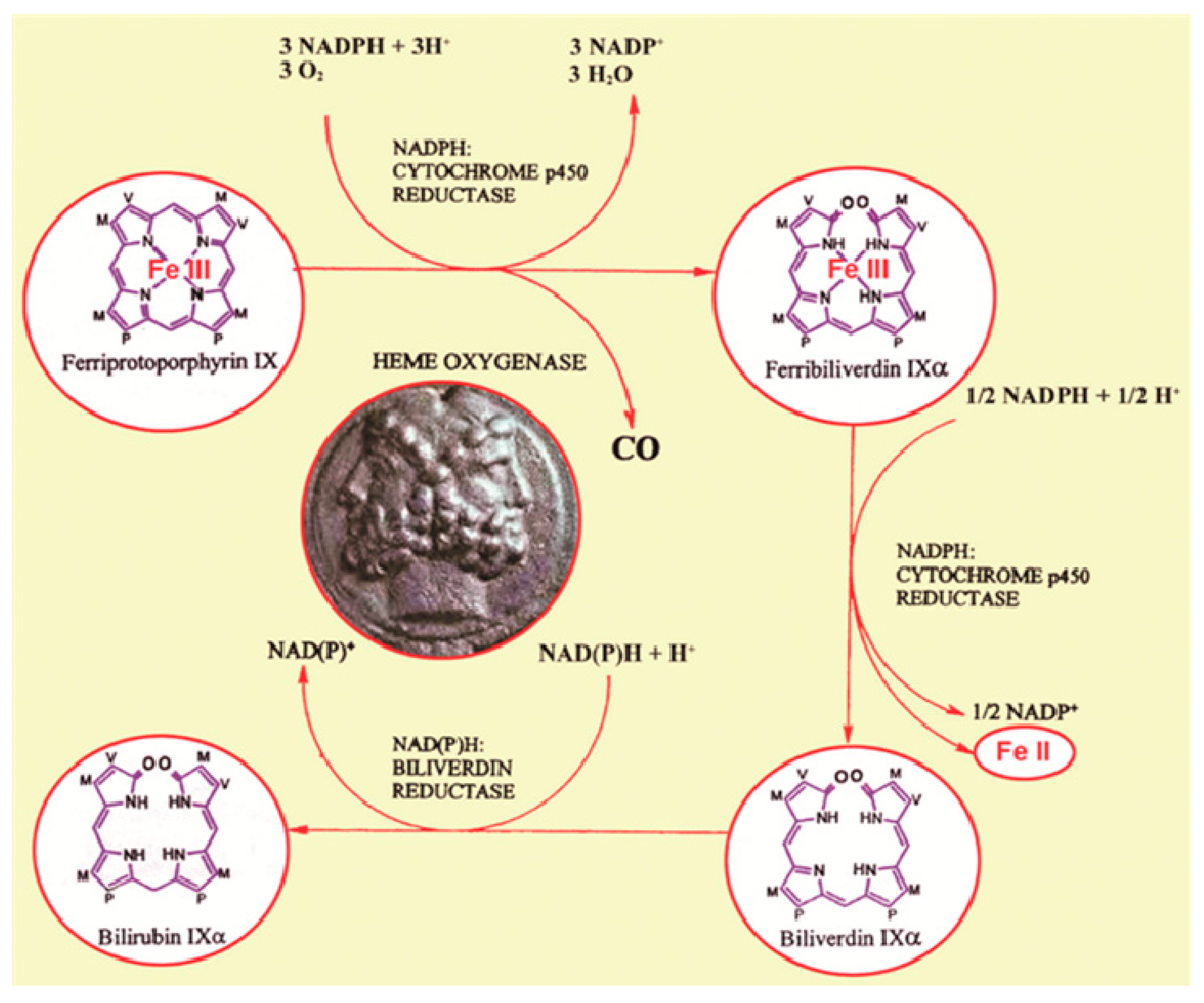
3. Heme Oxygenase-1
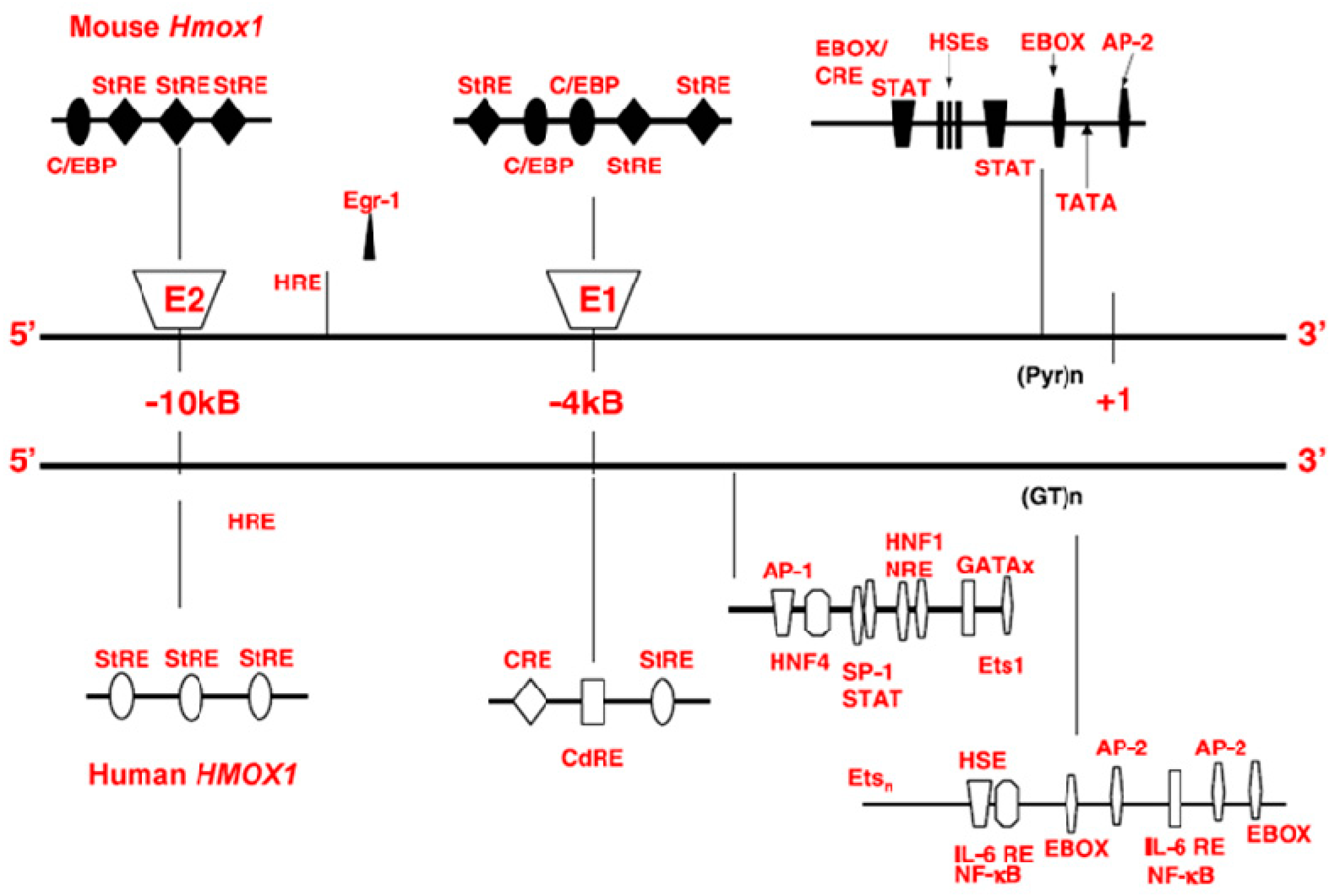
3.1. HO-1 Regulation and Physiology


3.2. HO-1 in Human Brain Aging and Disease
3.3. HO-1: Mediator of “Core” Neuropathology in Stressed Astroglia

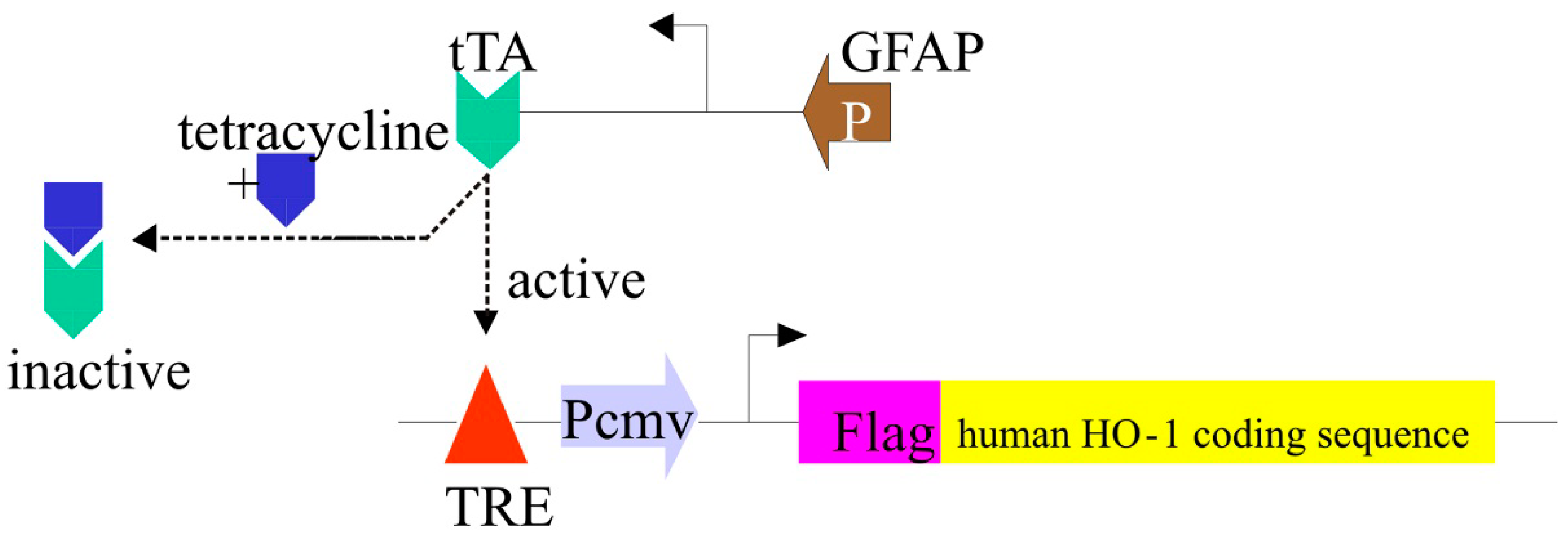
3.4. The GFAP.HMOX1 Transgenic Mouse
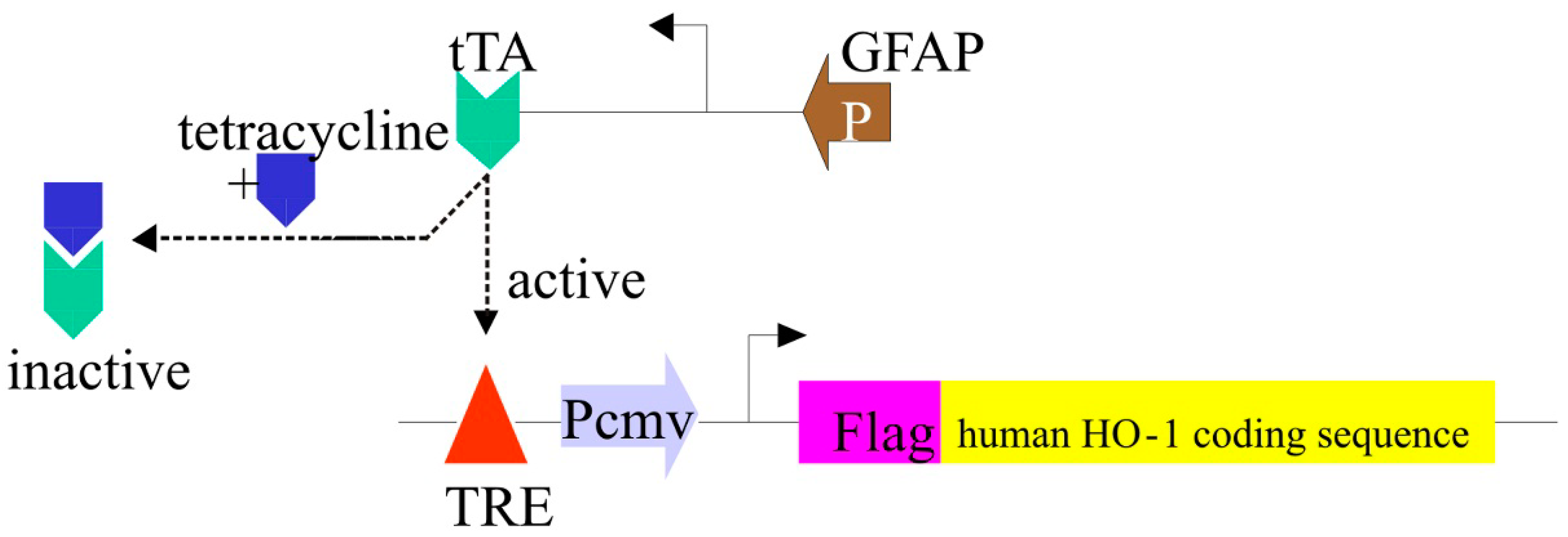

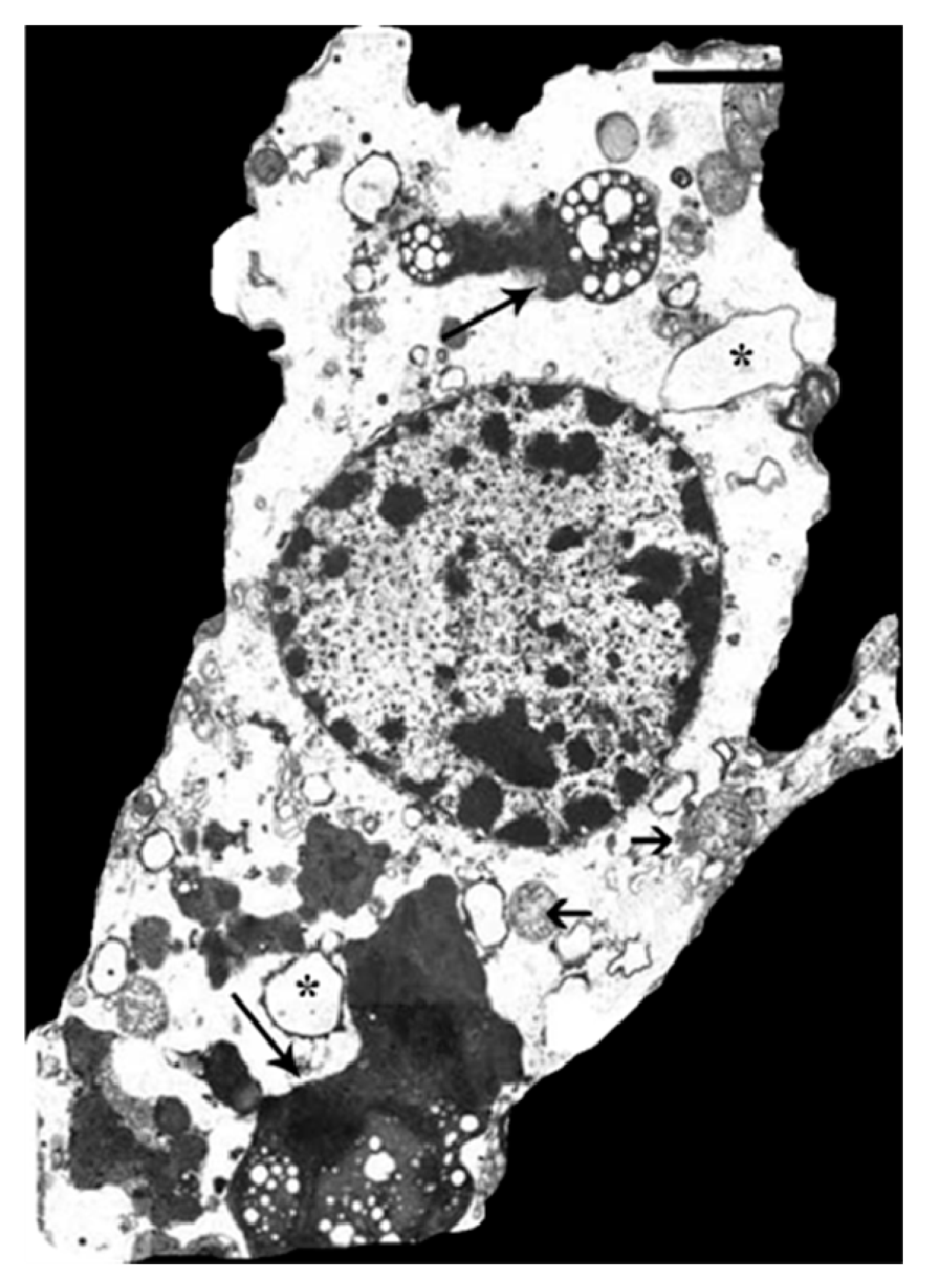
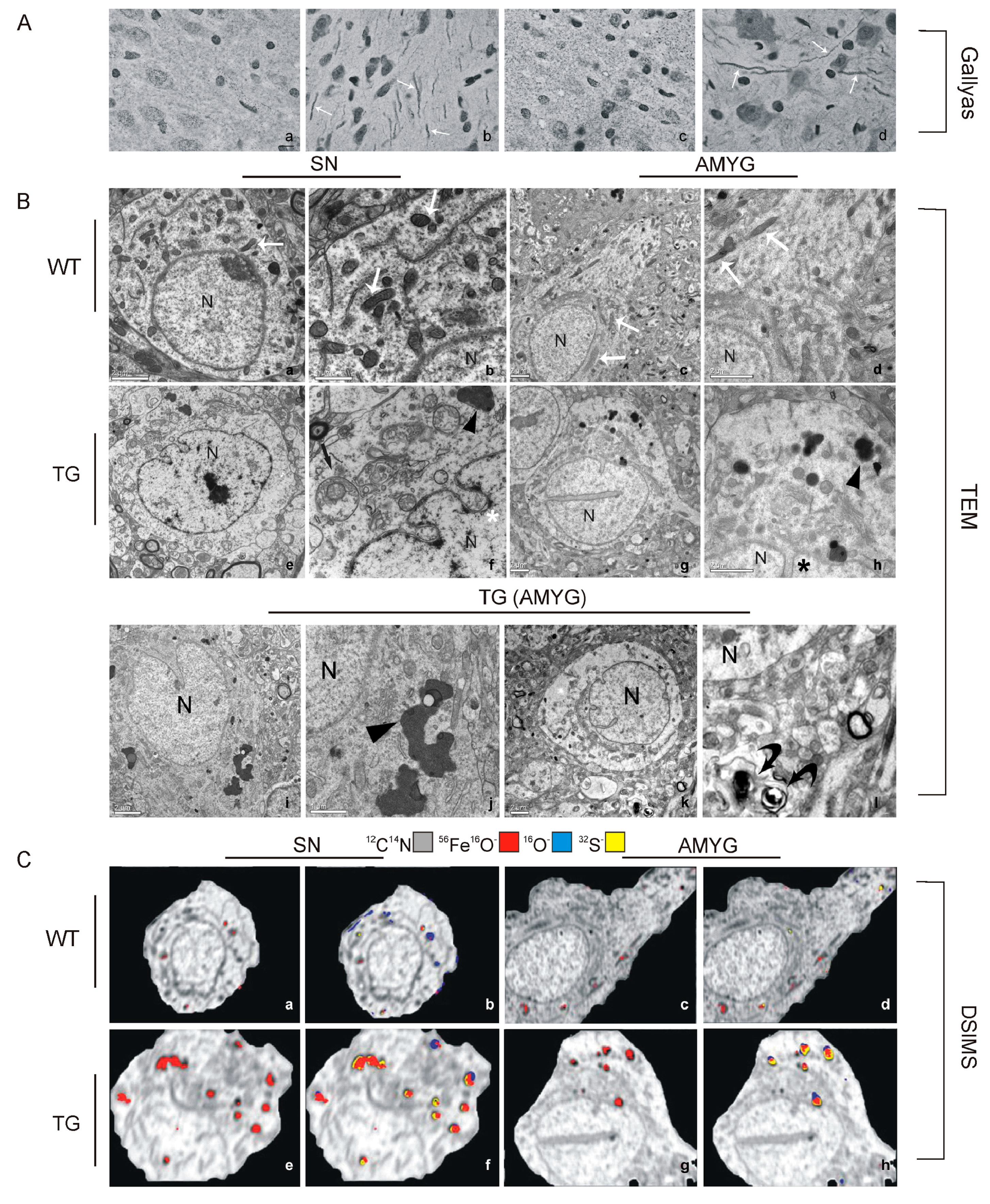
3.5. HO-1 Suppression in APPswe/PS1ΔE9 Transgenic Mice
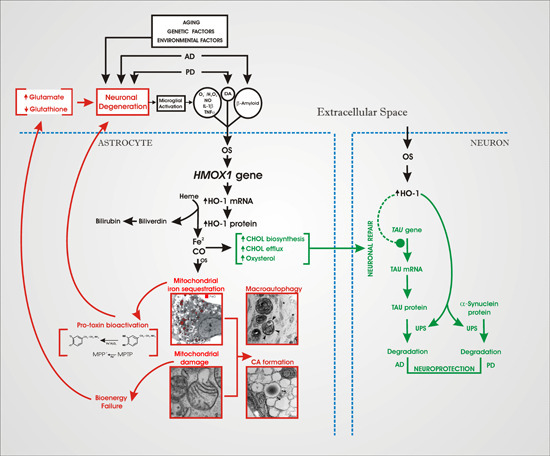
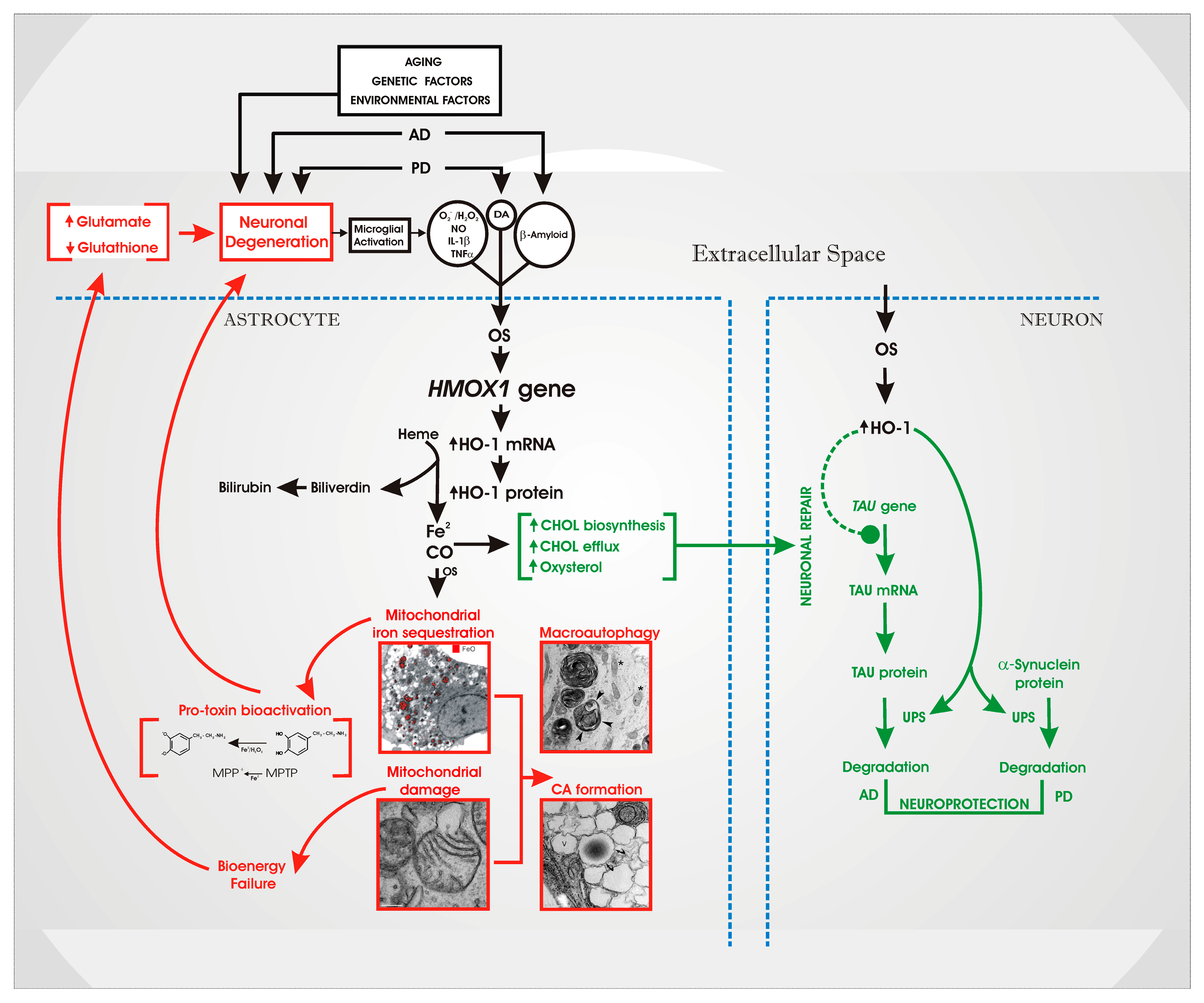
4. An “HO-1 Transducer” Model of Chronic Brain Disease
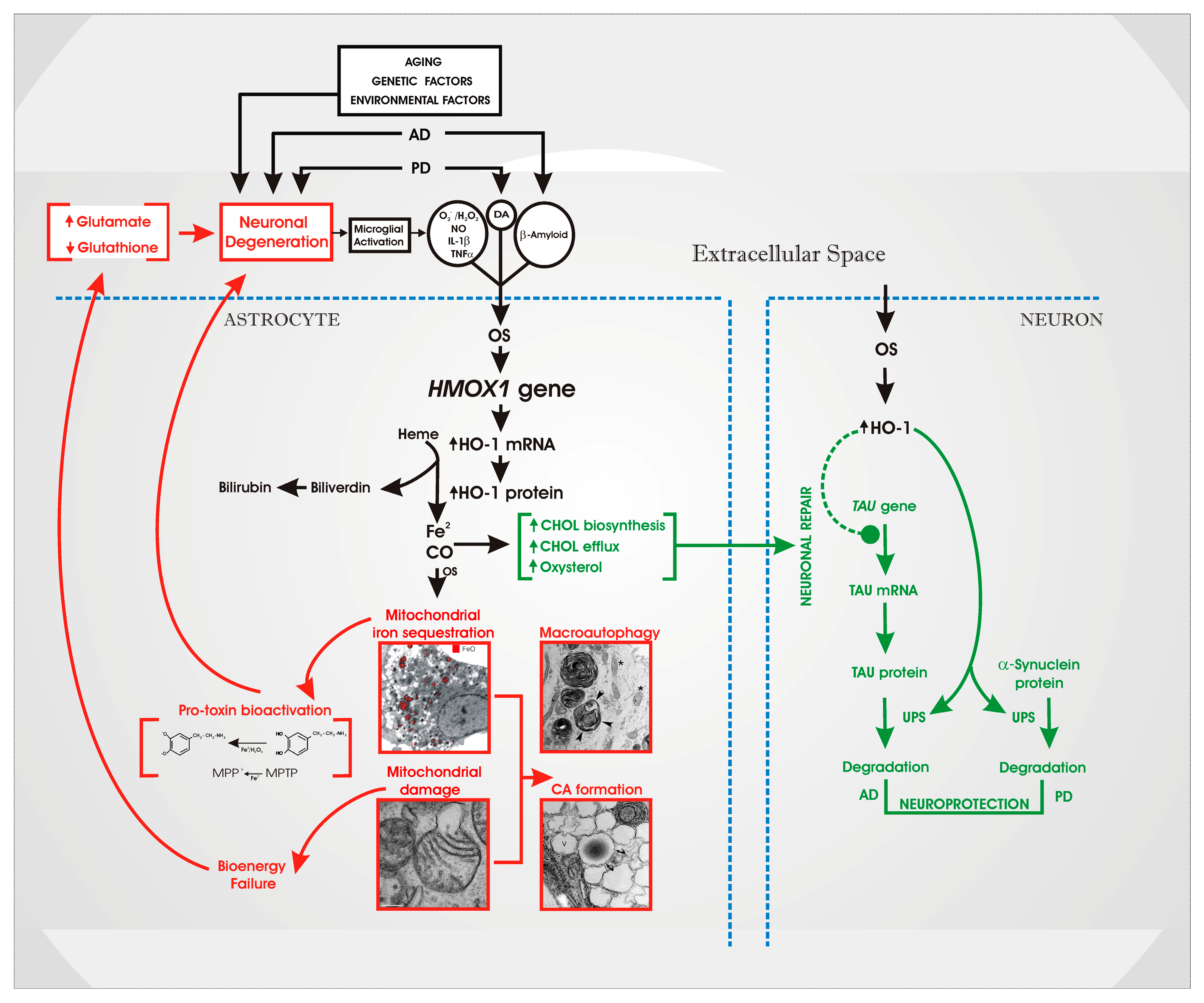
5. Therapeutic Implications
Acknowledgments
Conflicts of Interest
References
- Schipper, H.M. Brain iron deposition and the free radical-mitochondrial theory of ageing. Ageing Res. Rev. 2004, 3, 265–301. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial DNA deletion levels in alzheimer brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Tanno, Y.; Okuizumi, K.; Tsuji, S. Mtdna polymorphisms in japanese sporadic alzheimer’s disease. Neurobiol. Aging 1998, 19, S47–S51. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Tanji, K.; Hirano, M.; Vu, T.H.; DiMauro, S.; Schon, E.A. Mitochondrial involvement in alzheimer’s disease. Biochim. Biophys. Acta 1999, 1410, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in alzheimer disease. J. Neural Transm. 1998, 105, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [PubMed]
- Minoshima, S.; Giordani, B.; Berent, S.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Metabolic reduction in the posterior cingulate cortex in very early alzheimer’s disease. Ann. Neurol. 1997, 42, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, H.; Ogawa, M.; Yamauchi, H.; Yamaguchi, S.; Kimura, J.; Yonekura, Y.; Konishi, J. Altered cerebral energy metabolism in alzheimer’s disease: A pet study. J. Nucl. Med. 1994, 35, 1–6. [Google Scholar] [PubMed]
- Butterfield, D.A. Amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. The inflammatory response system of brain: Implications for therapy of alzheimer and other neurodegenerative diseases. Brain Res. Brain Res. Rev. 1995, 21, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Glial iron sequestration and neurodegeneration. In Astrocytes in Brain Aging and Neurodegeneration; Schipper, H.M., Ed.; R.G. Landes Co.: Austin, TX, USA, 1998; pp. 235–251. [Google Scholar]
- Sayre, L.M.; Smith, M.A.; Perry, G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr. Med. Chem. 2001, 8, 721–738. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox. Res. 1999, 1, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Z.H.; Ip, N.Y. The emerging role of autophagy in parkinson’s disease. Mol. Brain 2009, 2, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, P. Oxidative stress in parkinson’s disease. Ann. Neurol. 2003, S26–S38. [Google Scholar] [CrossRef]
- Hattori, N.; Tanaka, M.; Ozawa, T.; Mizuno, Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in parkinson’s disease. Ann. Neurol. 1991, 30, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Storm, T.; Rath, S.; Mohamed, S.A.; Bruse, P.; Kowald, A.; Oehmichen, M.; Meissner, C. Mitotic brain cells are just as prone to mitochondrial deletions as neurons: A large-scale single-cell PCR study of the human caudate nucleus. Exp. Gerontol. 2002, 37, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Bowen, B.C.; Block, R.E.; Sanchez-Ramos, J.; Pattany, P.M.; Lampman, D.A.; Murdoch, J.B.; Quencer, R.M. Proton MR spectroscopy of the brain in 14 patients with parkinson disease. Am. J. Neuroradiol. 1995, 16, 61–68. [Google Scholar] [PubMed]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Comparini, S.O.; Kim, R.W.; Kleinman, J.E. Staining intensity of brain iron in patients with schizophrenia: A postmortem study. J. Neuropsychiatry Clin. Neurosci. 1992, 4, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Kolomeets, N.S.; Uranova, N. Ultrastructural abnormalities of astrocytes in the hippocampus in schizophrenia and duration of illness: A postortem morphometric study. World J. Biol. Psychiatry 2010, 11, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; D'Agata, V.; Calabrese, V.; Pascale, A.; Colombrita, C.; Alkon, D.; Cavallaro, S. Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res. 2002, 954, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Baranano, D.E.; Snyder, S.H. Neural roles for heme oxygenase: Contrasts to nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 10996–11002. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, T.; Toyomura, K.; Kinoshita, T.; Morisawa, S. A beneficial role of bile pigments as an endogenous tissue protector: Anti-complement effects of biliverdin and conjugated bilirubin. Biochim. Biophys. Acta 1993, 1158, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Llesuy, S.F.; Tomaro, M.L. Heme oxygenase and oxidative stress. Evidence of involvement of bilirubin as physiological protector against oxidative damage. Biochim. Biophys. Acta 1994, 1223, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Dore, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Dennery, P.A. Regulation and role of heme oxygenase in oxidative injury. Curr. Top. Cell. Regul. 2000, 36, 181–199. [Google Scholar] [PubMed]
- Piantadosi, C.A.; Carraway, M.S.; Suliman, H.B. Carbon monoxide, oxidative stress, and mitochondrial permeability pore transition. Free Radic. Biol. Med. 2006, 40, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Frankel, D.; Mehindate, K.; Schipper, H.M. Role of heme oxygenase-1 in the regulation of manganese superoxide dismutase gene expression in oxidatively-challenged astroglia. J. Cell. Physiol. 2000, 185, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Hirose, W.; Ikematsu, K.; Tsuda, R. Age-associated increases in heme oxygenase-1 and ferritin immunoreactivity in the autopsied brain. Leg Med. (Tokyo) 2003, 5, S360–S366. [Google Scholar] [CrossRef]
- Song, W.; Zukor, H.; Liberman, A.; Kaduri, S.; Arvanitakis, Z.; Bennett, D.A.; Schipper, H.M. Astroglial heme oxygenase-1 and the origin of corpora amylacea in aging and degenerating neural tissues. Exp. Neurol. 2014, 254, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2013, 62C, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; di Domenico, F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med. 2004, 37, 1995–2011. [Google Scholar] [CrossRef] [PubMed]
- Ham, D.; Schipper, H.M. Heme oxygenase-1 induction and mitochondrial iron sequestration in astroglia exposed to amyloid peptides. Cell. Mol. Biol. 2000, 46, 587–596. [Google Scholar] [PubMed]
- Mehindate, K.; Sahlas, D.J.; Frankel, D.; Mawal, Y.; Liberman, A.; Corcos, J.; Dion, S.; Schipper, H.M. Proinflammatory cytokines promote glial heme oxygenase-1 expression and mitochondrial iron deposition: Implications for multiple sclerosis. J. Neurochem. 2001, 77, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Bernier, L.; Mehindate, K.; Frankel, D. Mitochondrial iron sequestration in dopamine-challenged astroglia: Role of heme oxygenase-1 and the permeability transition pore. J. Neurochem. 1999, 72, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Su, H.; Song, S.; Paudel, H.K.; Schipper, H.M. Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J. Cell. Physiol. 2006, 206, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Vaya, J.; Song, W.; Khatib, S.; Geng, G.; Schipper, H.M. Effects of heme oxygenase-1 expression on sterol homeostasis in rat astroglia. Free Radic. Biol. Med. 2007, 42, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Bernier, L.; Bernatchez, G.; Schipper, H. In Oxidative Stress Promotes Mitochondrial Iron Sequestration in Cultured Astroglia; Psychology Press: London, UK, 1996; p. 1495. [Google Scholar]
- McLaren, J.; Brawer, J.R.; Schipper, H.M. Iron content correlates with peroxidase activity in cysteamine-induced astroglial organelles. J. Histochem. Cytochem. 1992, 40, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
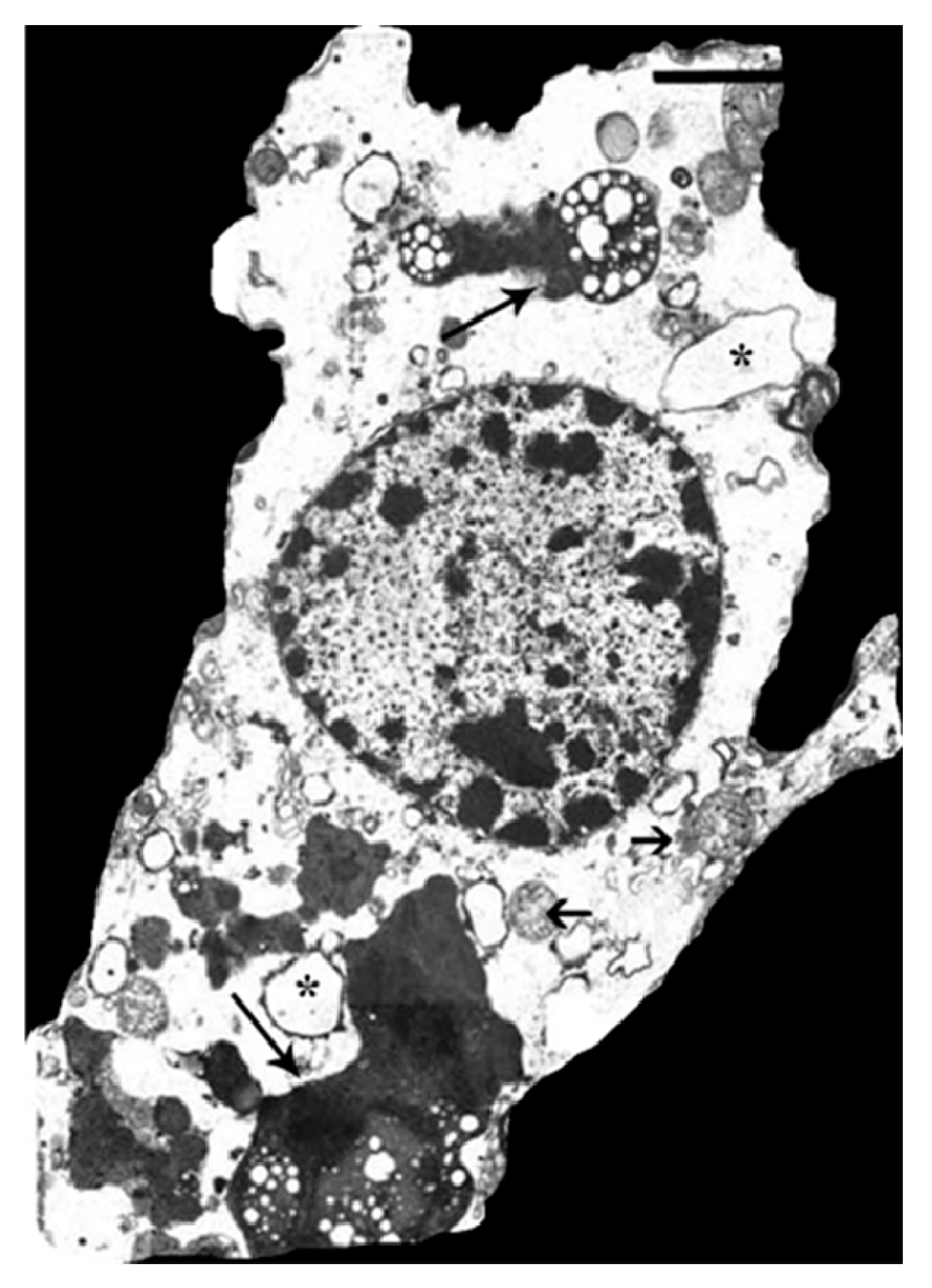
- Zukor, H.; Song, W.; Liberman, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.D.; Guerquin-Kern, J.L.; Schipper, H.M. Ho-1-mediated macroautophagy: A mechanism for unregulated iron deposition in aging and degenerating neural tissues. J. Neurochem. 2009, 109, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. The permeability transition pore. Control points of a cyclosporin a-sensitive mitochondrial channel involved in cell death. Biochim. Biophys. Acta 1996, 1275, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Cola, C.; Massari, S.; Colonna, R.; Bernardi, P. Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J. Biol. Chem. 1993, 268, 21939–21945. [Google Scholar] [PubMed]
- Linnane, A.W.; Zhang, C.; Baumer, A.; Nagley, P. Mitochondrial DNA mutation and the ageing process: Bioenergy and pharmacological intervention. Mutat. Res. 1992, 275, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.S.; Moozar, K.L.; Mehindate, K.; Schipper, H.M. A cellular stress model for the differential expression of glial lysosomal cathepsins in the aging nervous system. Exp. Neurol. 1997, 147, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Cissé, S. Mitochondrial constituents of corpora amylacea and autofluorescent astrocytic inclusions in senescent human brain. Glia 1995, 14, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, J.B. Corpora-amylacea and the family of polyglucosan diseases. Brain Res. Brain Res. Rev. 1999, 29, 265–295. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Song, W.; Schipper, H.M. Astroglia overexpressing heme oxygenase-1 predispose co-cultured PC12 cells to oxidative injury. J. Neurosci. Res. 2007, 85, 2186–2195. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Kotake, Y.; Janzen, E.G. Catechol oxidation by peroxidase-positive astrocytes in primary culture: An electron spin resonance study. J. Neurosci. 1991, 11, 2170–2176. [Google Scholar] [PubMed]
- Schipper, H.M. Mitochondrial iron deposition in aging astroglia: Mechanisms and disease implications. In Mitochondrial Ubiquinone (Coenzyme Q): Biochemical, Functional, Medical, and Therapeutic Aspects in Human Health And Disease; Ebadi, M.J., Chopra, R.K., Eds.; Prominent Press: Shepparton, Austria, 2001; pp. 267–280. [Google Scholar]
- Di Monte, D.A.; Schipper, H.M.; Hetts, S.; Langston, J.W. Iron-mediated bioactivation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in glial cultures. Glia 1995, 15, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Kontos, H.A.; Wei, E.P.; Ellis, E.F.; Jenkins, L.W.; Povlishock, J.T.; Rowe, G.T.; Hess, M.L. Appearance of superoxide anion radical in cerebral extracellular space during increased prostaglandin synthesis in cats. Circ. Res. 1985, 57, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Hascalovici, J.R.; Song, W.; Vaya, J.; Khatib, S.; Fuhrman, B.; Aviram, M.; Schipper, H.M. Impact of heme oxygenase-1 on cholesterol synthesis, cholesterol efflux and oxysterol formation in cultured astroglia. J. Neurochem. 2009, 108, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Hascalovici, J.R.; Vaya, J.; Khatib, S.; Holcroft, C.A.; Zukor, H.; Song, W.; Arvanitakis, Z.; Bennett, D.A.; Schipper, H.M. Brain sterol dysregulation in sporadic ad and mci: Relationship to heme oxygenase-1. J. Neurochem. 2009, 110, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Patel, A.; Qureshi, H.Y.; Han, D.; Schipper, H.M.; Paudel, H.K. The parkinson disease-associated a30p mutation stabilizes alpha-synuclein against proteasomal degradation triggered by heme oxygenase-1 over-expression in human neuroblastoma cells. J. Neurochem. 2009, 110, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zukor, H.; Lin, S.H.; Hascalovici, J.; Liberman, A.; Tavitian, A.; Mui, J.; Vali, H.; Tong, X.K.; Bhardwaj, S.K.; et al. Schizophrenia-like features in transgenic mice overexpressing human ho-1 in the astrocytic compartment. J. Neurosci. 2012, 32, 10841–10853. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zukor, H.; Lin, S.H.; Liberman, A.; Tavitian, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.D.; et al. Unregulated brain iron deposition in transgenic mice over-expressing hmox1 in the astrocytic compartment. J. Neurochem. 2012, 123, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S. The environment and susceptibility to schizophrenia. Prog. Neurobiol. 2011, 93, 23–58. [Google Scholar] [CrossRef] [PubMed]
- King, S.; St-Hilaire, A.; Heidkamp, D. Prenatal factors in schizophrenia. Curr. Dir. Psychol. Sci. 2010, 19, 209–213. [Google Scholar] [CrossRef]
- Wilson, C.; Terry, A.V., Jr. Neurodevelopmental animal models of schizophrenia: Role in novel drug discovery and development. Clin. Schizophr. Relat. Psychoses 2010, 4, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Liberman, A.; Schipper, H.M. Parkinsonian features in aging GFAP. Hmox1 transgenic mice overexpressing human HO-1 in the astroglial compartment. J. Parkinsons Dis. 2013, 3, 69–69. [Google Scholar] [PubMed]
- Gupta, A.; Lacoste, B.; Pistel, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; Song, W.; et al. Neurotherapeutic effects of novel ho-1 inhibitors in vitro and in a transgenic mouse model of alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Wang, D.; Li, W.; Zhang, L.; Jin, J.; Ma, N.; Zhou, L.; Nakajima, O.; Zhao, W.; Gao, X. Long-term overexpression of heme oxygenase 1 promotes tau aggregation in mouse brain by inducing tau phosphorylation. J. Alzheimers Dis. 2011, 26, 299–313. [Google Scholar] [PubMed]
- Kirkwood, T.B.; Rose, M.R. Evolution of senescence: Late survival sacrificed for reproduction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1991, 332, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.C. Pleiotropy, natural selection, and the evolution of senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- Drummond, G.S.; Kappas, A. Chemoprevention of severe neonatal hyperbilirubinemia. Semin. Perinatol. 2004, 28, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Kappas, A.; Drummond, G.S.; Galbraith, R.A. Prolonged clinical use of a heme oxygenase inhibitor: Hematological evidence for an inducible but reversible iron-deficiency state. Pediatrics 1993, 91, 537–539. [Google Scholar] [PubMed]
- Alaoui-Jamali, M.A.; Bismar, T.A.; Gupta, A.; Szarek, W.A.; Su, J.; Song, W.; Xu, Y.; Xu, B.; Liu, G.; Vlahakis, J.Z.; et al. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer Res. 2009, 69, 8017–8024. [Google Scholar] [CrossRef] [PubMed]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic polyconjugates for targeted in vivo delivery of sirna to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering rna to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Syapin, P.J. Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br. J. Pharmacol. 2008, 155, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Le, W.D.; Xie, W.J.; Appel, S.H. Protective role of heme oxygenase-1 in oxidative stress-induced neuronal injury. J. Neurosci. Res. 1999, 56, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Perry, G.; Abraham, N.G.; Dwyer, B.E.; Kutty, R.K.; Laitinen, J.T.; Petersen, R.B.; Smith, M.A. Overexpression of heme oxygenase in neuronal cells, the possible interaction with TAU. J. Biol. Chem. 2000, 275, 5395–5399. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Polevoda, B.; Coban, T.; Johnson, K.; Stoliar, S.; Huang, T.J.; Panahian, N.; Cory-Slechta, D.A.; McCoubrey, W.K., Jr. Neuronal overexpression of heme oxygenase-1 correlates with an attenuated exploratory behavior and causes an increase in neuronal nadph diaphorase staining. J. Neurochem. 1998, 70, 2057–2069. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Gunter, K.; Maines, M.D. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem. 2000, 75, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Panahian, N.; Yoshiura, M.; Maines, M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J. Neurochem. 1999, 72, 1187–1203. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Richmon, J.D.; Sato, M.; Sharp, F.R.; Panter, S.S.; Noble, L.J. Induction of heme oxygenase-1 (HO-1) in glia after traumatic brain injury. Brain Res. 1996, 736, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Vreman, H.J.; Wong, R.J.; Tjoa, T.; Yamauchi, T.; Noble-Haeusslein, L.J. Heme oxygenase-1 stabilizes the blood-spinal cord barrier and limits oxidative stress and white matter damage in the acutely injured murine spinal cord. J. Cereb. Blood Flow Metab. 2007, 27, 1010–1021. [Google Scholar] [PubMed]
- Ahmad, A.S.; Zhuang, H.; Dore, S. Heme oxygenase-1 protects brain from acute excitotoxicity. Neuroscience 2006, 141, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Ong, W.Y.; Go, M.L.; Garey, L.J. Heme oxygenase-1 activity after excitotoxic injury: Immunohistochemical localization of bilirubin in neurons and astrocytes and deleterious effects of heme oxygenase inhibition on neuronal survival after kainate treatment. J. Neurosci. Res. 2005, 80, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Hascalovici, J.R.; Song, W.; Liberman, A.; Vaya, J.; Khatib, S.; Holcroft, C.; Laferla, F.; Schipper, H.M. Neural ho-1/sterol interactions in vivo: Implications for alzheimer’s disease. Neuroscience 2014, 280C, 40–49. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schipper, H.M.; Song, W. A Heme Oxygenase-1 Transducer Model of Degenerative and Developmental Brain Disorders. Int. J. Mol. Sci. 2015, 16, 5400-5419. https://doi.org/10.3390/ijms16035400
Schipper HM, Song W. A Heme Oxygenase-1 Transducer Model of Degenerative and Developmental Brain Disorders. International Journal of Molecular Sciences. 2015; 16(3):5400-5419. https://doi.org/10.3390/ijms16035400
Chicago/Turabian StyleSchipper, Hyman M., and Wei Song. 2015. "A Heme Oxygenase-1 Transducer Model of Degenerative and Developmental Brain Disorders" International Journal of Molecular Sciences 16, no. 3: 5400-5419. https://doi.org/10.3390/ijms16035400