
Licochalcone A, a Polyphenol Present in Licorice, Suppresses UV-Induced COX-2 Expression by Targeting PI3K, MEK1, and B-Raf


 and
and 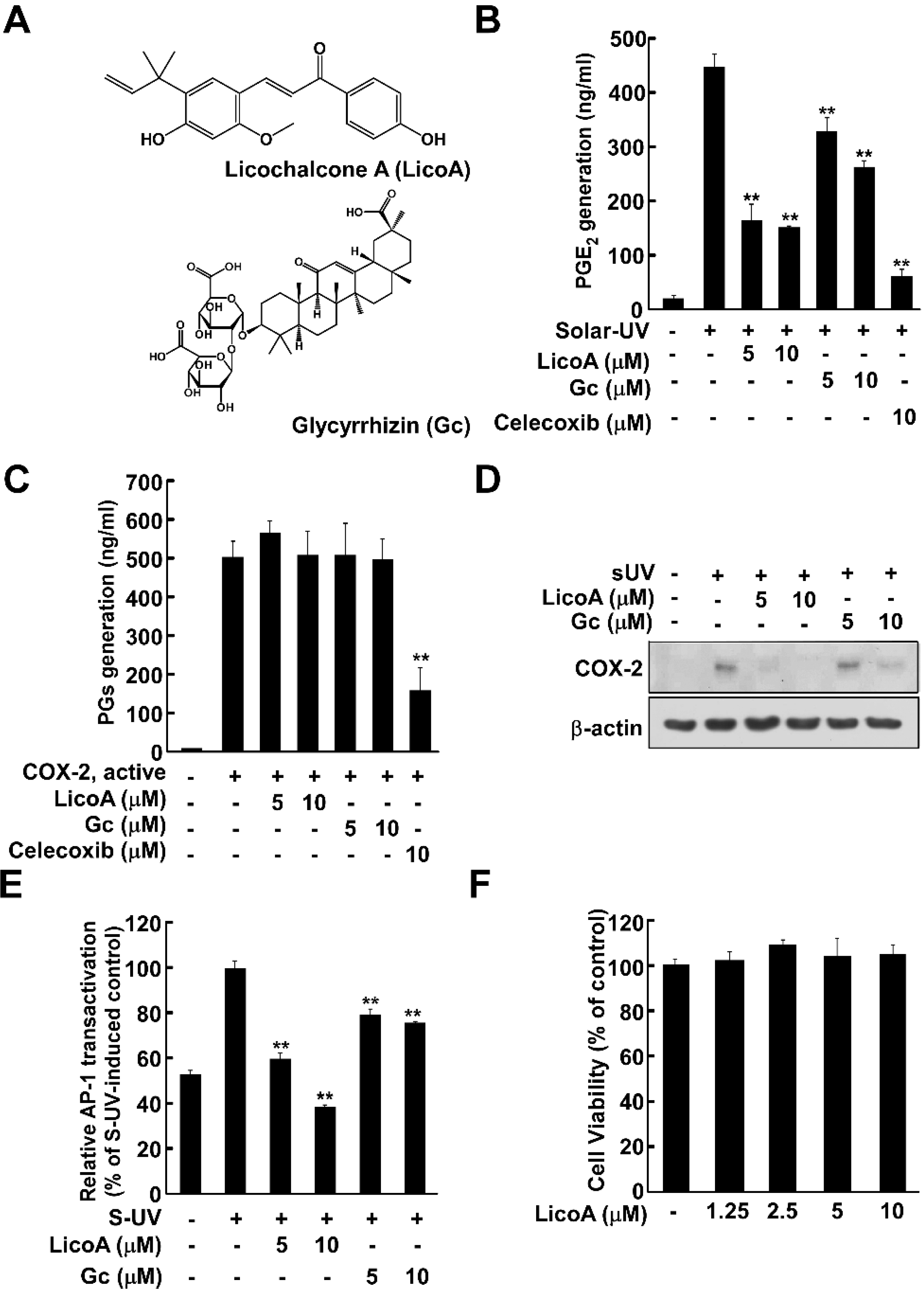{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Licochalcone A (LicoA) Inhibits Cyclooxygenase (COX)-2 Expression and Suppresses Solar Ultraviolet (sUV)-Induced prostaglandin E2 PGE2 Generation More Potently than Glycyrrhizin (Gc)
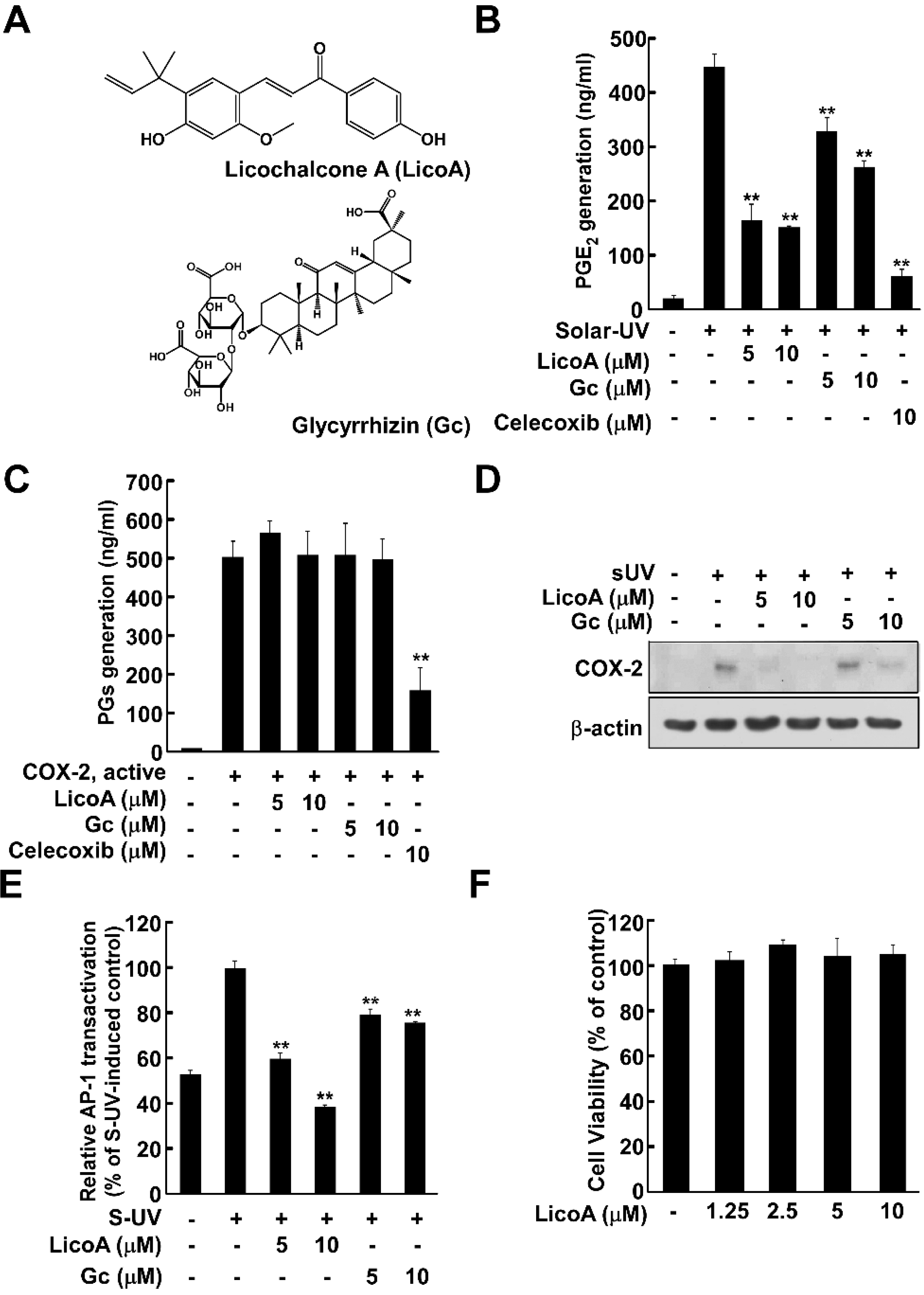
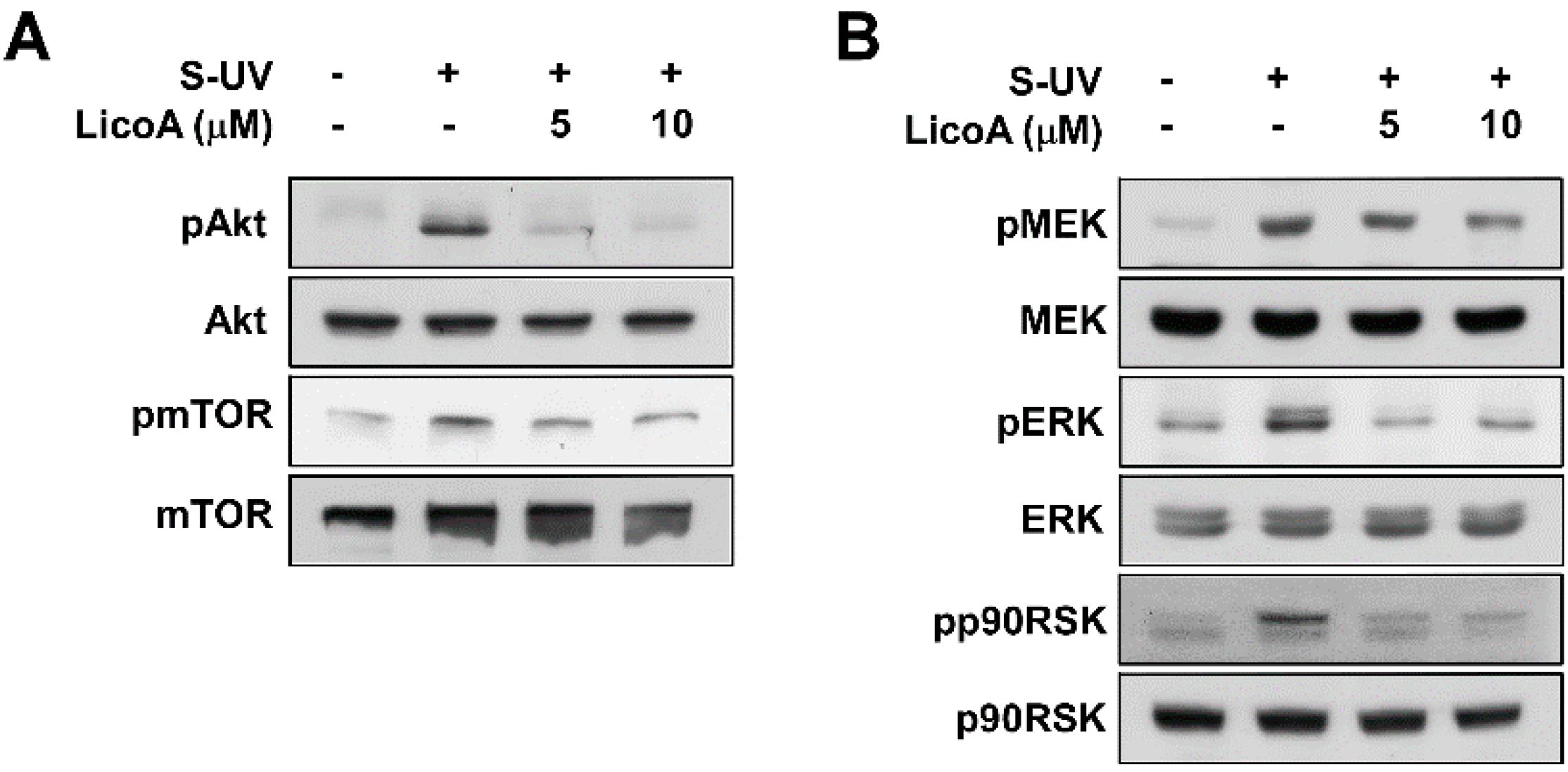
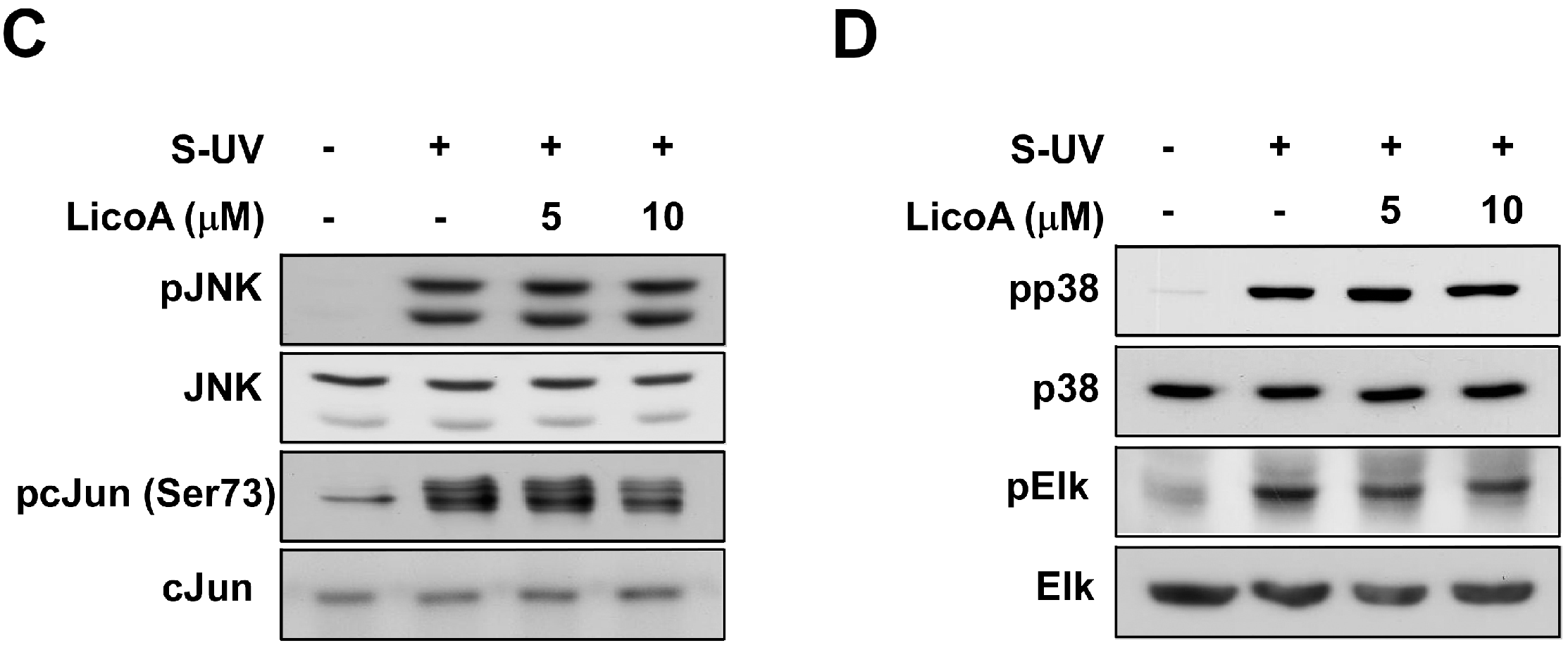
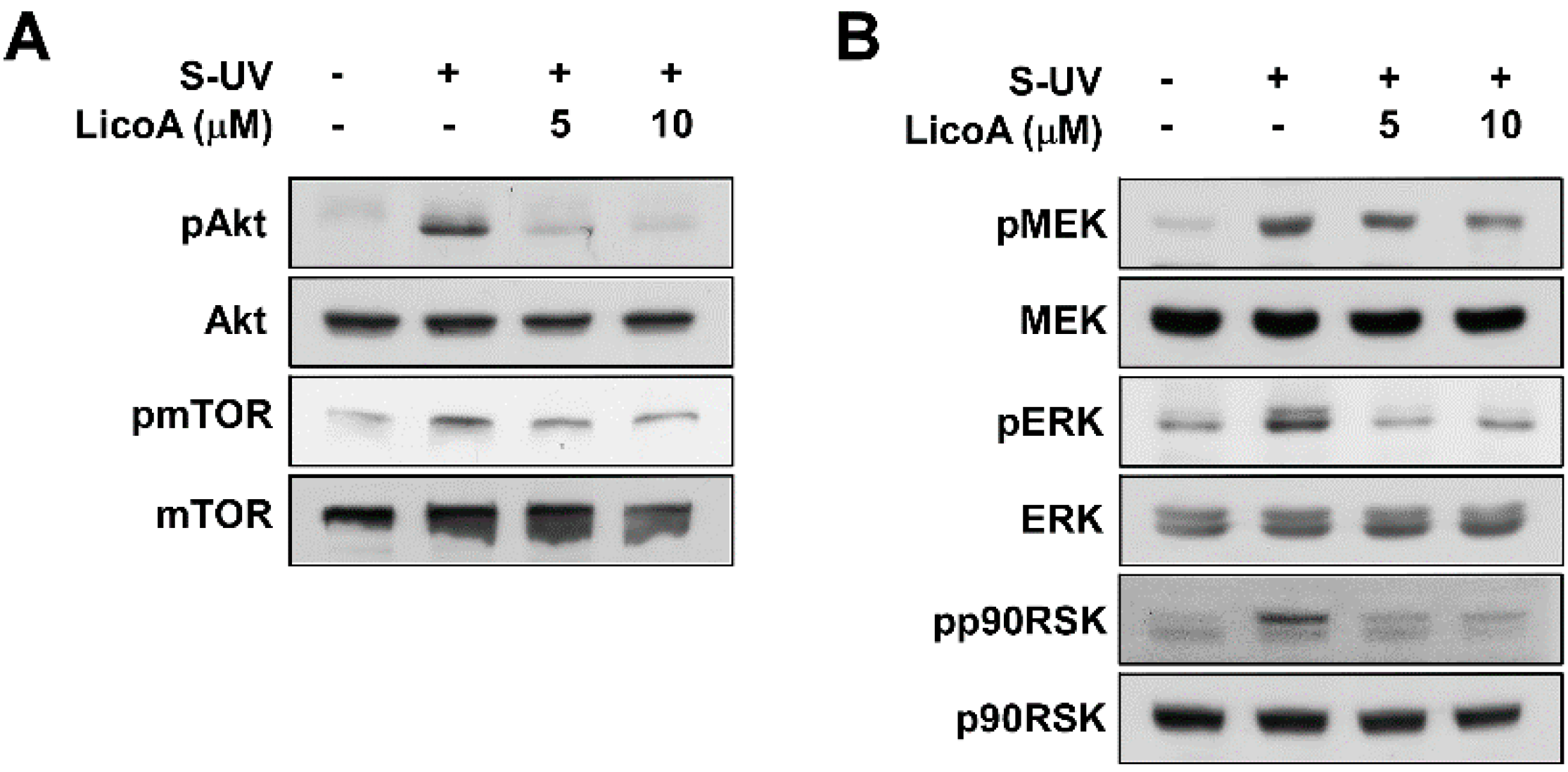
2.2. Lico A Inhibits sUV-Induced Phosphorylation of Akt/ mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase kinase (MEK)1/ extracellular signal-regulated kinases (ERKs)/p90 ribosomal protein S6 kinase (RSK) Pathways in HaCaT Cells

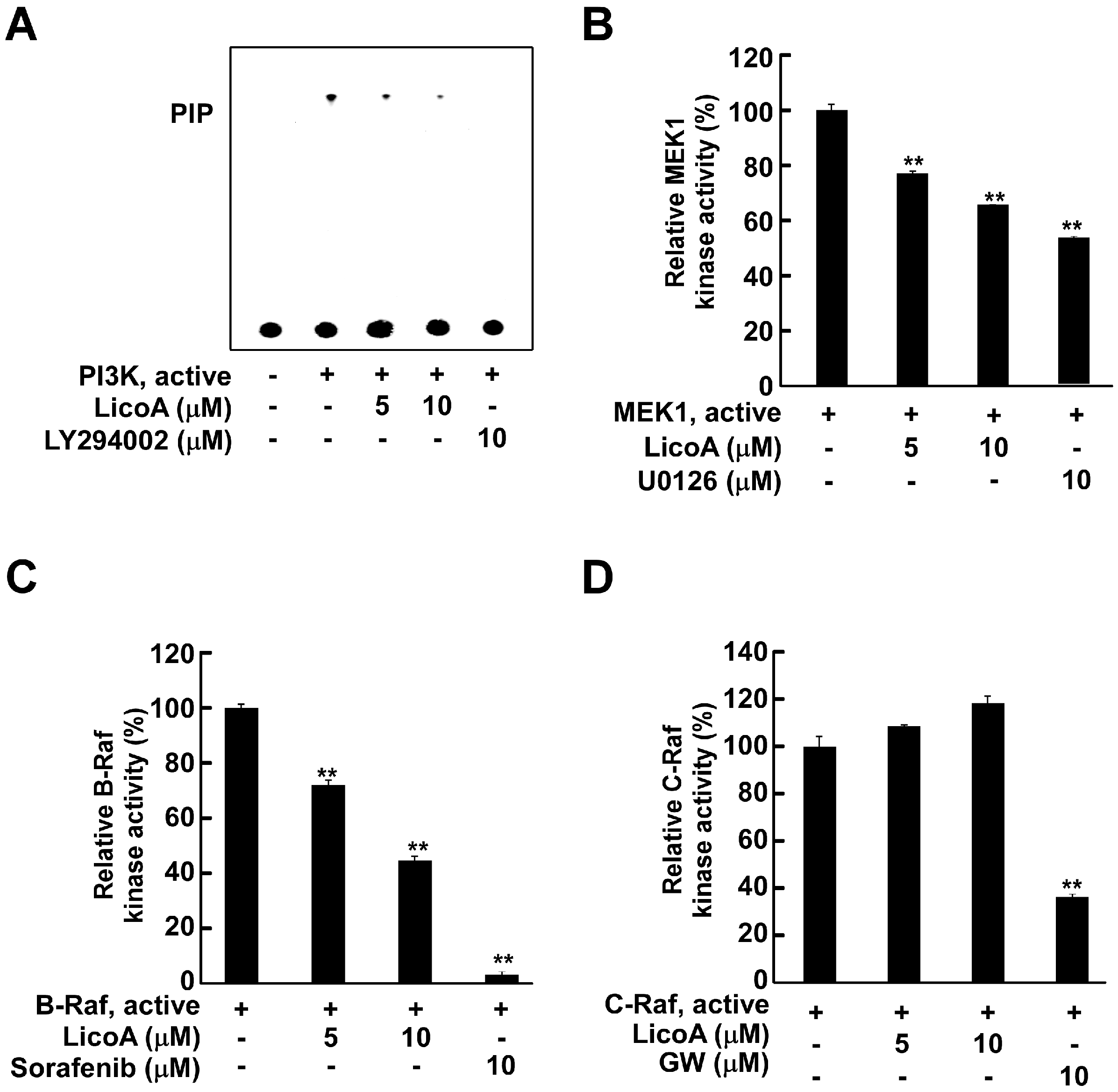
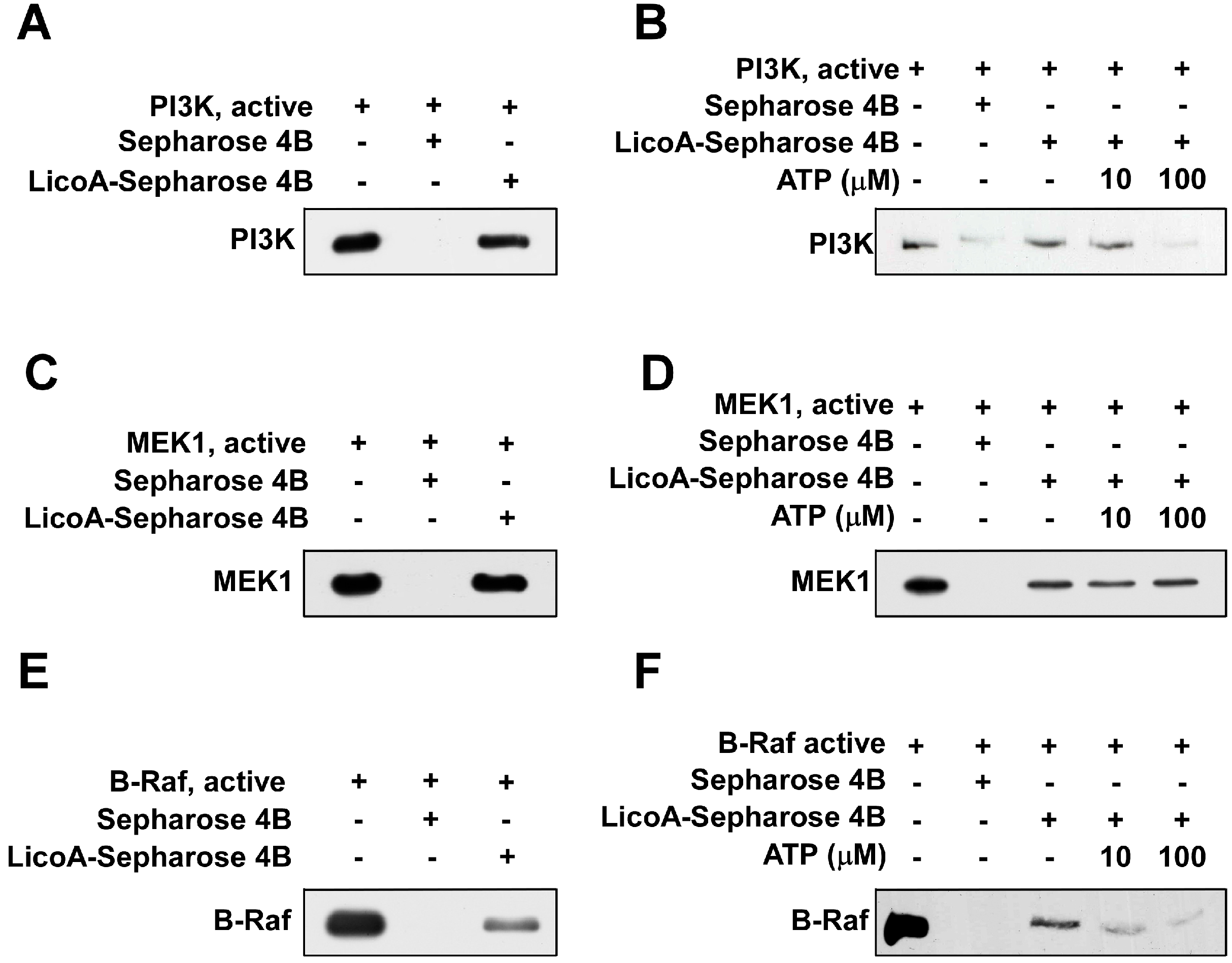
2.3. LicoA Inhibits phosphoinositide 3-kinase PI3K, MEK1, and B-Raf Kinase Activity

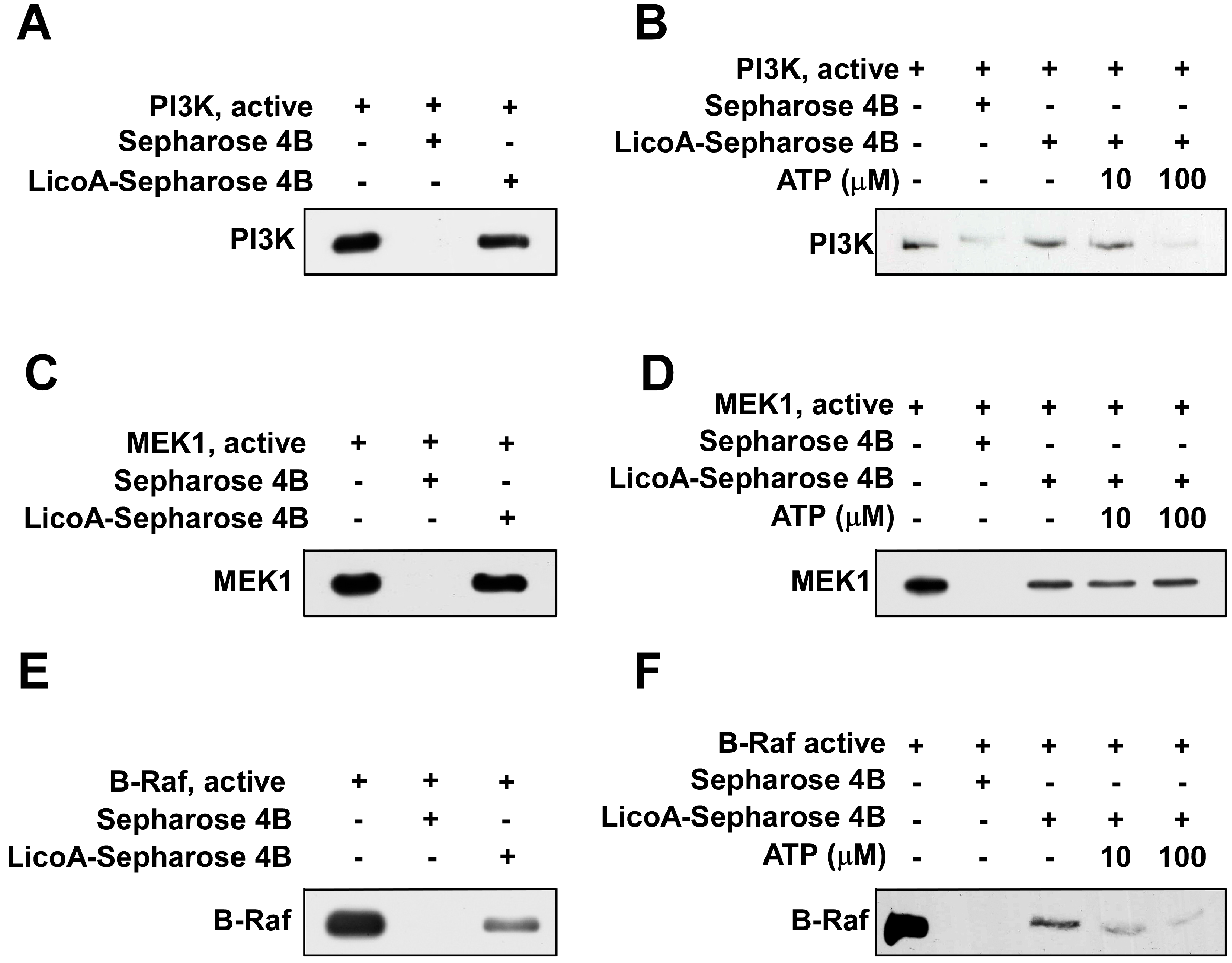
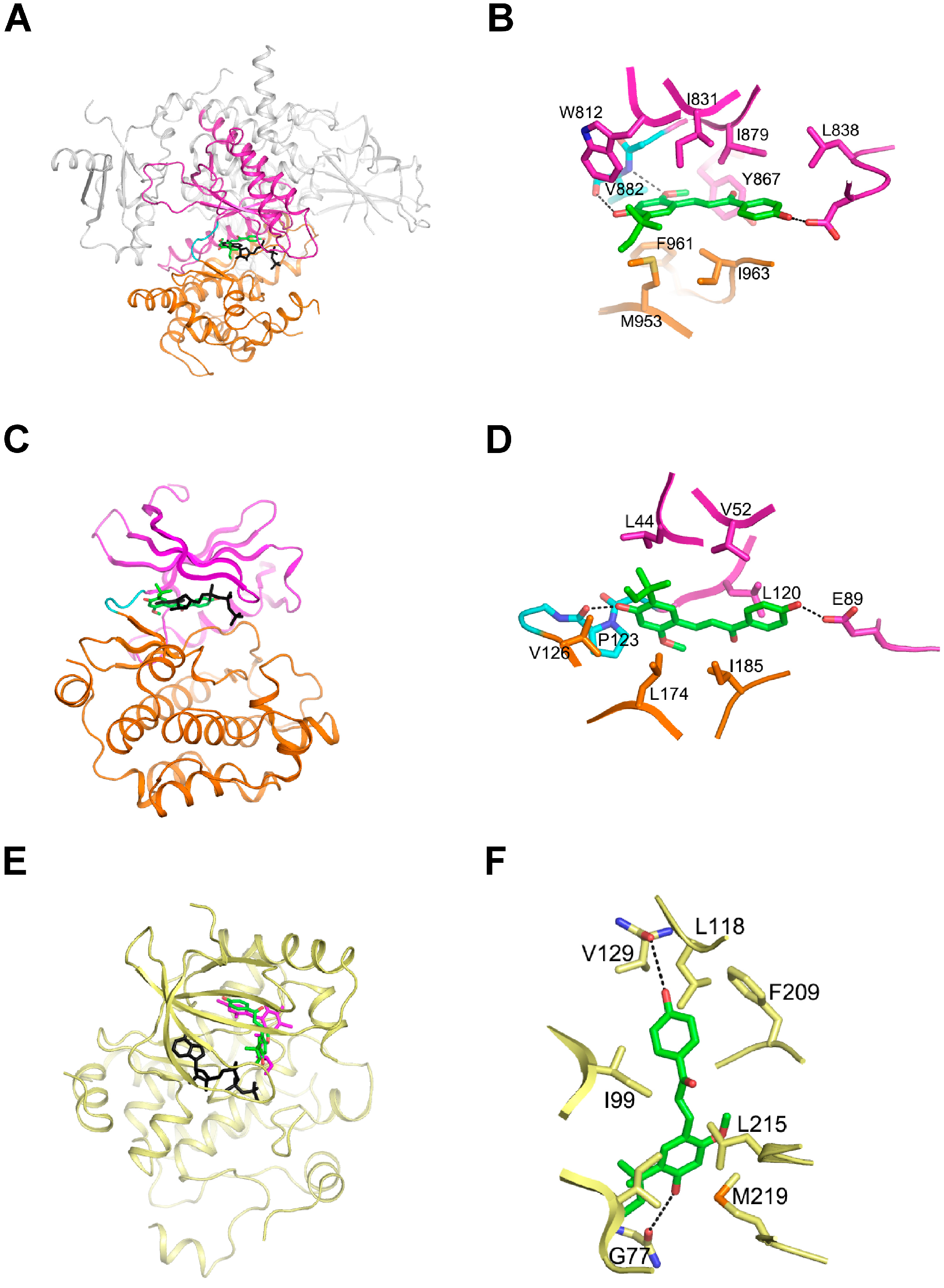
2.4. LicoA Directly Binds to PI3K and B-Raf in an ATP-Competitive Manner and Interacts Directly with MEK1 in an ATP Non-Competitive Manner
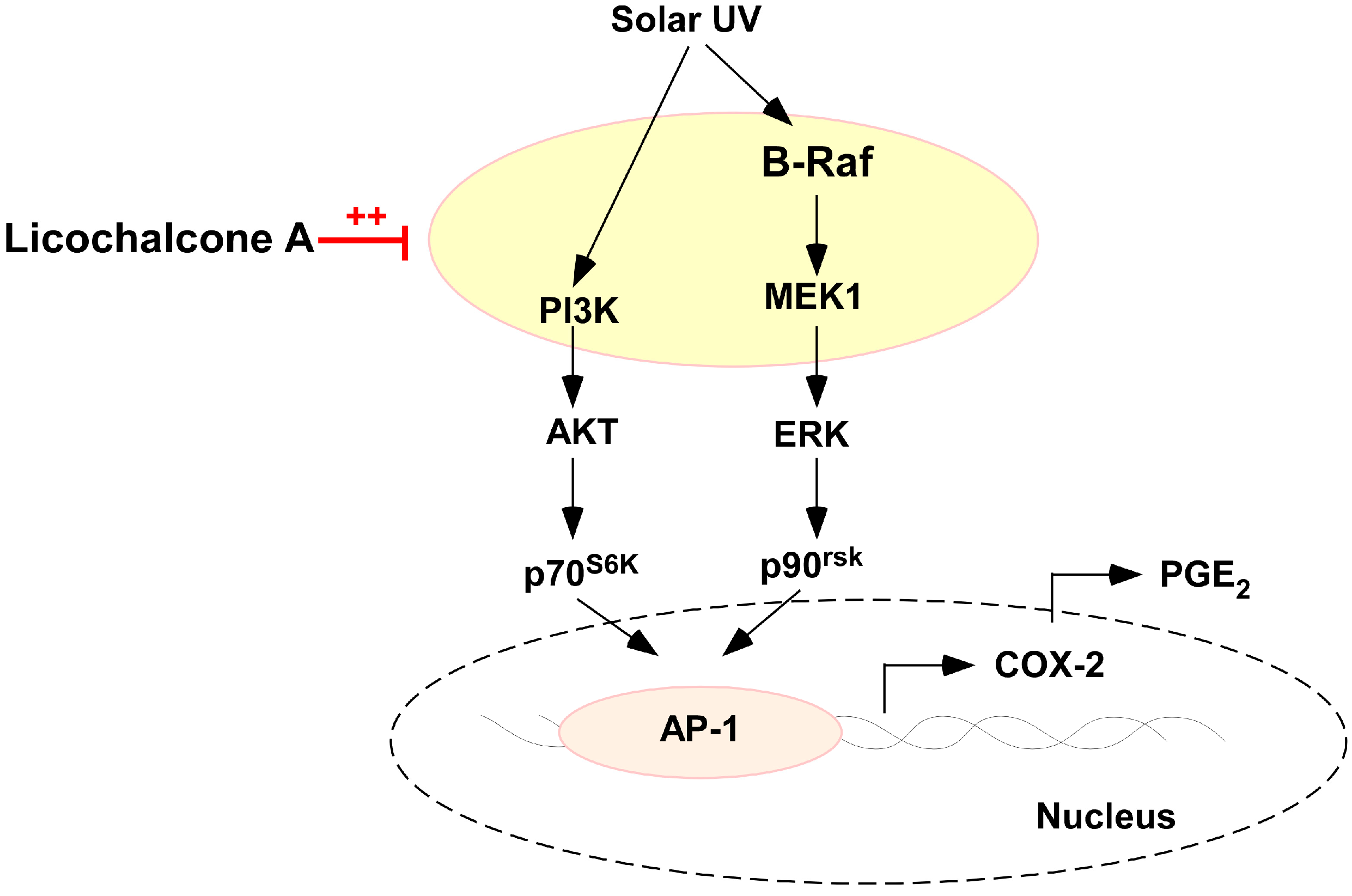
3. Discussion


3. Experimental Section
3.1. Materials
3.2. Cell Culture
3.3. sUV Irradiation
3.4. Cell Viability Assay
3.5. Western Blot Analysis
3.6. Prostaglandin E2 PGE2 Assay
3.7. Luciferase Assay for activator protein 1 AP-1 Transactivation
3.8. PI3K Assay
3.9. MEK1, B-Raf, and C-Raf Kinase Assays
3.10. Immunoprecipitation Assays
3.11. ATP and LicoA Competition Assays
3.12. Molecular Modeling
3.13. Statistical Analysis
Acknowledgments
Author Contributions
Abbreviations
| Gc | glycyrrhizin |
| LicoA | licochalcone A |
| sUV | solar ultraviolet |
| COX | cyclooxygenase |
| ATP | adenosine triphosphate |
| mTOR | mammalian target of rapamycin |
| PIP | phosphatidylinositol-3-phosphate |
| PI3K | phosphatidylinositide 3-kinase |
| PGE2 | prostaglandin E2 |
| AP-1 | activator protein 1 |
| ERK1/2 | extracellular signal-regulated kinases |
| RSK | p90 ribosomal protein S6 kinase |
| MEK | mitogen-activated protein kinase kinase |
| JNK | c-Jun N-terminal kinases |
| MAPKs | mitogen-activated protein kinases |
Conflicts of Interest
References
- Fukai, T.; Marumo, A.; Kaitou, K.; Kanda, T.; Terada, S.; Nomura, T. Anti-Helicobacter pylori flavonoids from licorice extract. Life Sci. 2002, 71, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lim, H.J.; Lee, K.S.; Lee, S.; Kwak, H.J.; Cha, J.H.; Park, H.Y. Anti-proliferative effect of licochalcone A on vascular smooth muscle cells. Biol. Pharm. Bull. 2008, 31, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Nixon, D.W. Licorice and cancer. Nutr. Cancer 2001, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, H.; Taneda, Y.; Chen, W.Z.; Kawanishi, T.; Hori, K.; Taniyama, T.; Kobayashi, M.; Ren, J.; Kitagawa, I. Variation of chemical constituents in processed licorice roots: Quantitative determination of saponin and flavonoid constituents in bark removed and roasted licorice roots. Yakugaku Zasshi 1999, 119, 945–955. [Google Scholar] [PubMed]
- Hayashi, R.; Maruyama, T.; Maruyama, K.; Yanagawa, S.; Tako, K.; Yanagisawa, N. Myotonic and repetitive discharges in hypokalemic myopathy associated with glycyrrhizin-induced hypochloremia. J. Neurol. Sci. 1992, 107, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Shintani, S.; Murase, H.; Tsukagoshi, H.; Shiigai, T. Glycyrrhizin (licorice)-induced hypokalemic myopathy. Report of 2 cases and review of the literature. Eur. Neurol. 1992, 32, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Stormer, F.C.; Reistad, R.; Alexander, J. Glycyrrhizic acid in liquorice—Evaluation of health hazard. Food Chem. Toxicol. 1993, 31, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Kagawa, H.; Yasuhara, T.; Okuda, T. Two new flavonoids and other constituents in licorice root: Their relative astringency and radical scavenging effects. Chem. Pharm. Bull. (Tokyo) 1988, 36, 2090–2097. [Google Scholar] [CrossRef]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E.; Fischer, S.M. Cyclo-oxygenase-2 plays a critical role in UV-induced skin carcinogenesis. Photochem. Photobiol. 2008, 84, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci. STKE 2003, 2003, RE2. [Google Scholar] [PubMed]
- Ley, R.D. Photoreactivation in humans. Proc. Natl. Acad. Sci. USA 1993, 90, 4337. [Google Scholar] [CrossRef] [PubMed]
- Jinlian, L.; Yingbin, Z.; Chunbo, W. p38 MAPK in regulating cellular responses to ultraviolet radiation. J. Biomed. Sci. 2007, 14, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.J. Health-promoting properties of common herbs. Am. J. Clin. Nutr. 1999, 70, 491S–499S. [Google Scholar] [PubMed]
- Armanini, D.; Scali, M.; Zennaro, M.C.; Karbowiak, I.; Wallace, C.; Lewicka, S.; Vecsei, P.; Mantero, F. The pathogenesis of pseudohyperaldosteronism from carbenoxolone. J. Endocrinol. Investig. 1989, 12, 337–341. [Google Scholar] [CrossRef]
- Soro, A.; Panarelli, M.; Holloway, C.D.; Fraser, R.; Kenyon, C.J. In vivo and in vitro effects of carbenoxolone on glucocorticoid receptor binding and glucocorticoid activity. Steroids 1997, 62, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, T.; Kobayashi, M.; Ito, M.; Pollard, R.B.; Suzuki, F. Glycyrrhizin improves the resistance of MAIDS mice to opportunistic infection of Candida albicans through the modulation of MAIDS-associated type 2 T cell responses. Clin. Immunol. 2000, 95, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Liu, Z.W.; Chen, C.P. Protective effects of Glycyrrhizae radix extract and its compounds in a renal hypoxia (ischemia)-reoxygenation (reperfusion) model. Phytomedicine 2000, 6, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Arase, Y.; Ikeda, K.; Murashima, N.; Chayama, K.; Tsubota, A.; Koida, I.; Suzuki, Y.; Saitoh, S.; Kobayashi, M.; Kumada, H. The long term efficacy of glycyrrhizin in chronic hepatitis C patients. Cancer 1997, 79, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, S.; Buhler, H.; Rudolf, M.; Weitzel, H.K.; Ragosch, V. Inhibition of 11 β-hydroxysteroid dehydrogenase activity enhances the antiproliferative effect of glucocorticosteroids on MCF-7 and ZR-75-1 breast cancer cells. J. Endocrinol. 1997, 155, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Kelloff, G.J.; Crowell, J.A.; Boone, C.W.; Steele, V.E.; Lubet, R.A.; Greenwald, P.; Alberts, D.S.; Covey, J.M.; Doody, L.A.; Knapp, G.G.; et al. Clinical development plan: 18β-Glycyrrhetinic acid. J. Cell. Biochem. Suppl. 1994, 20, 166–175. [Google Scholar]
- Wang, Z.Y.; Agarwal, R.; Khan, W.A.; Mukhtar, H. Protection against benzo[a]pyrene- and N-nitrosodiethylamine-induced lung and forestomach tumorigenesis in A/J mice by water extracts of green tea and licorice. Carcinogenesis 1992, 13, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, K.; Takido, M.; Takeuchi, M.; Nakagawa, S. Inhibitory effect of glycyrrhizin and caffeine on two-stage carcinogenesis in mice. Yakugaku Zasshi 1988, 108, 794–796. [Google Scholar] [PubMed]
- Agarwal, R.; Wang, Z.Y.; Mukhtar, H. Inhibition of mouse skin tumor-initiating activity of DMBA by chronic oral feeding of glycyrrhizin in drinking water. Nutr. Cancer 1991, 15, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Kobuke, T.; Inai, K.; Nambu, S.; Ohe, K.; Takemoto, T.; Matsuki, K.; Nishina, H.; Huang, I.B.; Tokuoka, S. Tumorigenicity study of disodium glycyrrhizinate administered orally to mice. Food Chem. Toxicol. 1985, 23, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Nishino, H.; Nishino, A.; Takayasu, J.; Hasegawa, T.; Iwashima, A.; Hirabayashi, K.; Iwata, S.; Shibata, S. Inhibition of the tumor-promoting action of 12-O-tetradecanoylphorbol-13-acetate by some oleanane-type triterpenoid compounds. Cancer Res. 1988, 48, 5210–5215. [Google Scholar] [PubMed]
- Wang, Z.Y.; Agarwal, R.; Zhou, Z.C.; Bickers, D.R.; Mukhtar, H. Inhibition of mutagenicity in Salmonella typhimurium and skin tumor initiating and tumor promoting activities in SENCAR mice by glycyrrhetinic acid: comparison of 18 α- and 18 β-stereoisomers. Carcinogenesis 1991, 12, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Johns, C. Glycyrrhizic acid toxicity caused by consumption of licorice candy cigars. CJEM 2009, 11, 94–96. [Google Scholar] [PubMed]
- Jia, S.S.; Ma, C.M.; Li, Y.H.; Hao, J.H. Glycosides of phenolic acid and flavonoids from the leaves of Glycyrrhiza uralensis Ficsh. Yao Xue Xue Bao 1992, 27, 441–444. [Google Scholar] [PubMed]
- Liu, Q.; Liu, Y. Application of 13CNMR to structural identification of the flavonoid glycosides. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 1990, 12, 359–364. [Google Scholar] [PubMed]
- Yang, L.; Liu, Y.L.; Lin, S.Q. HPLC analysis of flavonoids in the root of six Glycyrrhiza species. Yao Xue Xue Bao 1990, 25, 840–848. [Google Scholar] [PubMed]
- Szliszka, E.; Czuba, Z.P.; Mazur, B.; Sedek, L.; Paradysz, A.; Krol, W. Chalcones enhance TRAIL-induced apoptosis in prostate cancer cells. Int. J. Mol. Sci. 2009, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.Y.; Hao, M.; Yang, X.Y.; Ba, Q.; Li, M.; Ni, S.J.; Wang, L.S.; Du, X. Licochalcone A inhibits growth of gastric cancer cells by arresting cell cycle progression and inducing apoptosis. Cancer Lett. 2011, 302, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Yo, Y.T.; Shieh, G.S.; Hsu, K.F.; Wu, C.L.; Shiau, A.L. Licorice and licochalcone-A induce autophagy in LNCaP prostate cancer cells by suppression of Bcl-2 expression and the mTOR pathway. J. Agric. Food Chem. 2009, 57, 8266–8273. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, J.; Funakoshi-Tago, M.; Tago, K.; Mashino, T.; Inoue, H.; Sonoda, Y.; Kasahara, T. Licochalcone A significantly suppresses LPS signaling pathway through the inhibition of NF-κB p65 phosphorylation at serine 276. Cell Signal. 2009, 21, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Park, J.H.; Kim, D.H.; Kim, Y.H.; Shin, H.K.; Kim, J.K. Licochalcone A isolated from licorice suppresses lipopolysaccharide-stimulated inflammatory reactions in RAW264.7 cells and endotoxin shock in mice. J. Mol. Med. (Berl) 2008, 86, 1287–1295. [Google Scholar] [CrossRef]
- Muller-Decker, K.; Furstenberger, G. The cyclooxygenase-2-mediated prostaglandin signaling is causally related to epithelial carcinogenesis. Mol. Carcinog. 2007, 46, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Muller-Decker, K.; Neufang, G.; Berger, I.; Neumann, M.; Marks, F.; Furstenberger, G. Transgenic cyclooxygenase-2 overexpression sensitizes mouse skin for carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 12483–12488. [Google Scholar] [CrossRef] [PubMed]
- Tiano, H.F.; Loftin, C.D.; Akunda, J.; Lee, C.A.; Spalding, J.; Sessoms, A.; Dunson, D.B.; Rogan, E.G.; Morham, S.G.; Smart, R.C.; et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002, 62, 3395–3401. [Google Scholar] [PubMed]
- Kang, Y.J.; Wingerd, B.A.; Arakawa, T.; Smith, W.L. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J. Immunol. 2006, 177, 8111–8122. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Vandoros, G.P.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papavassiliou, A.G. NF-κB/PPARγ and/or AP-1/PPARγ “on/off” switches and induction of CBP in colon adenocarcinomas: Correlation with COX-2 expression. Int. J. Colorectal. Dis. 2007, 22, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.J.; Bowden, G.T. Ultraviolet B regulation of transcription factor families: Roles of nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr. Cancer Drug Targets 2007, 7, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Gonzales, M.; Inoue, H.; Bowden, G.T. Roles of Akt and glycogen synthase kinase 3β in the ultraviolet B induction of cyclooxygenase-2 transcription in human keratinocytes. Cancer Res. 2001, 61, 4329–4332. [Google Scholar] [PubMed]
- Chen, W.; Tang, Q.; Gonzales, M.S.; Bowden, G.T. Role of p38 MAP kinases and ERK in mediating ultraviolet-B induced cyclooxygenase-2 gene expression in human keratinocytes. Oncogene 2001, 20, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kwon, J.Y.; Seo, S.K.; Son, J.E.; Jung, S.K.; Min, S.Y.; Hwang, M.K.; Heo, Y.S.; Lee, K.W.; Lee, H.J. Cyanidin suppresses ultraviolet B-induced COX-2 expression in epidermal cells by targeting MKK4, MEK1, and Raf-1. Biochem. Pharmacol. 2010, 79, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K.; Kang, S. Kinase inhibitors: Vice becomes virtue. Cancer Cell 2006, 9, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Sebolt-Leopold, J.S.; English, J.M. Mechanisms of drug inhibition of signalling molecules. Nature 2006, 441, 457–462. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, N.R.; Kim, J.-E.; Park, J.S.; Kim, J.R.; Kang, H.; Lee, E.; Kang, Y.-G.; Son, J.E.; Seo, S.G.; Heo, Y.S.; et al. Licochalcone A, a Polyphenol Present in Licorice, Suppresses UV-Induced COX-2 Expression by Targeting PI3K, MEK1, and B-Raf. Int. J. Mol. Sci. 2015, 16, 4453-4470. https://doi.org/10.3390/ijms16034453
Song NR, Kim J-E, Park JS, Kim JR, Kang H, Lee E, Kang Y-G, Son JE, Seo SG, Heo YS, et al. Licochalcone A, a Polyphenol Present in Licorice, Suppresses UV-Induced COX-2 Expression by Targeting PI3K, MEK1, and B-Raf. International Journal of Molecular Sciences. 2015; 16(3):4453-4470. https://doi.org/10.3390/ijms16034453
Chicago/Turabian StyleSong, Nu Ry, Jong-Eun Kim, Jun Seong Park, Jong Rhan Kim, Heerim Kang, Eunjung Lee, Young-Gyu Kang, Joe Eun Son, Sang Gwon Seo, Yong Seok Heo, and et al. 2015. "Licochalcone A, a Polyphenol Present in Licorice, Suppresses UV-Induced COX-2 Expression by Targeting PI3K, MEK1, and B-Raf" International Journal of Molecular Sciences 16, no. 3: 4453-4470. https://doi.org/10.3390/ijms16034453
APA StyleSong, N. R., Kim, J.-E., Park, J. S., Kim, J. R., Kang, H., Lee, E., Kang, Y.-G., Son, J. E., Seo, S. G., Heo, Y. S., & Lee, K. W. (2015). Licochalcone A, a Polyphenol Present in Licorice, Suppresses UV-Induced COX-2 Expression by Targeting PI3K, MEK1, and B-Raf. International Journal of Molecular Sciences, 16(3), 4453-4470. https://doi.org/10.3390/ijms16034453