
Transcriptome Analysis of Methyl Jasmonate-Elicited Panax ginseng Adventitious Roots to Discover Putative Ginsenoside Biosynthesis and Transport Genes

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
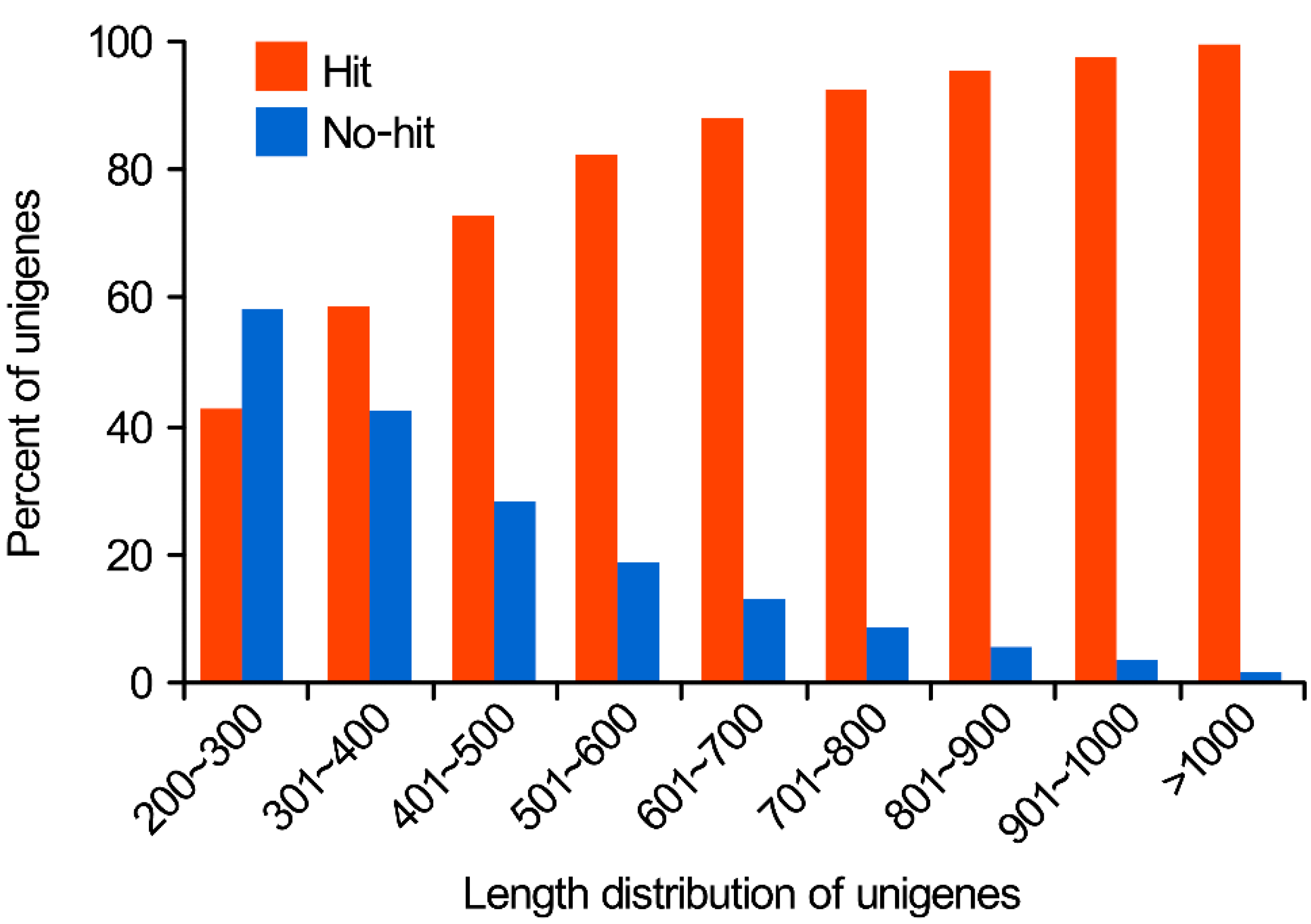
2.1.1. Sequencing Output and Transcriptome Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
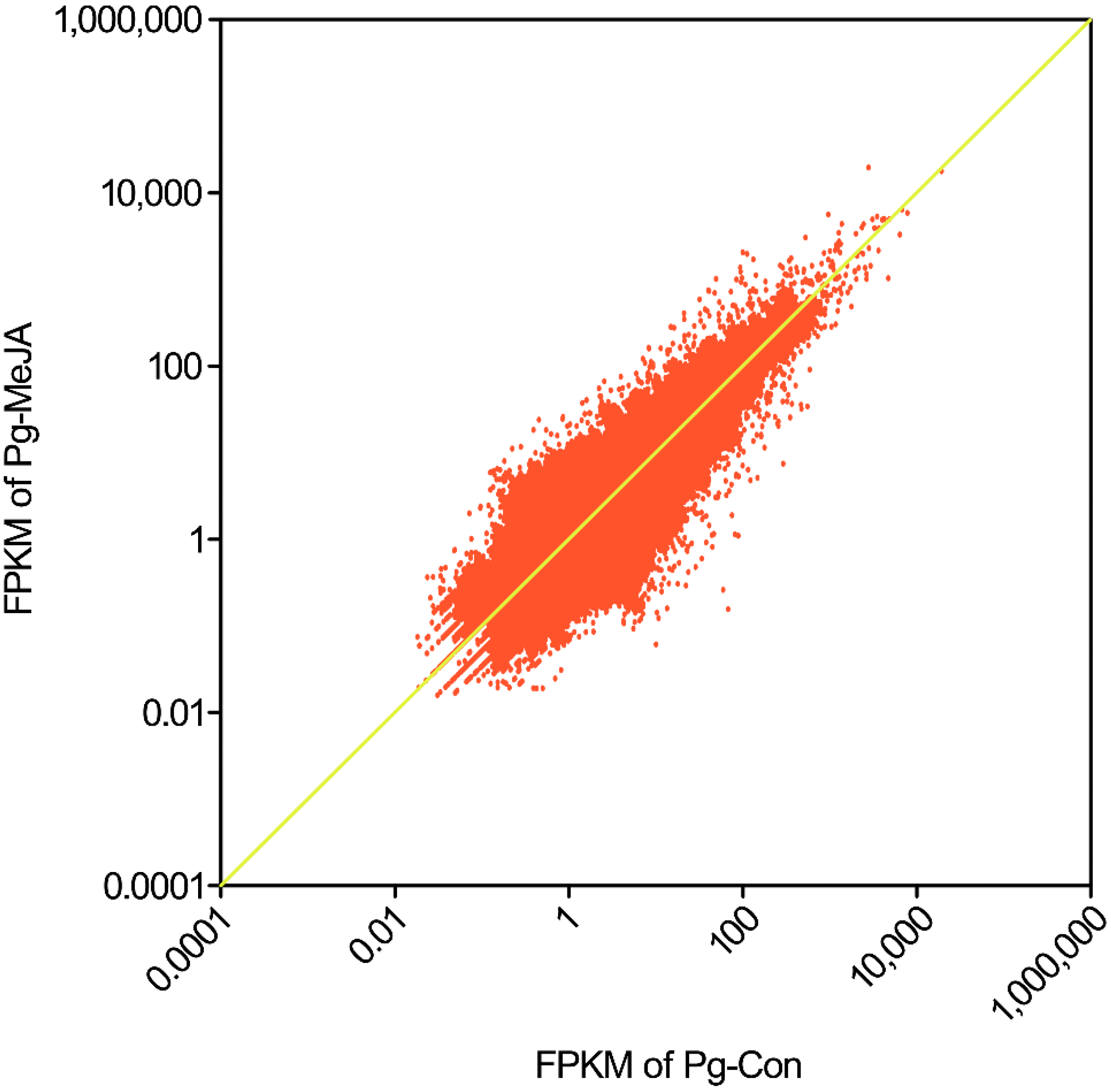{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length of Nucleotides (bp) | Contigs | Unigenes | All-unigenes | ||
|---|---|---|---|---|---|
| Pg-Con | Pg-MeJA | Pg-Con | Pg-MeJA | ||
| 0–199 | 85,621 | 82,631 | 14,443 | 12,401 | 0 |
| 200–299 | 16,761 | 17,177 | 12,719 | 13,661 | 13,923 |
| 300–399 | 9576 | 9502 | 9213 | 9269 | 9976 |
| 400–499 | 5430 | 5063 | 5909 | 5715 | 6472 |
| 500–599 | 3330 | 3359 | 4428 | 4400 | 4916 |
| 600–699 | 2581 | 2429 | 3568 | 3622 | 4180 |
| 700–799 | 1897 | 1806 | 3039 | 2882 | 3459 |
| 800–899 | 1611 | 1538 | 2738 | 2608 | 3089 |
| 900–999 | 1351 | 1270 | 2356 | 2283 | 2789 |
| 1000–1099 | 1129 | 1049 | 2081 | 2033 | 2476 |
| 1100–1199 | 969 | 910 | 1899 | 1870 | 2321 |
| 1200–1299 | 864 | 761 | 1642 | 1582 | 2006 |
| 1300–1399 | 753 | 670 | 1572 | 1435 | 1862 |
| 1400–1499 | 686 | 641 | 1387 | 1276 | 1731 |
| 1500–1599 | 593 | 544 | 1179 | 1218 | 1558 |
| 1600–1699 | 519 | 482 | 1111 | 1083 | 1441 |
| 1700–1799 | 496 | 384 | 997 | 932 | 1273 |
| 1800–1899 | 395 | 383 | 905 | 875 | 1189 |
| 1900–1999 | 322 | 285 | 688 | 639 | 920 |
| 2000–2099 | 257 | 234 | 597 | 537 | 748 |
| 2100–2199 | 248 | 193 | 481 | 425 | 638 |
| 2200–2299 | 194 | 159 | 478 | 340 | 571 |
| 2300–2399 | 177 | 154 | 357 | 306 | 482 |
| 2400–2499 | 119 | 109 | 255 | 296 | 407 |
| 2500–2599 | 120 | 96 | 272 | 237 | 374 |
| 2600–2699 | 108 | 99 | 239 | 223 | 311 |
| 2700–2799 | 88 | 64 | 184 | 154 | 242 |
| 2800–2899 | 81 | 56 | 173 | 133 | 223 |
| 2900–2999 | 56 | 57 | 125 | 133 | 192 |
| ≥3000 | 444 | 369 | 921 | 871 | 1326 |
| Total Number | 136,776 | 132,474 | 75,956 | 73,439 | 71,095 |
| Total length (bp) | 43,209,082 | 40,612,192 | 52,997,248 | 50,685,207 | 61,942,217 |
| Mean length (bp) | 316 | 307 | 698 | 690 | 871 |
| N50 (bp) | 525 | 488 | 1161 | 1130 | 1307 |
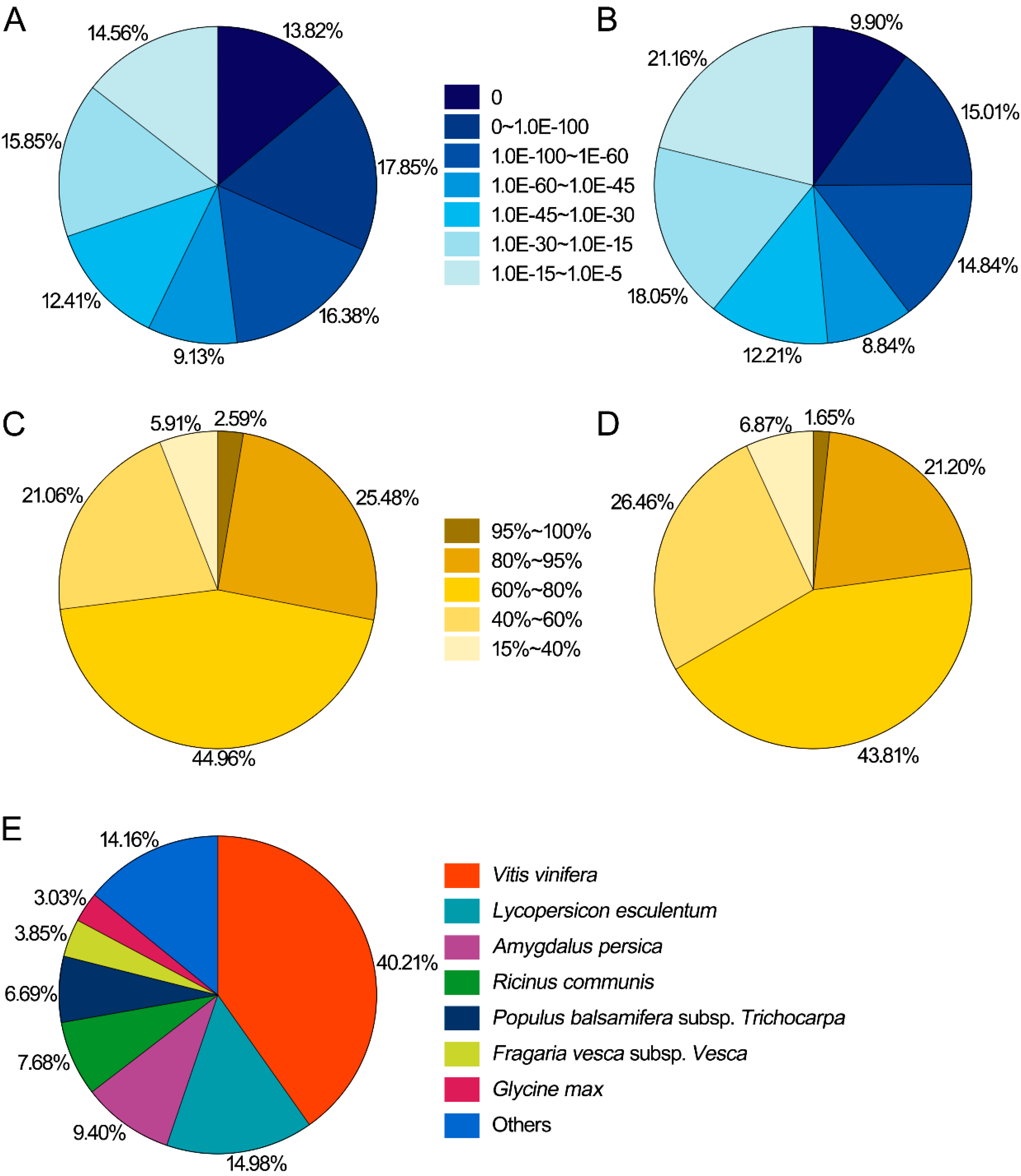
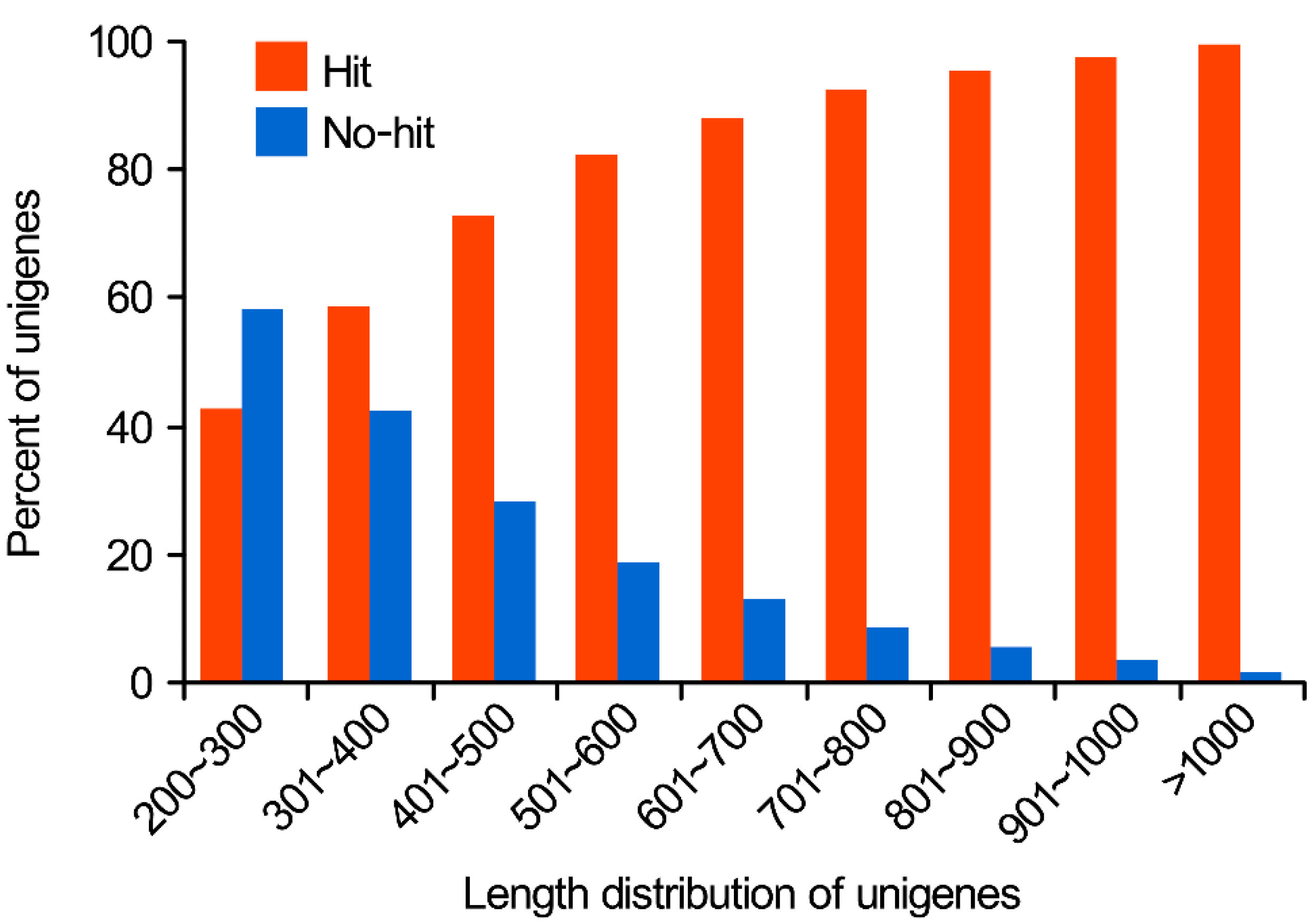
2.1.2. Functional Annotation of Predicted Proteins in Nr and Swiss-Prot Databases
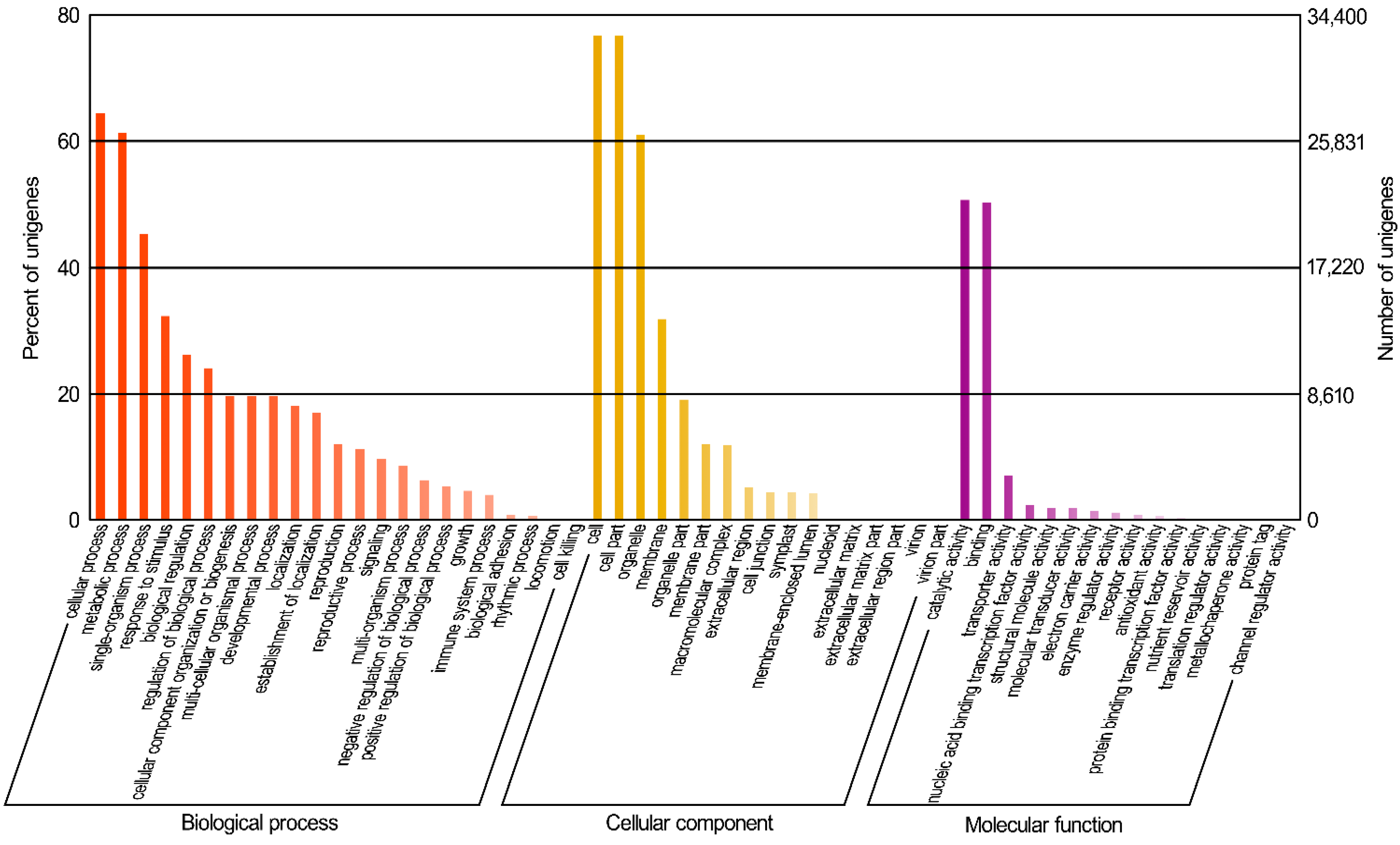
2.1.3. GO (Gene Ontology) Functional Classification


2.1.4. COG (Clusters of Orthologous Groups) Functional Classification
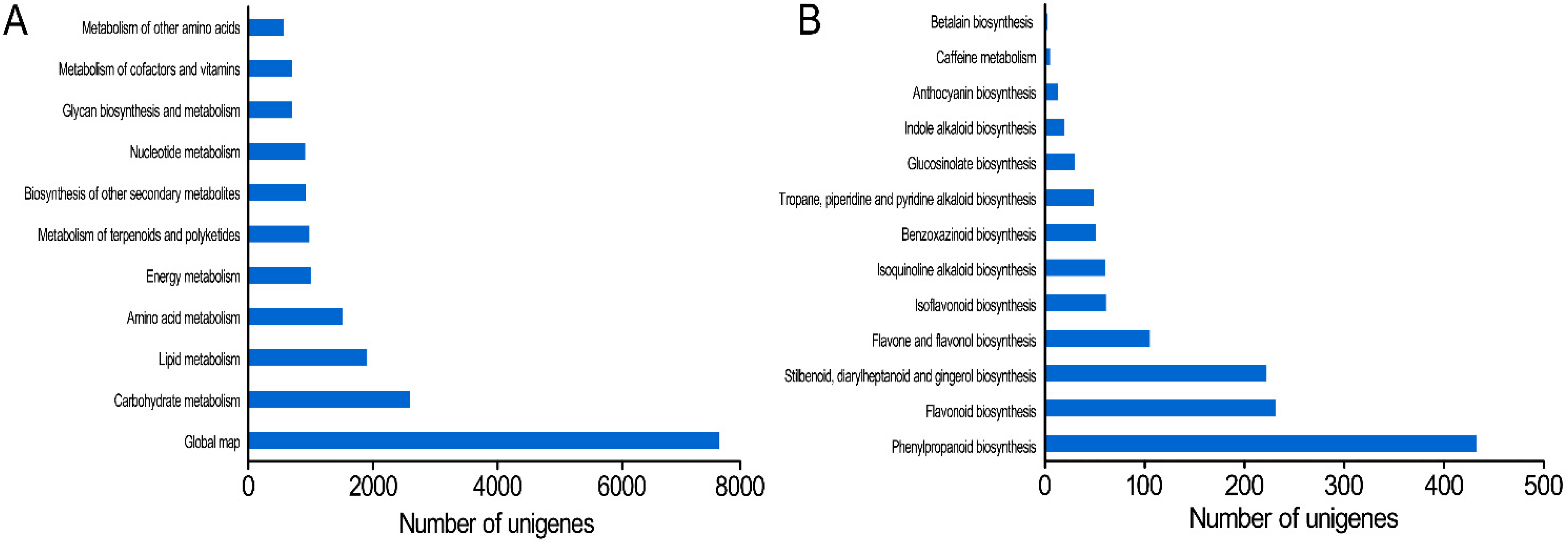
2.1.5. KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway Annotation


2.1.6. Gene Expression Profiling

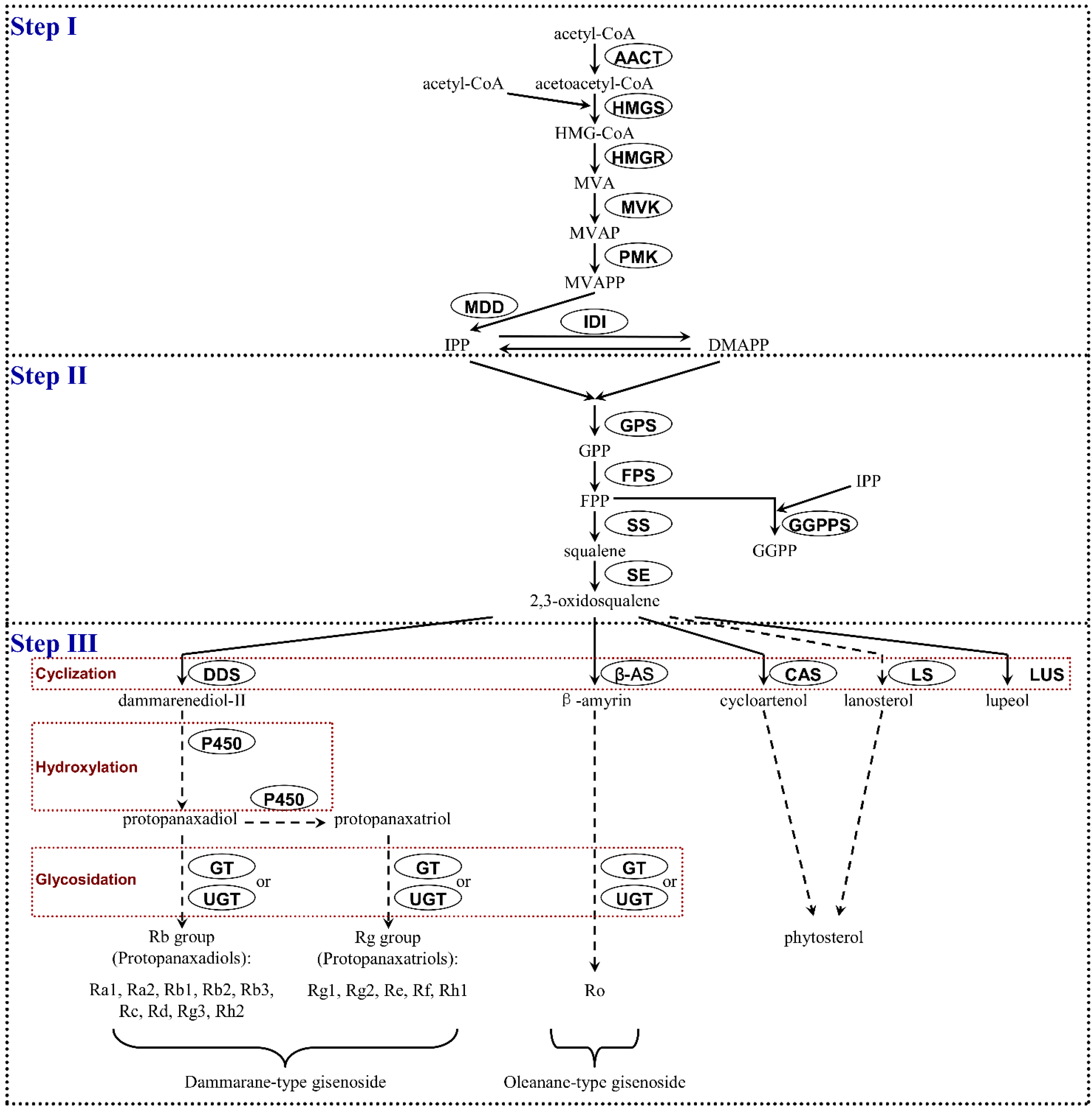
2.1.7. Identification of Candidate Genes Involved in Ginsenoside Biosynthesis
| Ginsenoside Biosynthesis Steps | Name of Enzymes | Number of Unigenes | Up-Regulated | Down-Regulated | Not DEGs |
|---|---|---|---|---|---|
| Step I | AACT | 3 | 1 | 2 | - |
| HMGS | 9 | 2 | 7 | - | |
| HMGR | 15 | 4 | 11 | - | |
| MVK | 20 | 14 | 6 | - | |
| PMK | 16 | 10 | 6 | - | |
| MDD | 2 | 1 | 1 | - | |
| IDI | 2 | 1 | 1 | - | |
| Step II | GPS | 24 | 8 | 16 | - |
| FPS | 1 | 1 | 0 | - | |
| GGPPS | 14 | 6 | 8 | - | |
| SS | 10 | 10 | 0 | - | |
| SE | 11 | 7 | 4 | - | |
| Step III | DDS | 1 | 1 | 0 | - |
| β-AS | 9 | 2 | 7 | - | |
| CAS | 17 | 1 | 16 | - | |
| LS | 2 | 0 | 2 | - | |
| P450 | 335 | 161 | 168 | 6 | |
| GT | 142 | 48 | 92 | 2 | |
| UGT | 116 | 71 | 45 | - |


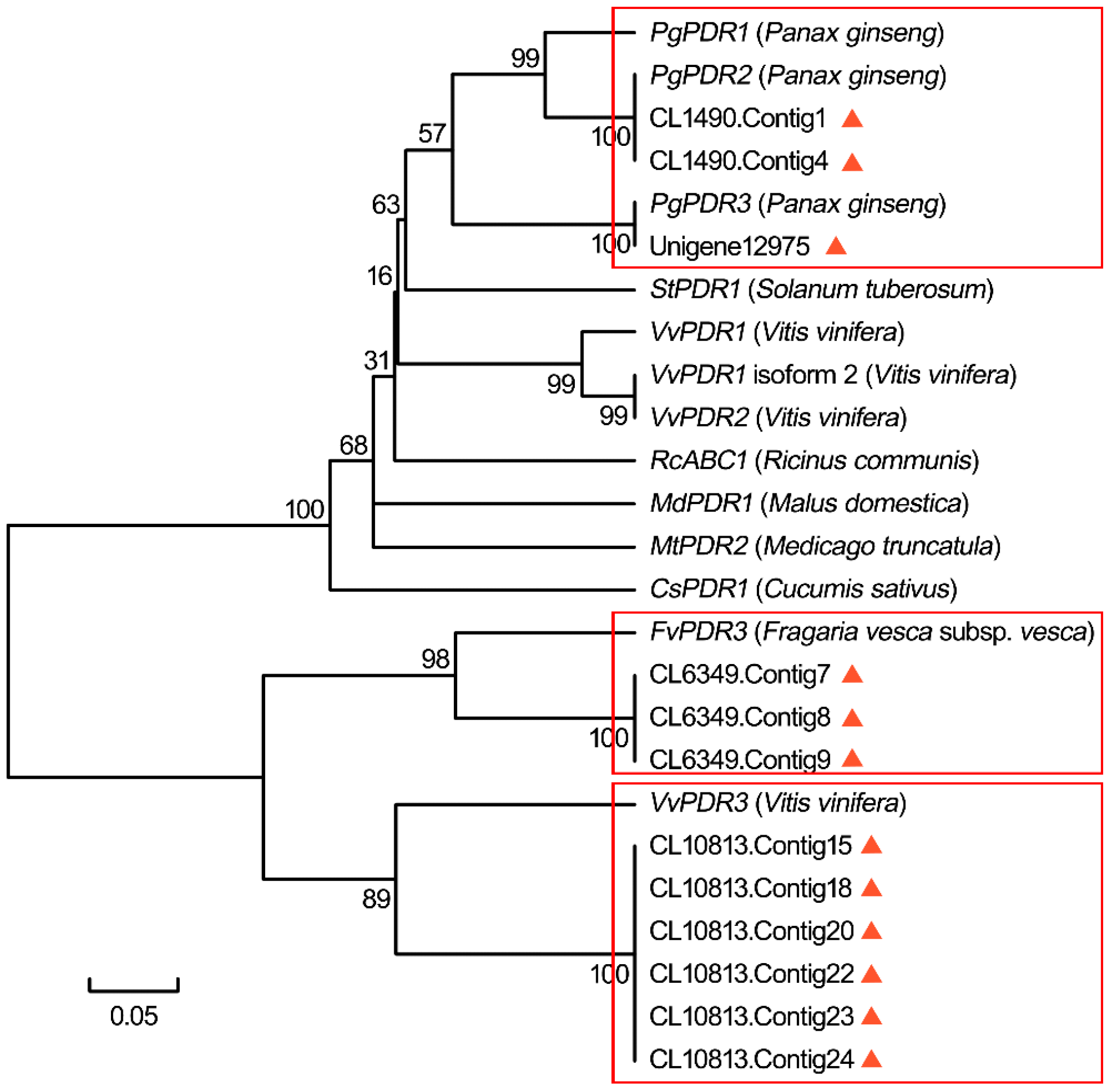
2.1.8. Identification of Candidate Genes Involved in Ginsenoside Transport
2.2. Discussion

3. Experimental Section
3.1. Preparation of Plant Materials
3.2. RNA Extraction and cDNA Library Construction for Sequencing
3.3. Data Filtering and Sequence Assembly
3.4. Protein Coding Sequence (CDS) Prediction
3.5. Unigene Annotation and Classification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wen, J.; Zimmer, E.A. Phylogeny and biogeography of Panax L. (the ginseng genus, araliaceae): Inferences from ITS sequences of nuclear ribosomal DNA. Mol. Phylogenet. Evol. 1996, 6, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.K. Brief introduction of Panax ginseng C.A. Meyer. J. Korean Med. Sci. 2001, 16 (Suppl.), S3–S5. [Google Scholar] [CrossRef]
- Yan, X.; Fan, Y.; Wei, W.; Wang, P.; Liu, Q.; Wei, Y.; Zhang, L.; Zhao, G.; Yue, J.; Zhou, Z. Production of bioactive ginsenoside compound K in metabolically engineered yeast. Cell Res. 2014, 24, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.F.; Zhang, J.T. New achievements in ginseng research and its future prospects. Chin. J. Integr. Med. 2009, 15, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Du, G.J.; Wang, C.Z.; Wen, X.D.; Calway, T.; Li, Z.; He, T.C.; Du, W.; Bissonnette, M.; Musch, M.W.; et al. Compound K, a ginsenoside metabolite, inhibits colon cancer growth via multiple pathways including p53-p21 interactions. Int. J. Mol. Sci. 2013, 14, 2980–2995. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, M.S.; Kim, C.T.; Kim, I.H.; Kim, Y. Ginsenoside Rg3 reduces lipid accumulation with AMP-activated protein kinase (AMPK) activation in HepG2 cells. Int. J. Mol. Sci. 2012, 13, 5729–5739. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Murthy, H.N.; Hahn, E.J.; Lee, H.L.; Paek, K.Y. Parameters affecting the extraction of ginsenosides from the adventitious roots of ginseng (Panax ginseng C.A. Meyer). Sep. Purif. Technol. 2007, 56, 401–406. [Google Scholar] [CrossRef]
- Chen, C.F.; Chiou, W.F.; Zhang, J.T. Comparison of the pharmacological effects of Panax ginseng and Panax quinquefolium. Acta Pharmacol. Sin. 2008, 29, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Sun, Y.Z.; Xu, J.; Luo, H.M.; Sun, C.; He, L.; Cheng, X.L.; Zhang, B.L.; Xiao, P.G. Strategies of the study on herb genome program. Acta Pharmacol. Sin. 2010, 45, 807–812. [Google Scholar]
- Choi, D.W.; Jung, J.; Ha, Y.I.; Park, H.W.; In, D.S.; Chung, H.J.; Liu, J.R. Analysis of transcripts in methyl jasmonate-treated ginseng hairy roots to identify genes involved in the biosynthesis of ginsenosides and other secondary metabolites. Plant Cell Rep. 2005, 23, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, S.; In, J.G.; Gayathri, S.; Kim, Y.J.; Yang, D.C. Generation and gene ontology based analysis of expressed sequence tags (EST) from a Panax ginseng C.A. Meyer roots. Mol. Biol. Rep. 2010, 37, 3465–3472. [Google Scholar] [CrossRef] [PubMed]
- Sathiyamoorthy, S.; In, J.G.; Gayathri, S.; Kim, Y.J.; Yang, D. Gene ontology study of methyl jasmonate-treated and non-treated hairy roots of Panax ginseng to identify genes involved in secondary metabolic pathway. Genetika 2010, 46, 932–939. [Google Scholar] [PubMed]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Jayakodi, M.; Lee, S.C.; Park, H.S.; Jang, W.; Lee, Y.S.; Choi, B.S.; Nah, G.J.; Kim, D.S.; Natesan, S.; Sun, C.; et al. Transcriptome profiling and comparative analysis of Panax ginseng adventitious roots. J. Ginseng Res. 2014, 38, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, Y.; Guo, X.; Sun, C.; Luo, H.; Song, J.; Li, Y.; Wang, L.; Qian, J.; Chen, S. Transcriptome analysis reveals ginsenosides biosynthetic genes, microRNAs and simple sequence repeats in Panax ginseng C.A. Meyer. BMC Genomics 2013, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Sun, C.; Sun, Y.; Wu, Q.; Li, Y.; Song, J.; Niu, Y.; Cheng, X.; Xu, H.; Li, C.; et al. Analysis of the transcriptome of Panax notoginseng root uncovers putative triterpene saponin-biosynthetic genes and genetic markers. BMC Genomics 2011, 12 (Suppl. 5), S5. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, Y.; Wu, Q.; Luo, H.; Sun, Y.; Song, J.; Lui, E.M.; Chen, S. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genomics 2010, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; In, J.G.; Kwon, Y.S.; Choi, Y.E. Regulation of ginsenoside and phytosterol biosynthesis by RNA interferences of squalene epoxidase gene in Panax ginseng. Phytochemistry 2010, 71, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhu, J.; Cao, H.Z.; An, Y.R.; Huang, J.J.; Chen, X.H.; Mohammed, N.; Afrin, S.; Luo, Z.Y. Molecular cloning and expression analysis of PDR1-like gene in ginseng subjected to salt and cold stresses or hormonal treatment. Plant Physiol. Biochem. 2013, 71, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Farmer, E.E. Plant biology: Jasmonate perception machines. Nature 2007, 448, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C. Jasmonates: An update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Hahn, E.J.; Murthy, H.N.; Paek, K.Y. Adventitious root growth and ginsenoside accumulation in Panax ginseng cultures as affected by methyl jasmonate. Biotechnol. Lett. 2004, 26, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.T.; Murthy, H.N.; Yu, K.W.; Hahn, E.J.; Paek, K.Y. Methyl jasmonate elicitation enhanced synthesis of ginsenoside by cell suspension cultures of Panax ginseng in 5-l balloon type bubble bioreactors. Appl. Microbiol. Biotechnol. 2005, 67, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Crouzet, J.; Trombik, T.; Fraysse, A.S.; Boutry, M. Organization and function of the plant pleiotropic drug resistance ABC transporter family. FEBS Lett. 2006, 580, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, J.; Zhu, J.; Xie, X.; Tang, Q.; Chen, X.; Luo, J.; Luo, Z. Isolation and characterization of a novel PDR-type ABC transporter gene PgPDR3 from Panax ginseng C.A. Meyer induced by methyl jasmonate. Mol. Biol. Rep. 2013, 40, 6195–6204. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X.; Li, Y. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genomics 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, H.; Kobayashi, Y.; Yamada, K.; Yokoyama, M.; Kusakari, K.; Tanaka, H. Efficient production of saikosaponins in Bupleurum falcatum root fragments combined with signal transducers. Appl. Microbiol. Biotechnol. 2001, 57, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Gundlach, H.; Muller, M.J.; Kutchan, T.M.; Zenk, M.H. Jasmonic acid is a signal transducer in elicitor-induced plant cell cultures. Proc. Natl. Acad. Sci. USA 1992, 89, 2389–2393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.L.; Cui, X.M.; Chen, Y.P.; Liang, Q. Key enzymes of triterpenoid saponin biosynthesis and the induction of their activities and gene expressions in plants. Nat. Prod. Commun. 2010, 5, 1147–1158. [Google Scholar] [PubMed]
- Zhao, S.; Wang, L.; Liu, L.; Liang, Y.; Sun, Y.; Wu, J. Both the mevalonate and the non-mevalonate pathways are involved in ginsenoside biosynthesis. Plant Cell Rep. 2014, 33, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Luo, H.; Li, Y.; Sun, Y.; Wu, Q.; Niu, Y.; Song, J.; Lv, A.; Zhu, Y.; Sun, C.; et al. 454 EST analysis detects genes putatively involved in ginsenoside biosynthesis in Panax ginseng. Plant Cell Rep. 2011, 30, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Song, J.; Sun, Y.; Suo, F.; Li, C.; Luo, H.; Liu, Y.; Li, Y.; Zhang, X.; Yao, H.; et al. Transcript profiles of Panax quinquefolius from flower, leaf and root bring new insights into genes related to ginsenosides biosynthesis and transcriptional regulation. Physiol. Plant 2010, 138, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhao, S. Progress in understanding of ginsenoside biosynthesis. Plant Biol. 2008, 10, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.T.; Bang, K.H.; Kim, Y.C.; Hyun, D.Y.; Kim, M.Y.; Cha, S.W. Upregulation of ginsenoside and gene expression related to triterpene biosynthesis in ginseng hairy root cultures elicited by methyl jasmonate. Plant Cell Tissue Organ Cult. 2009, 98, 25–33. [Google Scholar] [CrossRef]
- Xu, R.; Fazio, G.C.; Matsuda, S.P. On the origins of triterpenoid skeletal diversity. Phytochemistry 2004, 65, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kwon, Y.S.; Yang, D.C.; Jung, Y.R.; Choi, Y.E. Expression and RNA interference-induced silencing of the dammarenediol synthase gene in Panax ginseng. Plant Cell Physiol. 2006, 47, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhao, S.; Zhang, X. Antisense suppression of cycloartenol synthase results in elevated ginsenoside levels in Panax ginseng hairy roots. Plant Mol. Biol. Rep. 2009, 27, 298–304. [Google Scholar] [CrossRef]
- Nelson, D.R.; Ming, R.; Alam, M.; Schuler, M.A. Comparison of cytochrome P450 genes from six plant genomes. Trop. Plant Biol. 2008, 1, 216–235. [Google Scholar] [CrossRef]
- Coon, M.J. Cytochrome P450: Nature’s most versatile biological catalyst. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Gachon, C.M.; Langlois-Meurinne, M.; Saindrenan, P. Plant secondary metabolism glycosyltransferases: The emerging functional analysis. Trends Plant Sci. 2005, 10, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, K. ABC transporters involved in the transport of plant secondary metabolites. FEBS Lett. 2006, 580, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Zhang, R.; Cao, H.Z.; Luo, Z.Y. Plant pleiotropic drug resistance transporters: Transport mechanism, gene expression, and function. J. Integr. Plant Biol. 2014, 56, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, T.; Kohlen, W.; Sasse, J.; Borghi, L.; Schlegel, M.; Bachelier, J.B.; Reinhardt, D.; Bours, R.; Bouwmeester, H.J.; Martinoia, E. A petunia ABC protein controls strigolactone-dependent symbiotic signalling and branching. Nature 2012, 483, 341–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, P.; Brutnell, T.P. Exploring plant transcriptomes using ultra high-throughput sequencing. Brief. Funct. Genomics 2010, 9, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Lee, B.S.; In, J.G.; Sun, H.; Yoon, J.H.; Yang, D.C. Comparative analysis of expressed sequence tags (ESTs) of ginseng leaf. Plant Cell Rep. 2006, 25, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.D.; Park, H.W.; Hahn, Y.; Hur, C.G.; In, D.S.; Chung, H.J.; Liu, J.R.; Choi, D.W. Discovery of genes for ginsenoside biosynthesis by analysis of ginseng expressed sequence tags. Plant Cell Rep. 2003, 22, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.A.; Zhai, J.; Nandety, R.S.; McCormick, K.P.; Zeng, J.; Mejia, D.; Meyers, B.C. Short-read sequencing technologies for transcriptional analyses. Annu. Rev. Plant Biol. 2009, 60, 305–333. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Zody, M.C. Advancing RNA-Seq analysis. Nat. Biotechnol. 2010, 28, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Liu, B.; Wang, X.; Liu, L.; Lun, Y.; Li, X.; Ning, A.; Cao, J.; Huang, M. De novo characterization of Lentinula edodes C(91–3) transcriptome by deep Solexa sequencing. Biochem. Biophys. Res. Commun. 2013, 431, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, L.; Fan, X.; Bian, Y. De novo assembly of Auricularia polytricha transcriptome using Illumina sequencing for gene discovery and SSR marker identification. PLoS One 2014, 9, e91740. [Google Scholar] [CrossRef] [PubMed]
- Vijay, N.; Poelstra, J.W.; Kunstner, A.; Wolf, J.B. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol. Ecol. 2013, 22, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Trick, M.; Long, Y.; Meng, J.; Bancroft, I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 2009, 7, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, Z.; Zakrzewska, A.; Agoston, V.C.; Ordas, A.; Racz, P.; Mink, M.; Spaink, H.P.; Meijer, A.H. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol. Immunol. 2009, 46, 2918–2930. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Austin, R.S.; Zhou, S.; Brown, D. The root transcriptome for North American ginseng assembled and profiled across seasonal development. BMC Genomics 2013, 14, 564. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, P.; Luo, D.; Liu, W.; Wang, Y. Exploiting Illumina sequencing for the development of 95 novel polymorphic EST-SSR markers in common vetch (Vicia sativa subsp. sativa). Molecules 2014, 19, 5777–5789. [Google Scholar] [CrossRef] [PubMed]
- Ono, N.N.; Britton, M.T.; Fass, J.N.; Nicolet, C.M.; Lin, D.; Tian, L. Exploring the transcriptome landscape of pomegranate fruit peel for natural product biosynthetic gene and SSR marker discovery. J. Integr. Plant Biol. 2011, 53, 800–813. [Google Scholar] [CrossRef]
- Lee, M.H.; Jeong, J.H.; Seo, J.W.; Shin, C.G.; Kim, Y.S.; In, J.G.; Yang, D.C.; Yi, J.S.; Choi, Y.E. Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol. 2004, 45, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, M.; Toyoda, K.; Shiraishi, T.; Inagaki, Y.; Ichinose, Y. cDNA cloning and characterization of tobacco ABC transporter: NtPDR1 is a novel elicitor-responsive gene. FEBS Lett. 2002, 518, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Bienert, M.D.; Siegmund, S.E.; Drozak, A.; Trombik, T.; Bultreys, A.; Baldwin, I.T.; Boutry, M. A pleiotropic drug resistance transporter in Nicotiana tabacum is involved in defense against the herbivore Manduca sexta. Plant J. 2012, 72, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Bultreys, A.; Trombik, T.; Drozak, A.; Boutry, M. Nicotiana plumbaginifolia plants silenced for the ATP-binding cassette transporter gene NpPDR1 show increased susceptibility to a group of fungal and oomycete pathogens. Mol. Plant Pathol. 2009, 10, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]
- Fits, L.V.D.; Memelink, J. ORCA3, a jasmonate-responsive transcriptional regulator of plant primary and secondary metabolism. Science 2000, 289, 295–297. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, H.; Nuruzzaman, M.; Xiu, H.; Huang, J.; Wu, K.; Chen, X.; Li, J.; Wang, L.; Jeong, J.-H.; Park, S.-J.; et al. Transcriptome Analysis of Methyl Jasmonate-Elicited Panax ginseng Adventitious Roots to Discover Putative Ginsenoside Biosynthesis and Transport Genes. Int. J. Mol. Sci. 2015, 16, 3035-3057. https://doi.org/10.3390/ijms16023035
Cao H, Nuruzzaman M, Xiu H, Huang J, Wu K, Chen X, Li J, Wang L, Jeong J-H, Park S-J, et al. Transcriptome Analysis of Methyl Jasmonate-Elicited Panax ginseng Adventitious Roots to Discover Putative Ginsenoside Biosynthesis and Transport Genes. International Journal of Molecular Sciences. 2015; 16(2):3035-3057. https://doi.org/10.3390/ijms16023035
Chicago/Turabian StyleCao, Hongzhe, Mohammed Nuruzzaman, Hao Xiu, Jingjia Huang, Kunlu Wu, Xianghui Chen, Jijia Li, Li Wang, Ji-Hak Jeong, Sun-Jin Park, and et al. 2015. "Transcriptome Analysis of Methyl Jasmonate-Elicited Panax ginseng Adventitious Roots to Discover Putative Ginsenoside Biosynthesis and Transport Genes" International Journal of Molecular Sciences 16, no. 2: 3035-3057. https://doi.org/10.3390/ijms16023035
APA StyleCao, H., Nuruzzaman, M., Xiu, H., Huang, J., Wu, K., Chen, X., Li, J., Wang, L., Jeong, J.-H., Park, S.-J., Yang, F., Luo, J., & Luo, Z. (2015). Transcriptome Analysis of Methyl Jasmonate-Elicited Panax ginseng Adventitious Roots to Discover Putative Ginsenoside Biosynthesis and Transport Genes. International Journal of Molecular Sciences, 16(2), 3035-3057. https://doi.org/10.3390/ijms16023035