
Antioxidant, Anti-Tyrosinase and Anti-Inflammatory Activities of Oil Production Residues from Camellia tenuifloria

 ,
,
Abstract
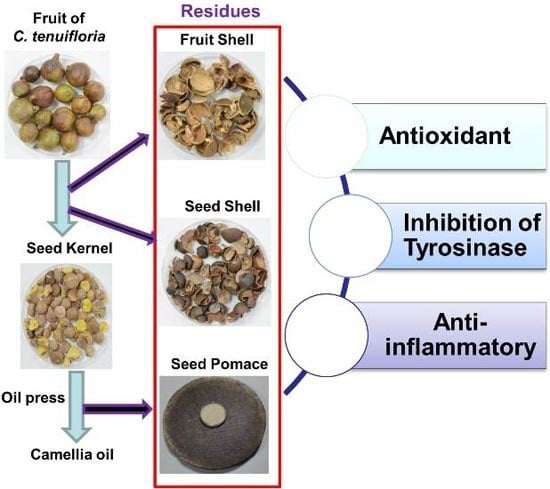
:
1. Introduction
2. Results
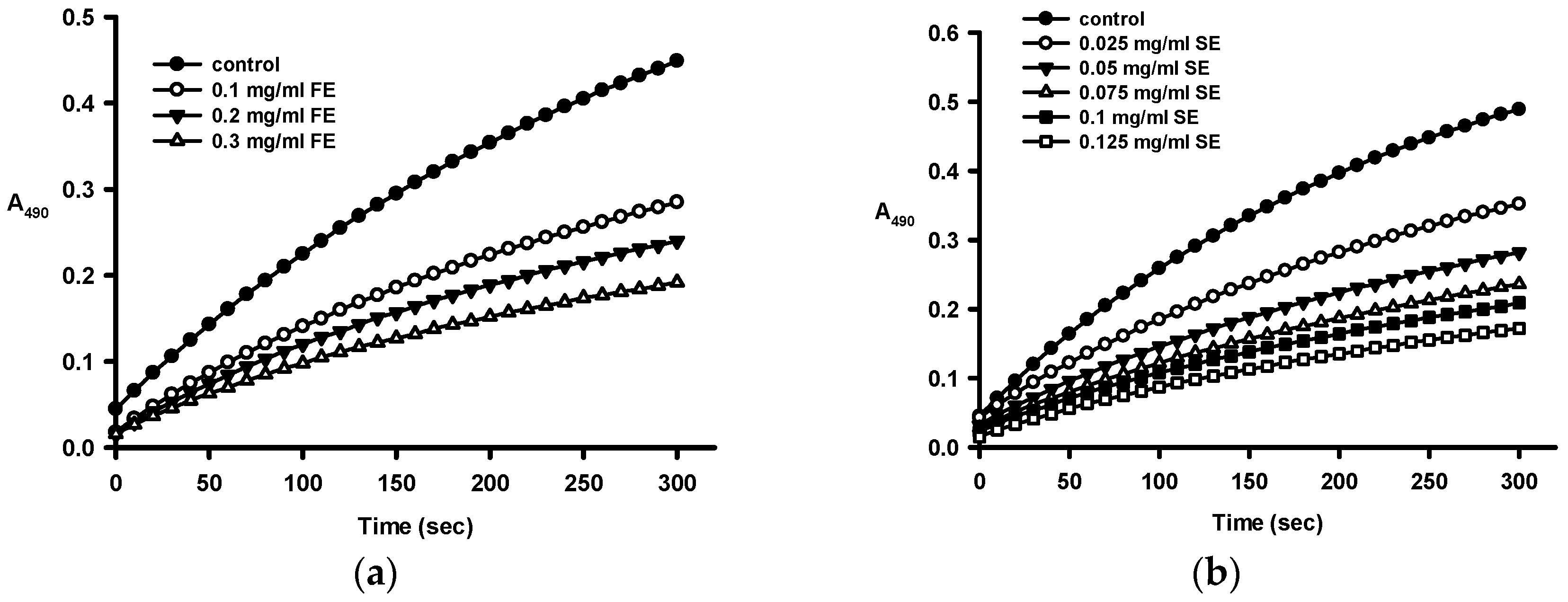
2.1. Antioxidant Activities of Crude Ethanol Extracts and Different Partition Fractions of Fruit Shell, Seed Shell, and Seed Pomace of C. tenuifloria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | IC50 for DPPH Scavenging (μg/mL) a | Total Phenolic Content (mg GAE/g dw) c |
|---|---|---|
| Fruit Shell | ||
| Crude ethanol extract (FE) | 19.74 ± 0.19 | 107.37 ± 3.54 |
| n-Hexane fraction (FH) | ND b | ND b |
| Methanol fraction (FM) | 7.34 ± 0.89 | 266.79 ± 1.85 |
| n-Butanol fraction (FB) | 13.18 ± 0.75 | 129.13 ± 2.55 |
| Aqueous fraction (FA) | 27.25 ± 1.30 | 51.85 ± 3.16 |
| Seed Shell | ||
| Crude ethanol extract (SE) | 14.30 ± 1.01 | 91.42 ± 1.47 |
| n-Hexane fraction (SH) | ND b | ND b |
| Methanol fraction (SM) | 5.47 ± 0.28 | 266.30 ± 7.29 |
| n-Butanol fraction (SB) | 15.55 ± 0.10 | 106.95 ± 3.09 |
| Aqueous fraction (SA) | 5.82 ± 0.09 | 88.90 ± 5.71 |
| Seed Pomace | ||
| Crude ethanol extract (PE) | 84.96 ± 2.75 | 62.40 ± 3.26 |
| n-Hexane fraction (PH) | ND b | ND b |
| Methanol fraction (PM) | 14.38 ± 0.23 | 120.56 ± 2.16 |
| n-Butanol fraction (PB) | 170.99 ± 16.69 | 43.34 ± 0.27 |
| Aqueous fraction (PA) | 218.03 ± 23.12 | 6.26 ± 1.6 |
| α-Tocopherol (control) | 11.89 ± 1.14 |
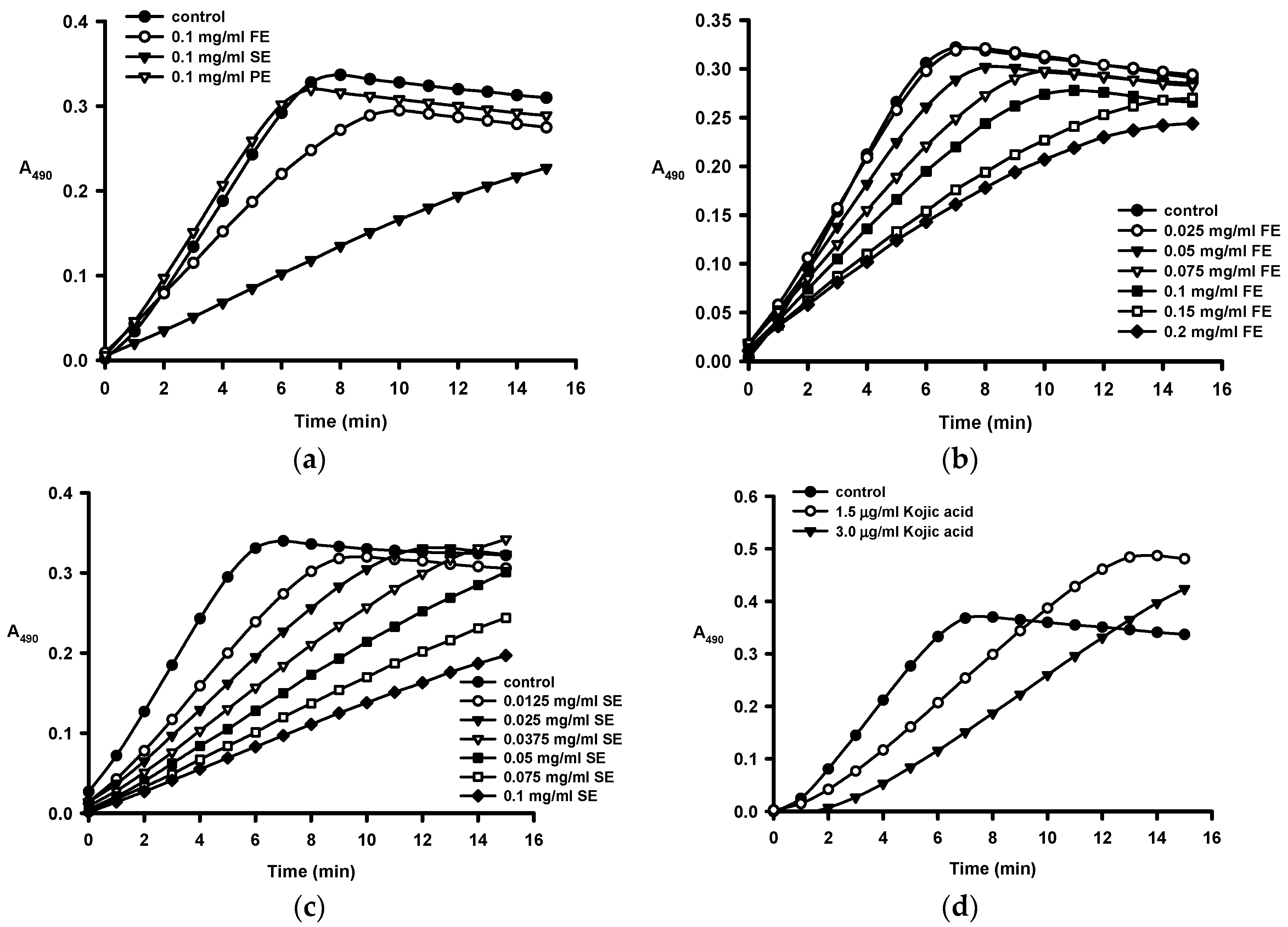
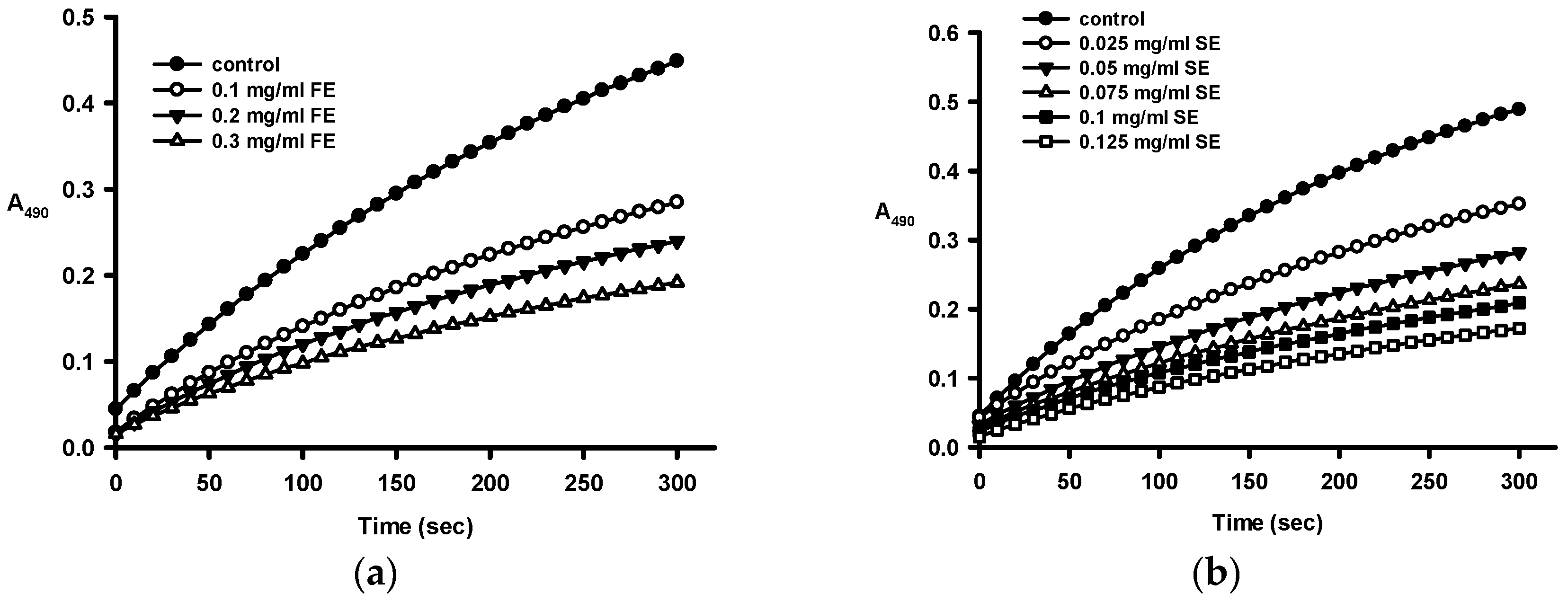
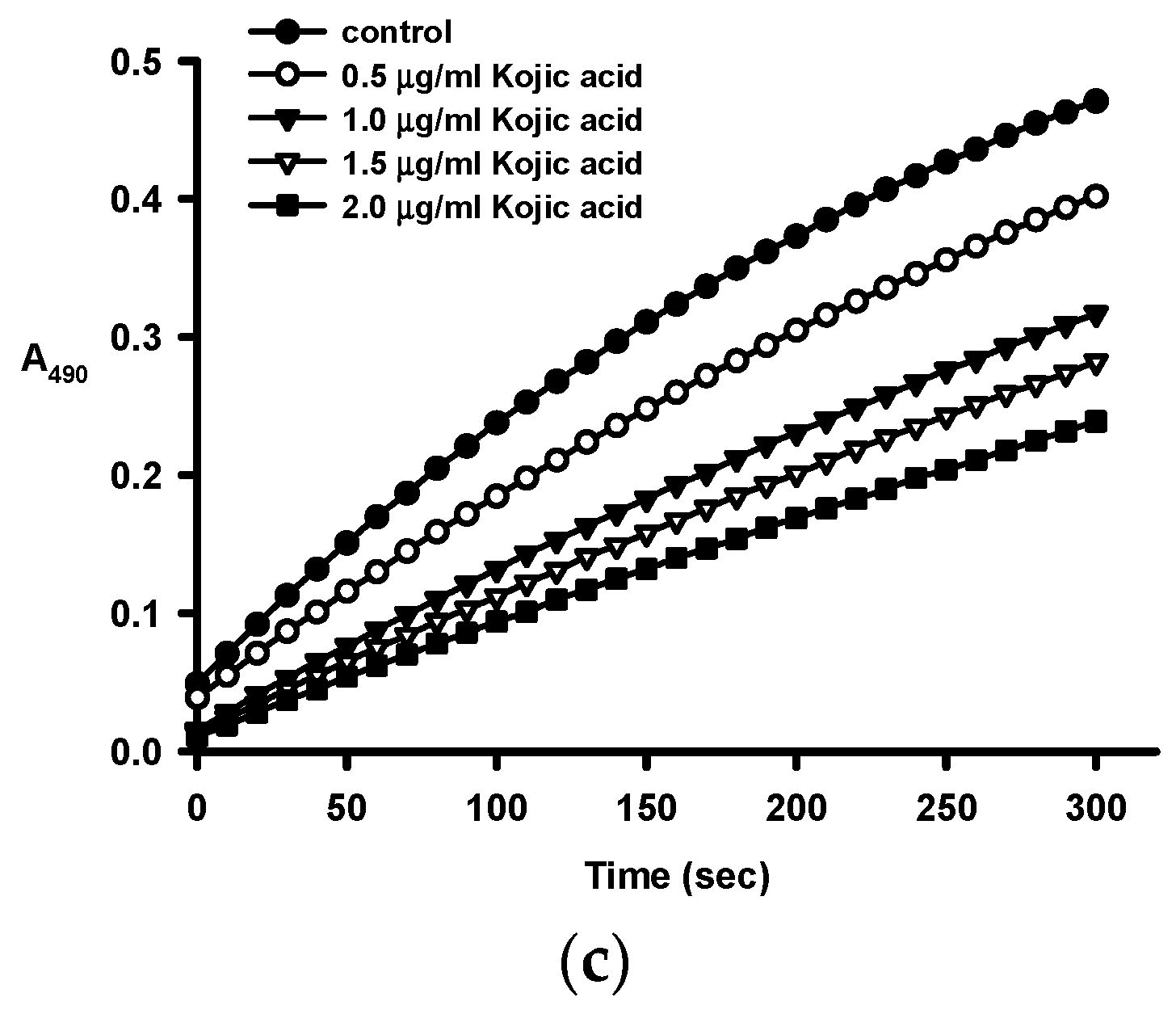
2.2. Anti-Tyrosinase Activities of Crude Ethanol Extracts and Different Partition Fractions of Fruit Shell, Seed Shell, and Seed Pomace of C. tenuifloria


| Samples | IC50 for Monophenolase (μg/mL) a | IC50 for Diphenolase (μg/mL) a |
|---|---|---|
| Fruit Shell | ||
| Ethanol crude extract (FE) | 112.1 ± 2.2 | 307 ± 9 |
| n-Hexane fraction (FH) | ND b | ND b |
| Methanol fraction (FM) | 539 ± 13 | 256 ± 18 |
| n-Butanol fraction (FB) | 70.0 ± 5.5 | 219 ± 12 |
| Aqueous fraction (FA) | 276.7 ± 9.1 | 675 ± 26 |
| Seed Shell | ||
| Ethanol crude extract (SE) | 36.1 ± 0. 2 | 58.9 ± 2.5 |
| n-Hexane fraction (SH) | ND b | ND b |
| Methanol fraction (SM) | 61.5 ± 0.3 | 94.6 ± 3.0 |
| n-Butanol fraction (SB) | 32.7 ± 0.4 | 62.8 ± 0.9 |
| Aqueous fraction (SA) | 33.7 ± 0.2 | 93.9 ± 3.5 |
| Kojic acid (control) | 2.81 ± 0.01 | 1.63 ± 0.09 |
2.3. Anti-Inflammatory Activities of Residues from C. tenuifloria
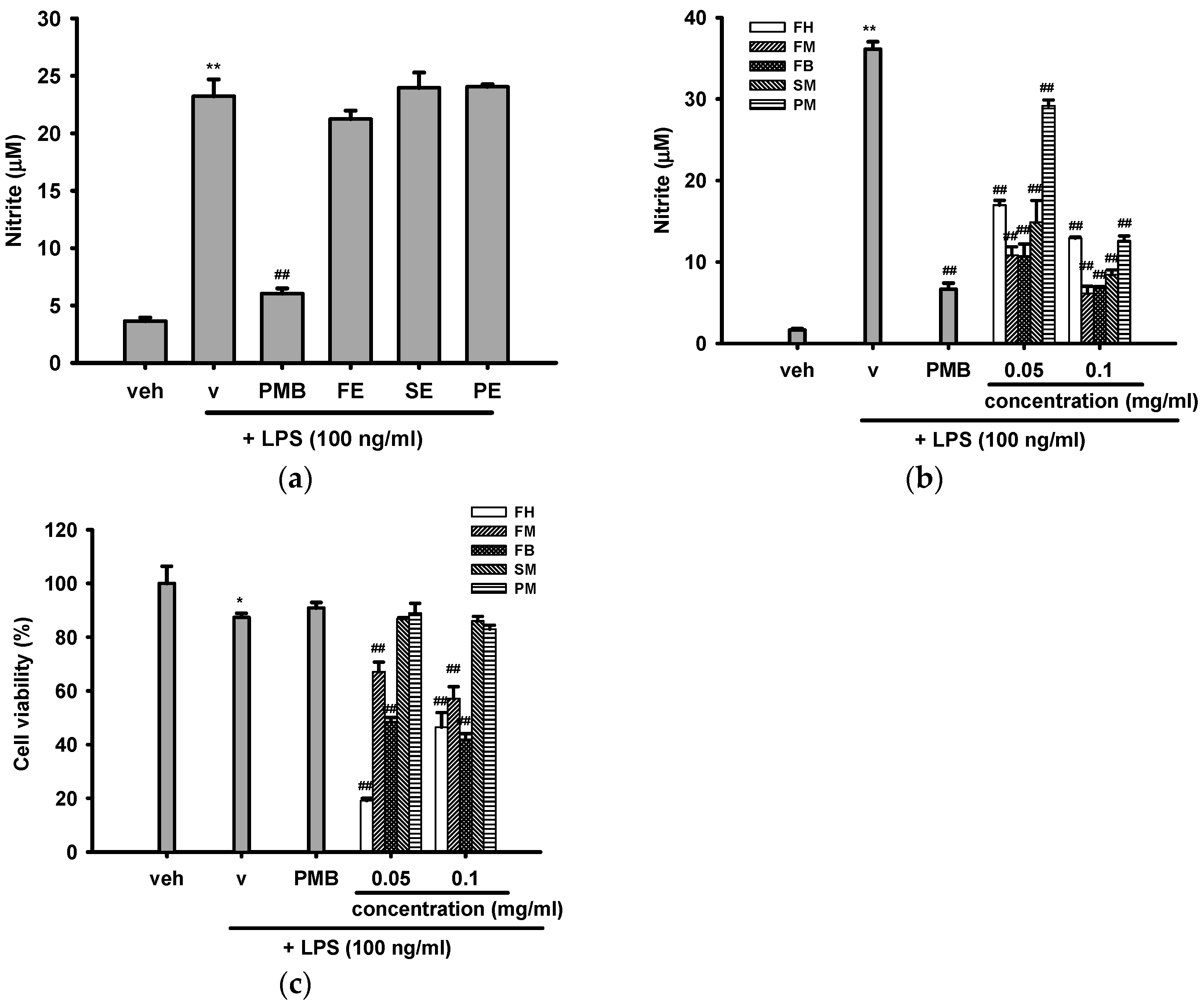
2.3.1. Effects of Crude Ethanol Extracts and Different Partition Fractions of Fruit Shell, Seed Shell, and Seed Pomace of C. tenuifloria on LPS-Induced Nitric Oxide (NO) Production and Cytotoxicity in RAW 264.7 Cells
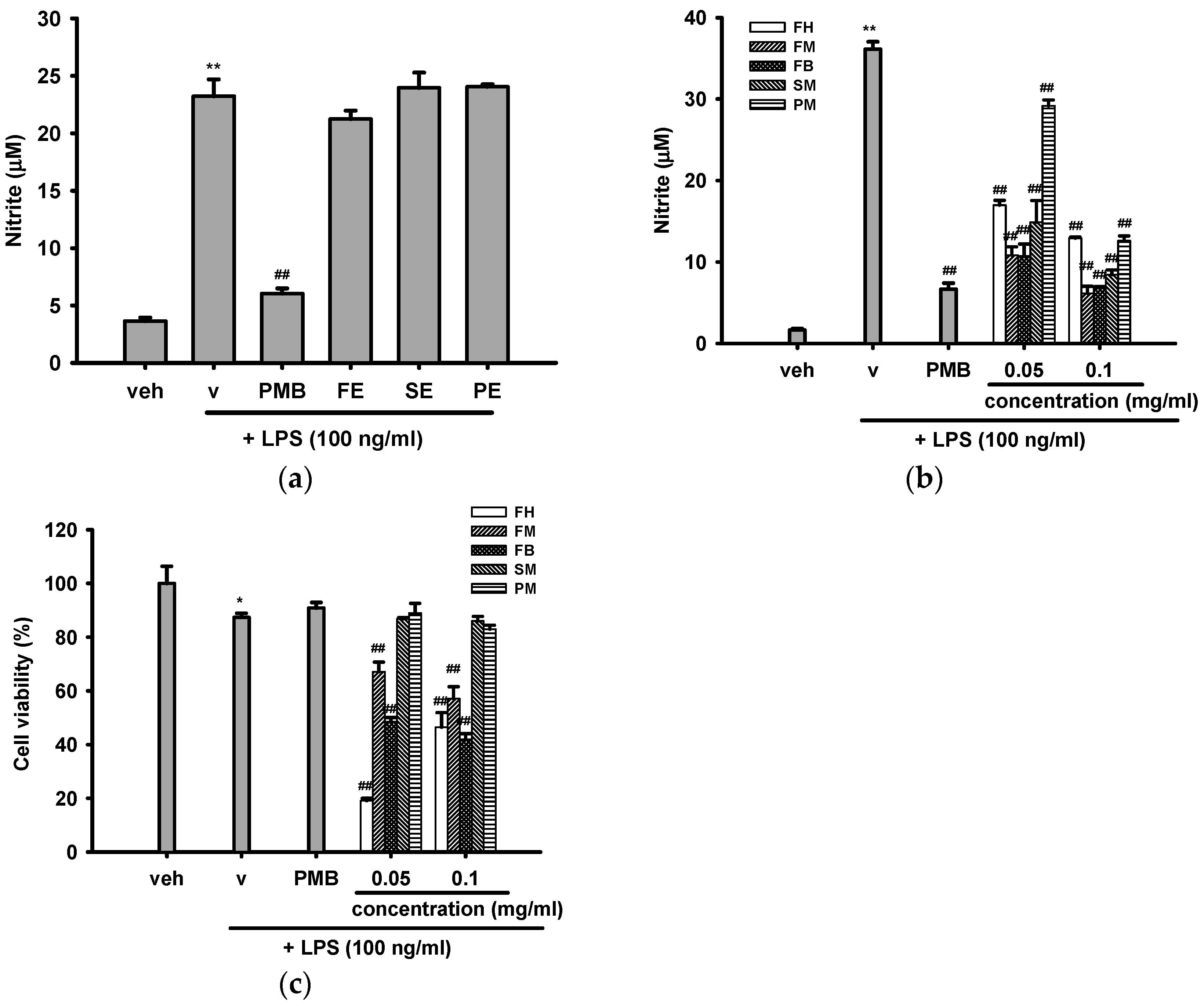
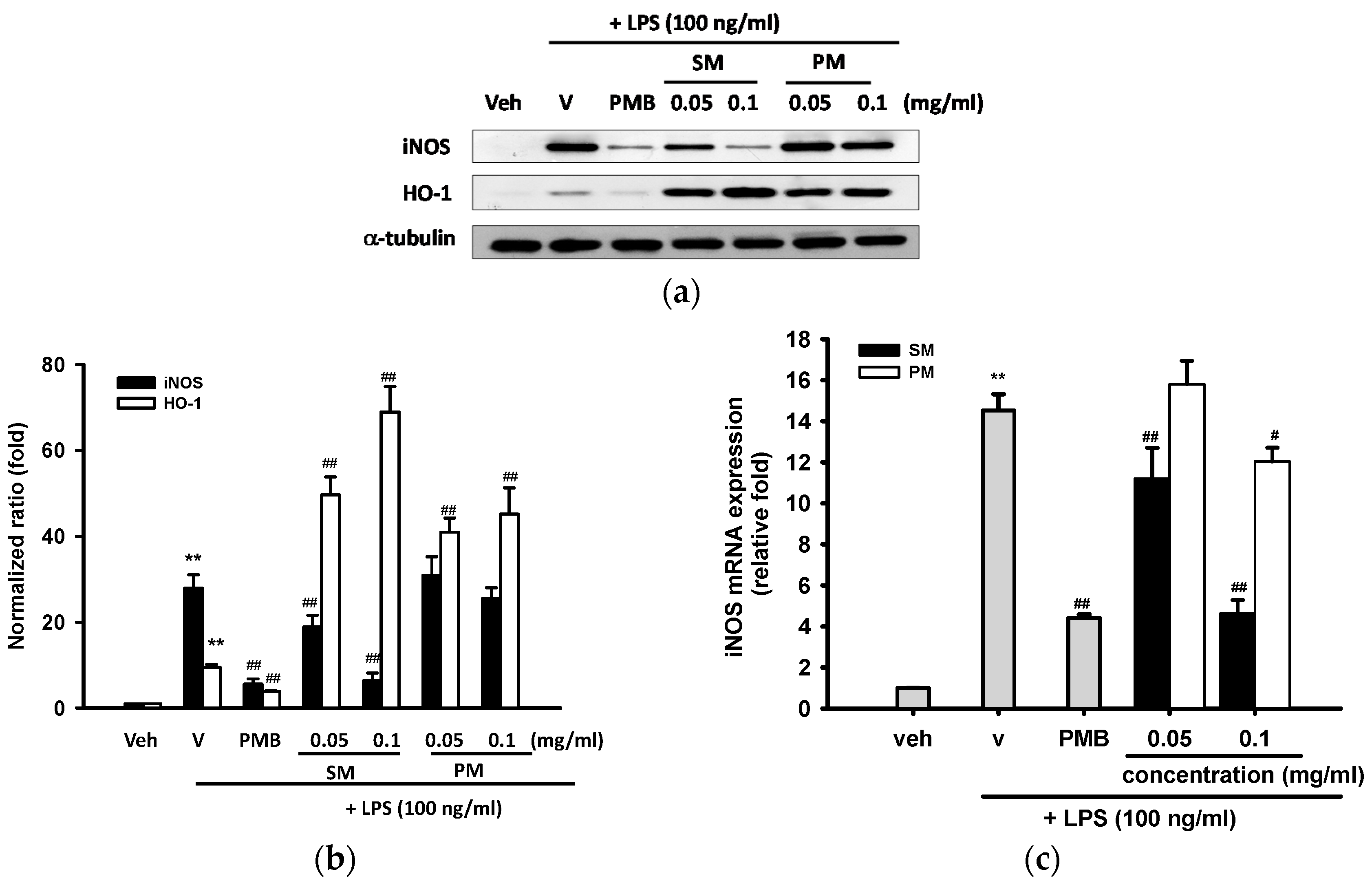
2.3.2. Effects of Methanol Fractions of Seed Shell (SM) and Seed Pomace (PM) on LPS-Mediated Inducible Nitric Oxide Synthase (iNOS) Expression
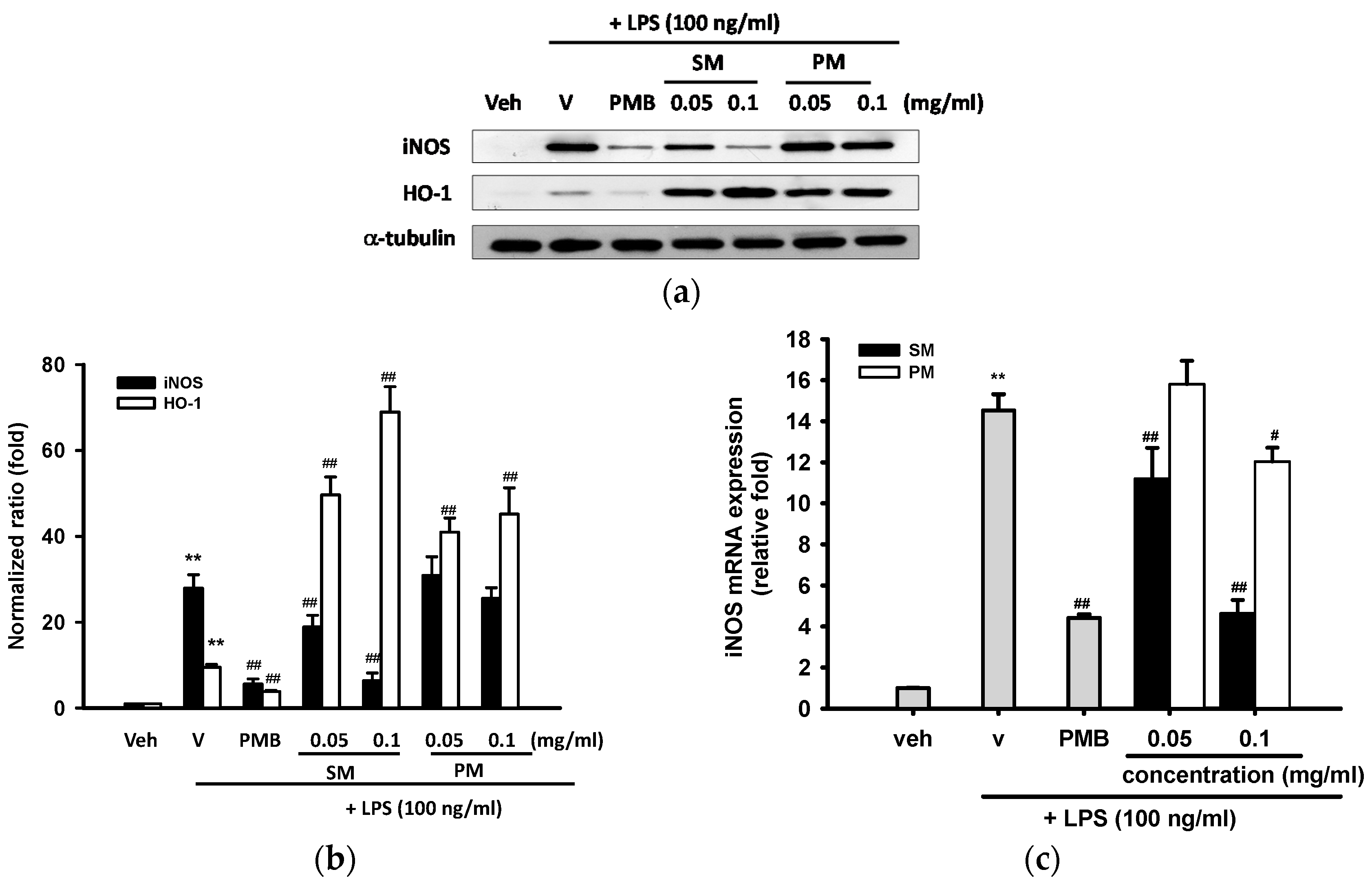
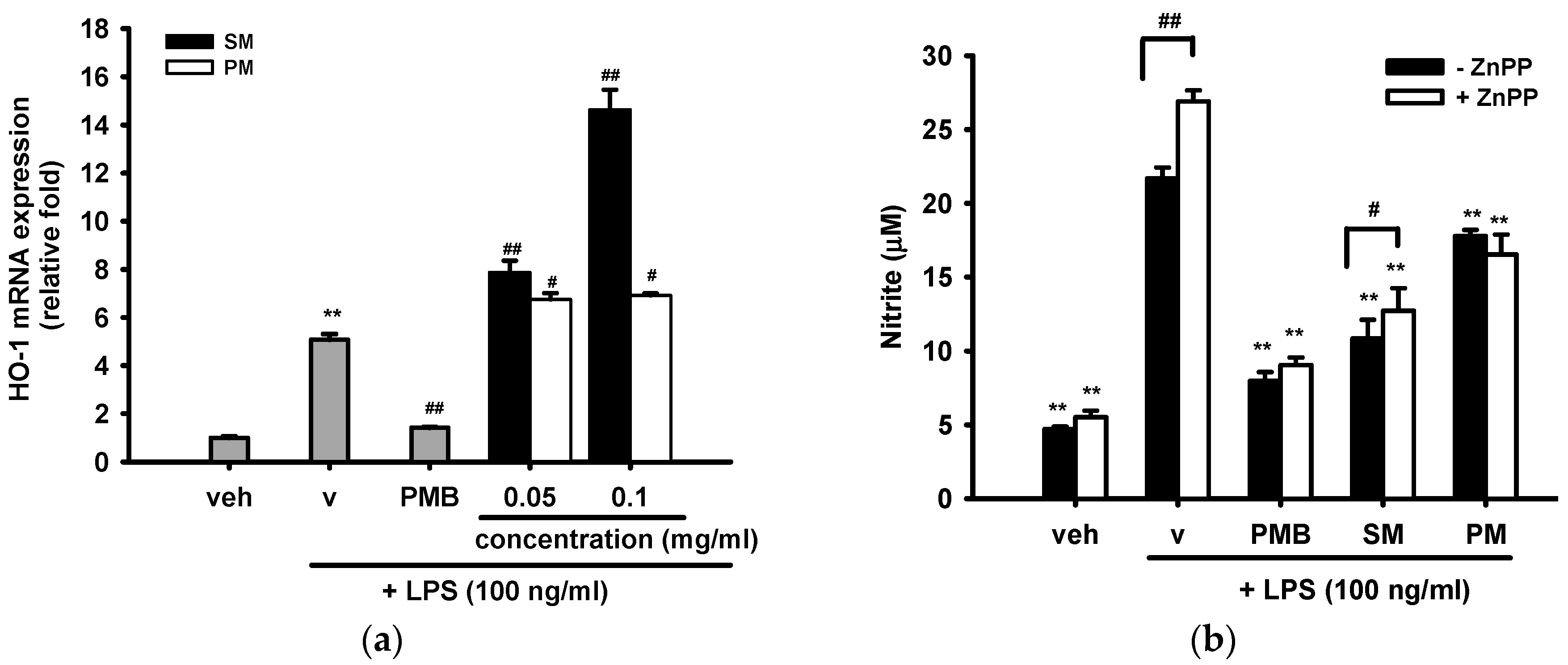
2.3.3. Effects of Methanol Fractions of Seed Shell (SM) and Seed Pomace (PM) on LPS-Mediated Heme Oxygenase 1 (HO-1) Expression

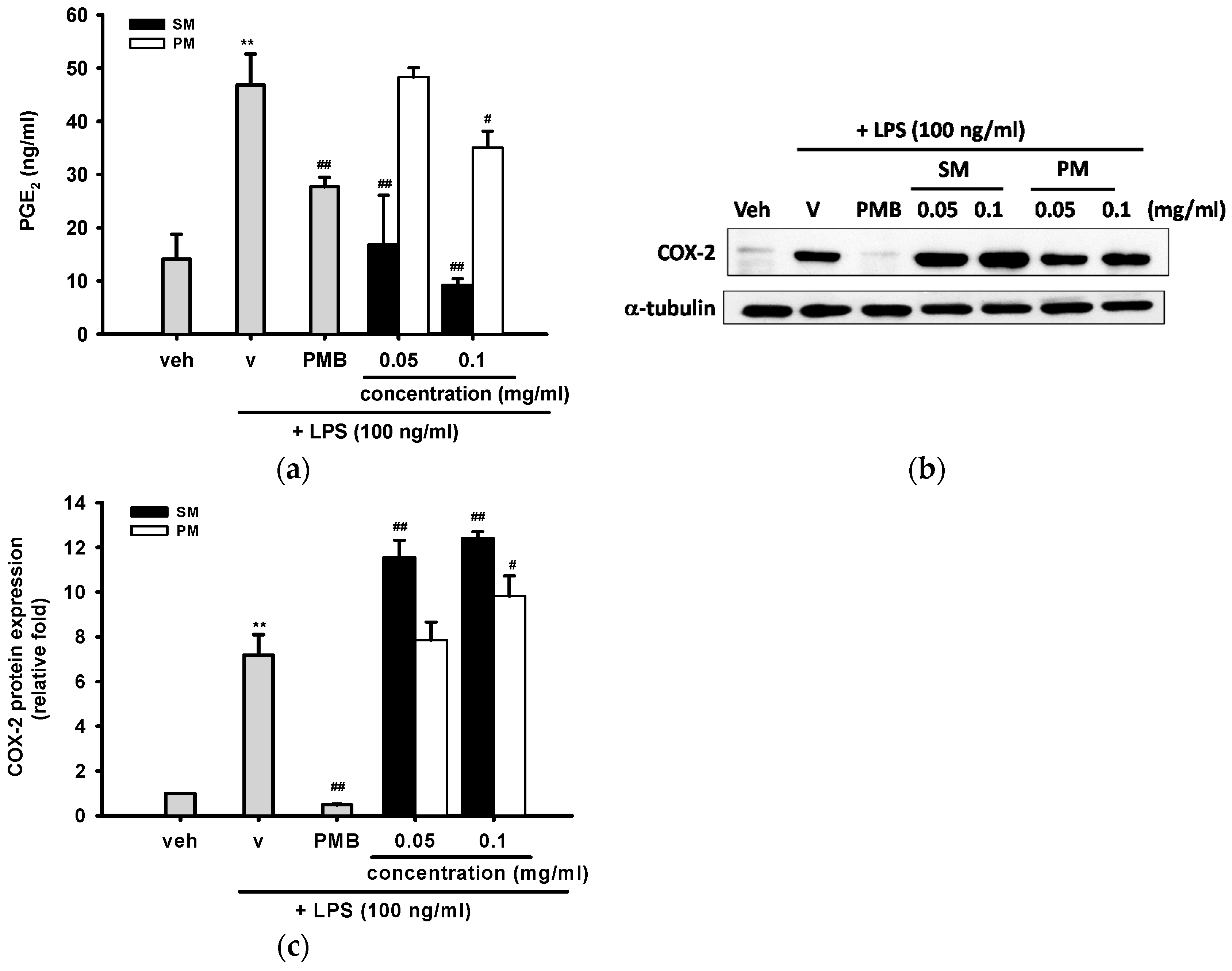
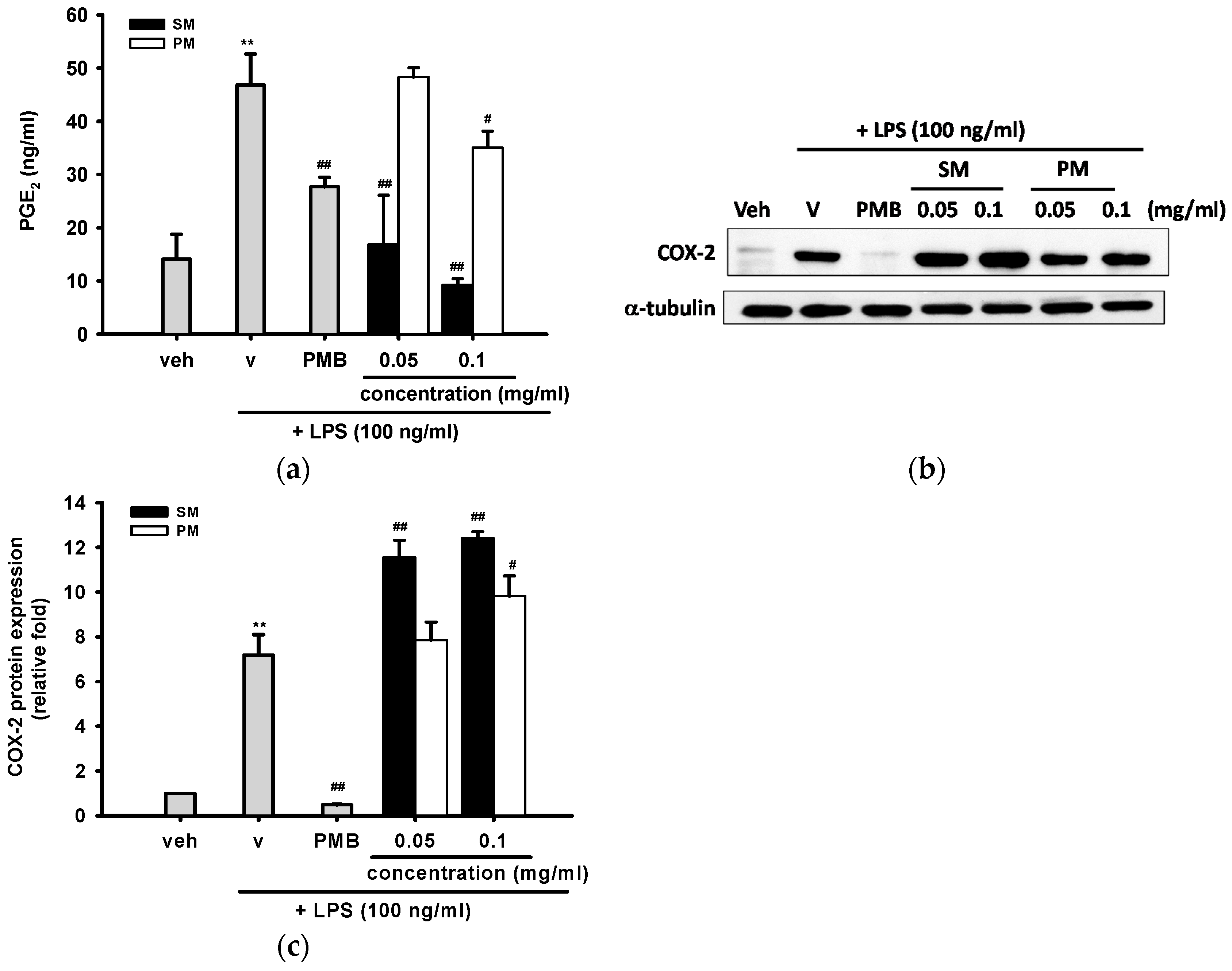
2.3.4. The Inhibitory Effects of Methanol Fractions of Seed Shell (SM) and Seed Pomace (PM) on LPS-induced Prostaglandin E2 (PGE2) Release and Expression of Cyclooxygenase-2 (COX-2)
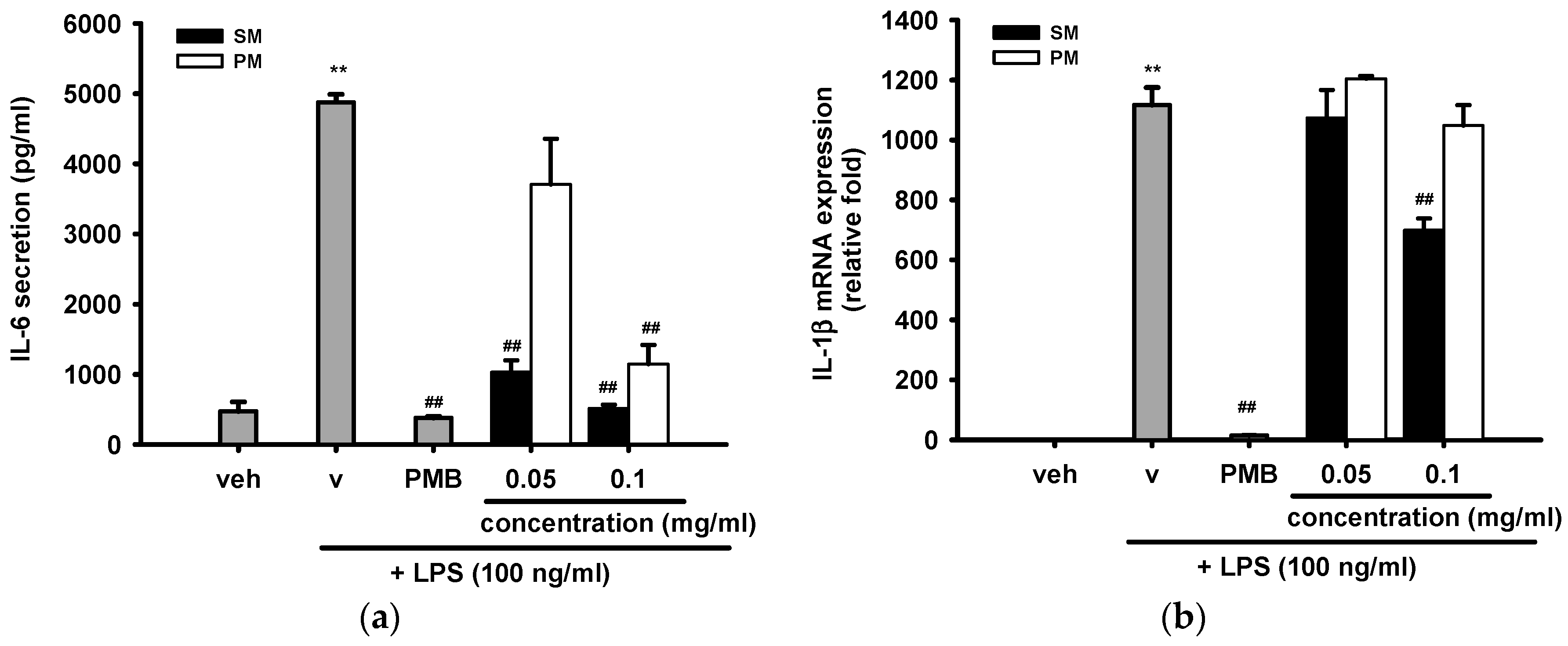
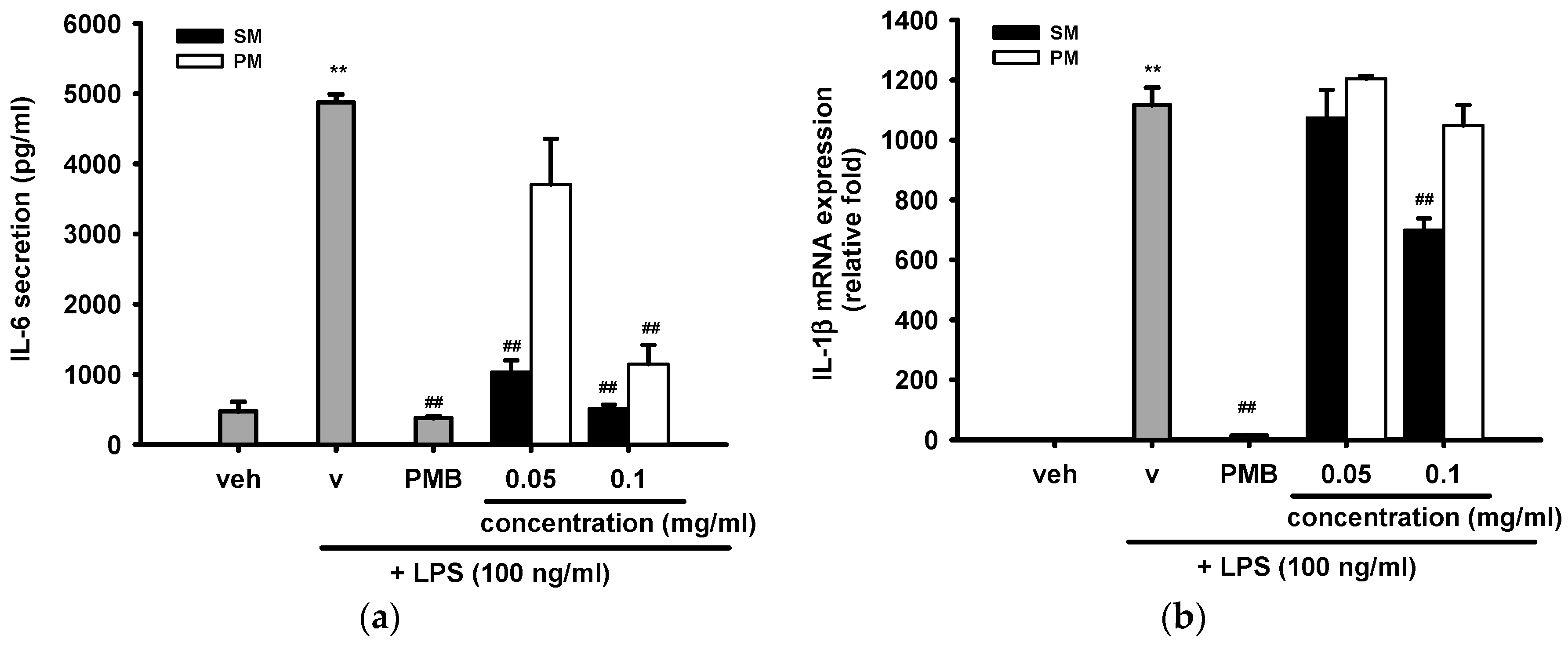
2.3.5. The Inhibitory Effects of Methanol Fractions of Seed Shell (SM) and Seed Pomace (PM) on LPS-Induced IL-6 Release and IL-1β mRNA Expression
3. Discussion
4. Materials and Methods
4.1. Materials
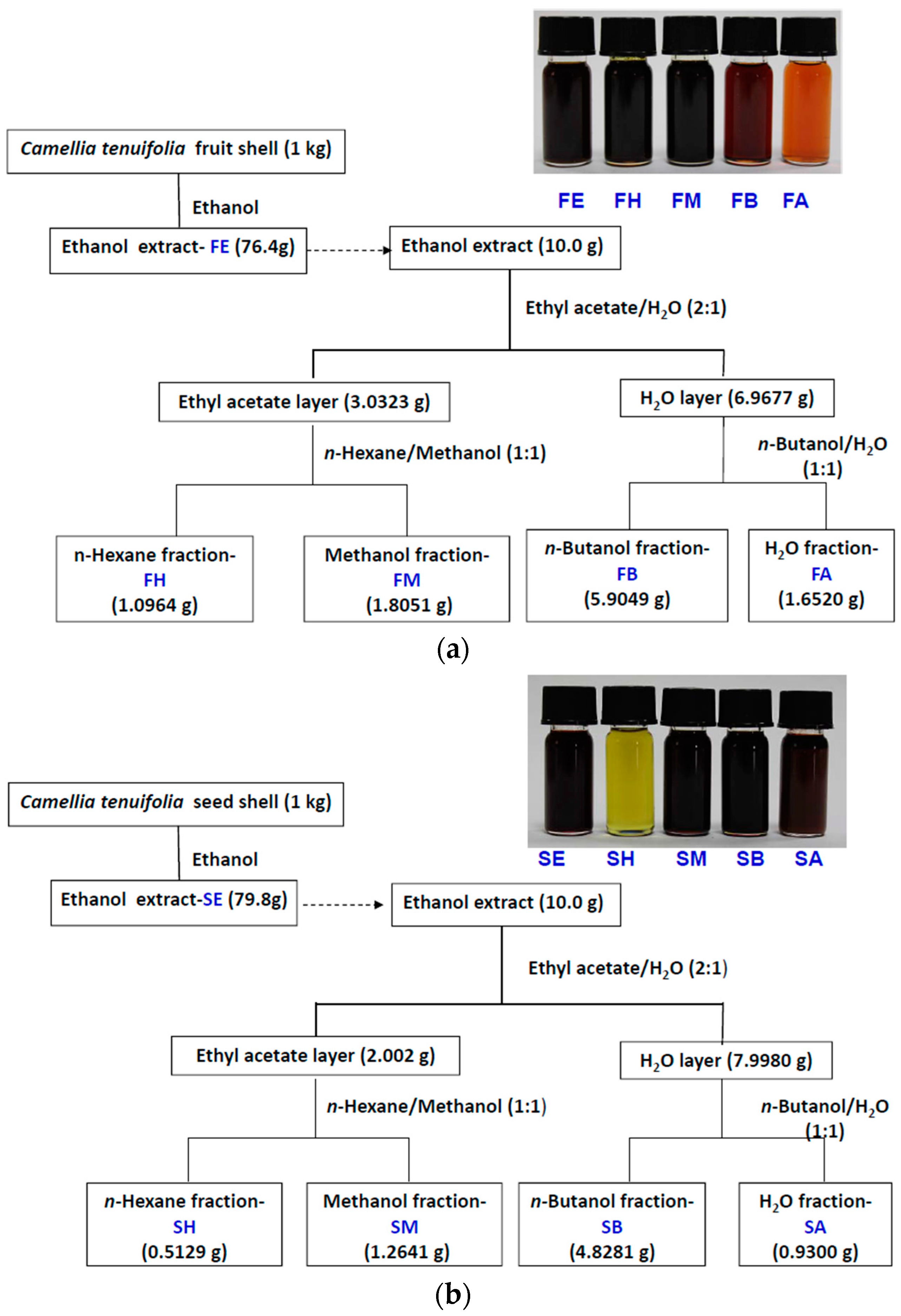
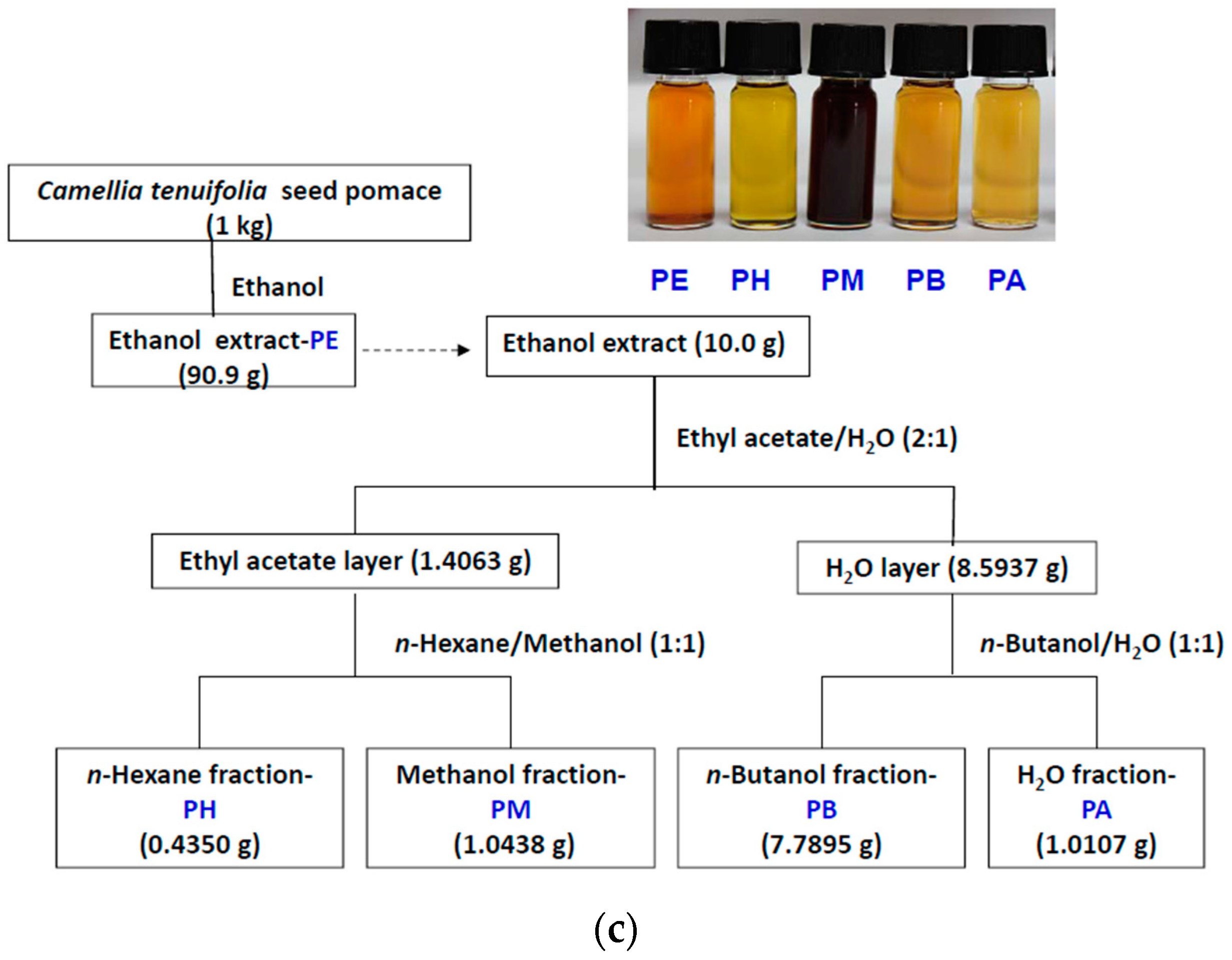
4.2. Cold Percolation Extraction and Partition of Fruit Shell, Seed Shell, and Seed Pomace of C. tenuifloria
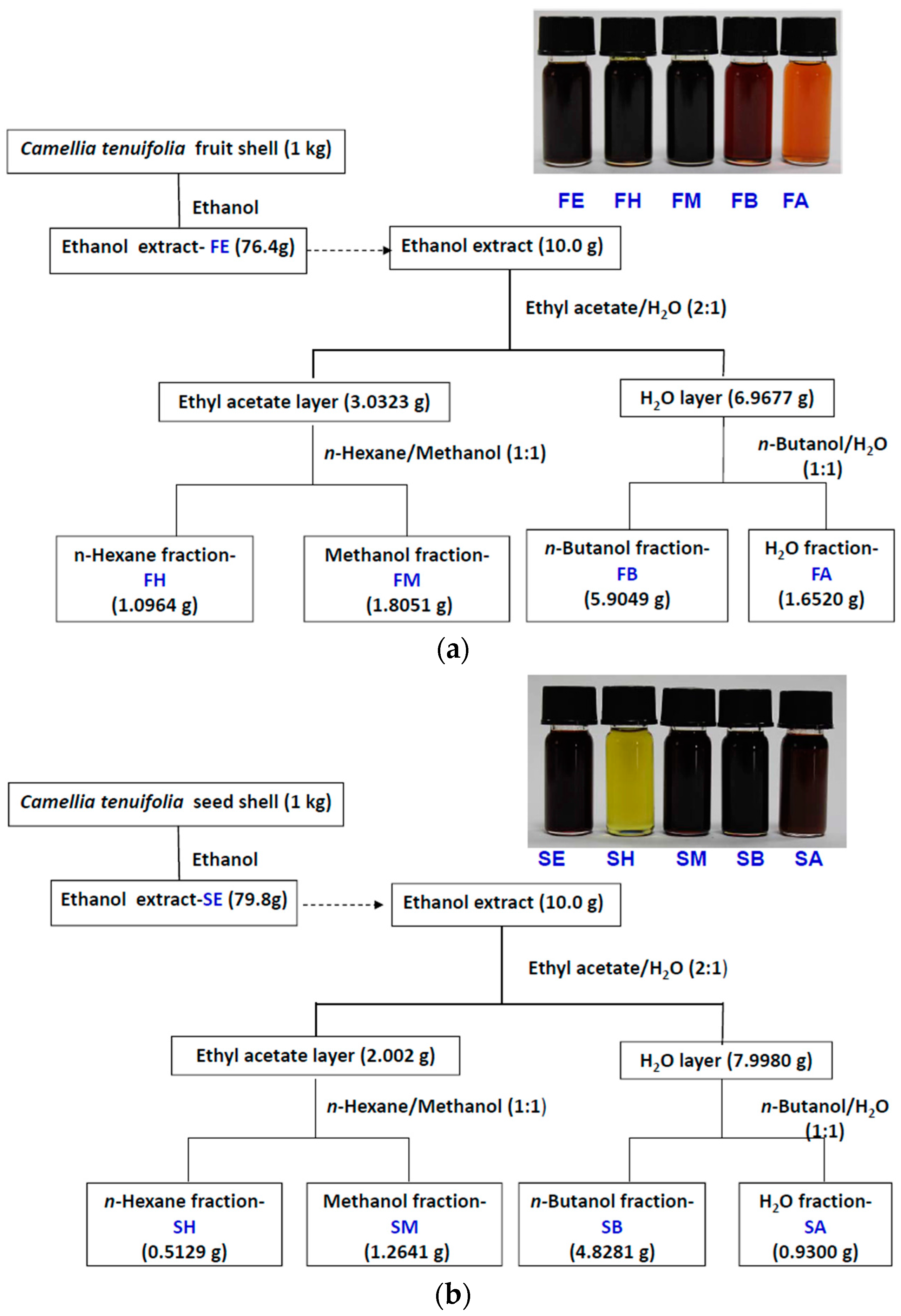
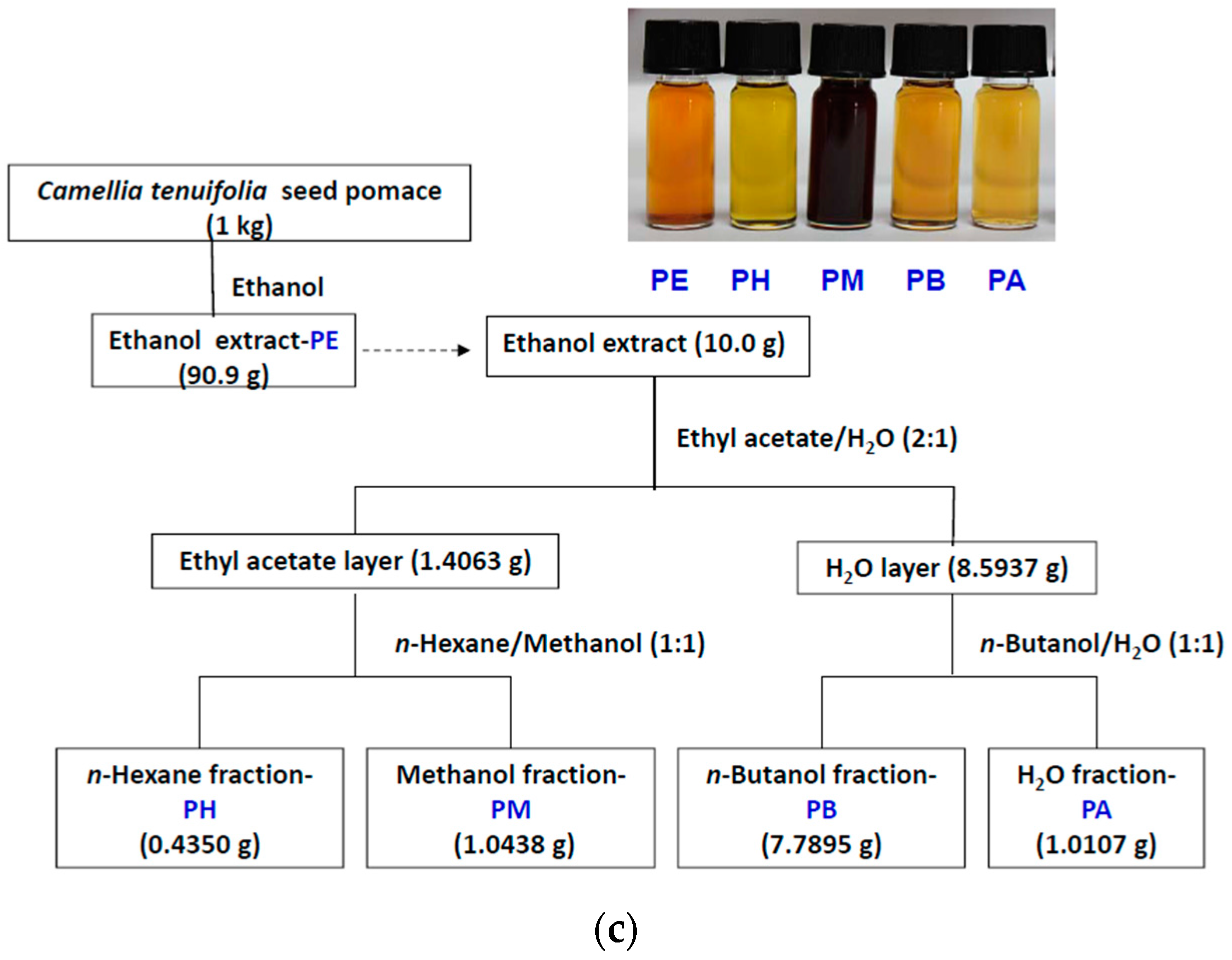
4.3. DPPH Scavenging Capacities
4.4. Folin–Ciocalteu Assay
4.5. Mushroom Tyrosinase Inhibition Assay and IC50 Determination
4.6. Culture and Measurement of Nitrite, Prostaglandin E2 (PGE2) and IL-6 Release in RAW 264.7 Cells
4.7. Cell Viability
4.8. Western Blotting Analysis
| Antibody | Company | Catalog Number |
|---|---|---|
| α-tubulin | Sigma | T6199 |
| NOS | Cell Signaling | 2977 |
| COX-2 | Santa Cruz | Sc-166475 |
| HO-1 | Stressgen | SPA-895 |
4.9. RNA Extraction and Reverse Transcription Real-Time PCR
| Gene | Primers | Amplicon (bp) |
|---|---|---|
| β-actin | GGCTGTATTCCCCTCCATCG, CCAGTTGGTAACAATGCCATGT | 154 |
| iNOS | GTTCTCAGCCCAACAATACAAGA, GTGGACGGGTCGATGTCAC | 127 |
| HO-1 | AAGCCGAGAATGCTGAGTTCA, GCCGTGTAGATATGGTACAAGGA | 100 |
| IL-1β | TTCAGGCAGGCAGTATCACTC, GAAGGTCCACGGGAAAGACAC | 75 |
| COX-2 | TGAGCAACTATTCCAAACCAGC, GCACGTAGTCTTCGATCACTATC | 74 |
| mPGES-1 | ATGAGGCTGCGGAAGAAGG, GCCGAGGAAGAGGAAAGGATAG | 150 |
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chang, H.T. A Taxonomy of the Genus Camellia; Editorial Staff of the Journal of Sun Yatsen University, Ed.; Sun Yatsen University: Guangzhou, China, 1981. [Google Scholar]
- Ming, T. Monograph of the genus' camellia'; Yunnan Science and Technology Press: Kunming, China, 2000. [Google Scholar]
- Vela, P.; Salinero, C.; Sainz, M.J. Phenological growth stages of Camellia japonica. Ann. Appl. Biol. 2013, 162, 182–190. [Google Scholar] [CrossRef]
- Su, M.H.; Shih, M.C.; Lin, K.H. Chemical composition of seed oils in native Taiwanese Camellia species. Food. Chem. 2014, 156, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-M.; Yang, J.-C.; Chen, D.-S.; Chuang, Y.-C.; Eugene, I.; Wang, C.; Lee, Y.-L. Effects of roasting camellia tenuifolia seeds with/without shells on the quality of seed oil. J. Taiwan Agric. Res. 2014, 63, 17–29. [Google Scholar]
- Du, L.C.; Wu, B.L.; Chen, J.M. Flavonoid triglycosides from the seeds of Camellia oleifera abel. Chin. Chem. Lett. 2008, 19, 1315–1318. [Google Scholar] [CrossRef]
- Lee, C.P.; Yen, G.C. Antioxidant activity and bioactive compounds of tea seed (Camellia oleifera abel.) oil. J. Agric. Food Chem. 2006, 54, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, L.; Gao, Y.; Li, B.; Tu, Y. Anti-inflammatory activity of total flavonoids from seeds of Camellia oleifera abel. Acta Biochim. Biophys. Sin. 2014, 46, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Guo, Y.; Luo, Y.-T.; Wang, Y.-F. Isolation and free radical scavenging activities of a novel biflavonoid from the shells of Camellia oleifera abel. Fitoterapia 2012, 83, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xing, H.; Chen, X. Anti-inflammatory and analgesic activities of the hydrolyzed sasanquasaponins from the defatted seeds of Camellia oleifera. Arch. Pharm. Res. 2013, 36, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Yang, S.L.; Han, Y.Y.; Zhao, L.; Lu, G.L.; Xia, T.; Gao, L.P. Qualitative and quantitative analysis of triterpene saponins from tea seed pomace (Camellia oleifera abel) and their activities against bacteria and fungi. Molecules 2014, 19, 7568–7580. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-L.; Nie, S.-P.; Huang, D.-F.; Li, C.; Xie, M.-Y.; Wan, Y. Antimicrobial activity of saponin-rich fraction from Camellia oleifera cake and its effect on cell viability of mouse macrophage RAW 264.7. J. Sci. Food Agric. 2012, 92, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ning, Y. Antioxidant and antitumor activities of the polysaccharide from seed cake of Camellia oleifera abel. Int. J. Biol. Macromol. 2012, 51, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Guo, Y.; Luo, Y.T. Anti-inflammatory and analgesic activities of a novel biflavonoid from shells of Camellia oleifera. Int. J. Mol. Sci. 2012, 13, 12401–12411. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.C.; Yu, C.W.; Yen, P.L.; Lin, H.Y.; Chang, S.T.; Hsu, F.L.; Liao, V.H. Antioxidant activity, delayed aging, and reduced amyloid-β toxicity of methanol extracts of tea seed pomace from Camellia tenuifolia. J. Agric. Food Chem. 2014, 62, 10701–10707. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar] [PubMed]
- Liu, R.H. Health benefits of fruit and vegetables are from additive and synergistic combinations of phytochemicals. Am. J. Clin. Nutr. 2003, 78, 517–520. [Google Scholar]
- Joseph, J.A.; Casadesus, G.; Smith, M.A.; Perry, G.; Shukitt-Hale, B. Chapter 20—nutrients and food constituents in cognitive decline and neurodegenerative disease. In Nutrition in the Prevention and Treatment of Disease, 3th ed.; Coulston, A.M., Ferruzzi, M.G., Boushey, C.J., Eds.; Elsevier Academic Press: Waltham, MA, USA, 2013; pp. 373–390. [Google Scholar]
- Seifried, H.E.; Pilch, S.M. Chapter 18—antioxidants in health and disease. In Nutrition in the Prevention and Treatment of Disease, 3th ed.; Coulston, A.M., Ferruzzi, M.G., Boushey, C.J., Eds.; Elsevier Academic Press: Waltham, MA, USA, 2013; pp. 319–339. [Google Scholar]
- Neergheen, V.S.; Bahorun, T.; Taylor, E.W.; Jen, L.S.; Aruoma, O.I. Targeting specific cell signaling transduction pathways by dietary and medicinal phytochemicals in cancer chemoprevention. Toxicology 2010, 278, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ferrer, A.; Rodriguez-Lopez, J.N.; Garcia-Canovas, F.; Garcia-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Loizzo, M.R.; Tundis, R.; Menichini, F. Natural and synthetic tyrosinase inhibitors as antibrowning agents: An update. Compr. Rev. Food Sci. Food. Saf. 2012, 11, 378–398. [Google Scholar] [CrossRef]
- Rescigno, A.; Sollai, F.; Pisu, B.; Rinaldi, A.; Sanjust, E. Tyrosinase inhibition: General and applied aspects. J. Enzym. Inhib. Med. Chem. 2002, 17, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell. Mol. Life Sci. 2005, 62, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.A.; Dorf, M.E. Interleukin-6 production by murine macrophage cell lines P388D1 and J774A.1: Stimulation requirements and kinetics. Cell. Immunol. 1990, 128, 555–568. [Google Scholar] [CrossRef]
- Gomes, R.N.; Teixeira-Cunha, M.G.; Figueiredo, R.T.; Almeida, P.E.; Alves, S.C.; Bozza, P.T.; Bozza, F.A.; Bozza, M.T.; Zimmerman, G.A.; Castro-Faria-Neto, H.C. Bacterial clearance in septic mice is modulated by MCP-1/CCL2 and nitric oxide. Shock 2013, 39, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, R.; Lahti, A.; Kankaanranta, H.; Moilanen, E. Nitric oxide production and signaling in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.Y.; Sharma, V.K.; Sharma, N. Mushroom tyrosinase: Recent prospects. J. Agric. Food Chem. 2003, 51, 2837–2853. [Google Scholar] [CrossRef] [PubMed]
- Xaus, J.; Comalada, M.; Valledor, A.F.; Lloberas, J.; Lopez-Soriano, F.; Argiles, J.M.; Bogdan, C.; Celada, A. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-α. Blood 2000, 95, 3823–3831. [Google Scholar] [PubMed]
- Wu, M.L.; Ho, Y.C.; Lin, C.Y.; Yet, S.F. Heme oxygenase-1 in inflammation and cardiovascular disease. Am. J. Cardiovasc. Dis. 2011, 1, 150–158. [Google Scholar] [PubMed]
- Lin, H.Y.; Juan, S.H.; Shen, S.C.; Hsu, F.L.; Chen, Y.C. Inhibition of lipopolysaccharide-induced nitric oxide production by flavonoids in RAW 264.7 macrophages involves heme oxygenase-1. Biochem. Pharmacol. 2003, 66, 1821–1832. [Google Scholar] [CrossRef]
- Lee, C.J.; Lee, S.S.; Chen, S.C.; Ho, F.M.; Lin, W.W. Oregonin inhibits lipopolysaccharide-induced iNOS gene transcription and upregulates HO-1 expression in macrophages and microglia. Br. J. Pharmacol. 2005, 146, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chiou, S.Y.; Shen, Y.T.; Yen, F.T.; Ding, H.Y.; Wu, M.J. Anti-inflammatory effect and mechanism of the green fruit extract of solanum integrifolium poir. Biomed. Res. Int. 2014, 2014, 953873. [Google Scholar] [PubMed]
- Korotkova, M.; Jakobsson, P.J. Characterization of microsomal prostaglandin e synthase 1 inhibitors. Basic Clin. Pharmacol. Toxicol. 2014, 114, 64–69. [Google Scholar] [CrossRef] [PubMed]
- John-Aryankalayil, M.; Palayoor, S.T.; Cerna, D.; Falduto, M.T.; Magnuson, S.R.; Coleman, C.N. Ns-398, ibuprofen, and cyclooxygenase-2 RNA interference produce significantly different gene expression profiles in prostate cancer cells. Mol. Cancer Ther. 2009, 8, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Godambe, S.A.; Chaplin, D.D.; Bellone, C.J. Regulation of IL-1 gene expression: Differential responsiveness of murine macrophage lines. Cytokine 1993, 5, 327–335. [Google Scholar] [CrossRef]
- Bravo, L. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutr. Rev. 1998, 56, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-G.; AG Koffas, M. Bioavailability and recent advances in the bioactivity of flavonoid and stilbene compounds. Curr. Org. Chem. 2010, 14, 1727–1751. [Google Scholar] [CrossRef]
- Chen, J.-H.; Wu, H.-Y.; Liau, B.-C.; Chang, C.-M.J.; Jong, T.-T.; Wu, L.-C. Identification and evaluation of antioxidants defatted camellia oleifera seeds by isopropanol salting-out pretreatment. Food. Chem. 2010, 121, 1246–1254. [Google Scholar] [CrossRef]
- Yao, Y.; Luong, T.N.; Lepik, M.; Aftab, N.; Fong, V.H.; Vieira, A. Synergism of antioxidant phytochemicals: Comparisons among purified polyphenols and dietary-plant extracts. In XXVIII International Horticultural Congress on Science and Horticulture for People (IHC2010); International Symposium: Leuven, Belgium, 2010; pp. 121–127. [Google Scholar]
- Bisswanger, H. Enzyme assays. Perspect. Sci. 2014, 1, 41–55. [Google Scholar] [CrossRef]
- Sato, K.; Toriyama, M. The inhibitory effect of non-steroidal anti-inflammatory drugs (NSAIDs) on the monophenolase and diphenolase activities of mushroom tyrosinase. Int. J. Mol. Sci. 2011, 12, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.H. Molecular design of tyrosinase inhibitors: A critical review of promising novel inhibitors from synthetic origins. Pure Appl. Chem. 2007, 79, 2277–2295. [Google Scholar] [CrossRef]
- Chanput, W.; Mes, J.; Vreeburg, R.A.; Savelkoul, H.F.; Wichers, H.J. Transcription profiles of LPS-stimulated THP-1 monocytes and macrophages: A tool to study inflammation modulating effects of food-derived compounds. Food Funct. 2010, 1, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Denis, M. Human monocytes/macrophages: NO or no NO? J. Leukoc. Biol. 1994, 55, 682–684. [Google Scholar] [PubMed]
- Schneemann, M.; Schoedon, G. Species differences in macrophage NO production are important. Nat. Immunol. 2002, 3, 102. [Google Scholar] [CrossRef] [PubMed]
- Schneemann, M.; Schoedon, G.; Hofer, S.; Blau, N.; Guerrero, L.; Schaffner, A. Nitric oxide synthase is not a constituent of the antimicrobial armature of human mononuclear phagocytes. J. Infect. Dis. 1993, 167, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Kmoníčková, E.; Melkusová, P.; Farghali, H.; Holý, A.; Zídek, Z. Nitric oxide production in mouse and rat macrophages: A rapid and efficient assay for screening of drugs immunostimulatory effects in human cells. Nitric Oxide 2007, 17, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Srisook, K.; Cha, Y.N. Biphasic induction of heme oxygenase-1 expression in macrophages stimulated with lipopolysaccharide. Biochem. Pharmacol. 2004, 68, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Kinneer, K.; Ye, J.; Chen, B.J. Inhibition of nuclear factor kappab by phenolic antioxidants: Interplay between antioxidant signaling and inflammatory cytokine expression. Mol. Pharmacol. 2003, 64, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Jayasooriya, R.G.; Lee, K.T.; Lee, H.J.; Choi, Y.H.; Jeong, J.W.; Kim, G.Y. Anti-inflammatory effects of beta-hydroxyisovalerylshikonin in BV2 microglia are mediated through suppression of the PI3K/AKT/NF-KB pathway and activation of the Nrf2/HO-1 pathway. Food Chem. Toxicol. 2013, 65, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Miller, I.; Renz, M.; Whited, T.; Kim, L.; Adams, B.; Dudley, R.; Kinder, D. COX-2 is induced by the COX-2 selective inhibitors celecoxib and etodolac and the non-selective inhibitor ibuprofen in several human tumor cell lines (397.8). FEBS J. 2014, 28, 397. [Google Scholar]
- Ivanov, A.I.; Romanovsky, A.A. Prostaglandin E2 as a mediator of fever: Synthesis and catabolism. Front. Biosci. 2004, 9, 1977–1993. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, T.; Fujikawa, K.; Nishio, Y.; Mukaida, N.; Matsushima, K.; Kishimoto, T.; Akira, S. Transcription factors NF-IL6 and NF-Kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad. Sci. USA 1993, 90, 10193–10197. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, C.J.; Alley, E.W.; Raval, P.; Snowman, A.M.; Snyder, S.H.; Russell, S.W.; Murphy, W.J. Macrophage nitric oxide synthase gene: Two upstream regions mediate induction by interferon γ and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1993, 90, 9730–9734. [Google Scholar] [CrossRef] [PubMed]
- Sharma, O.P.; Bhat, T.K. DPPH antioxidant assay revisited. Food. Chem. 2009, 113, 1202–1205. [Google Scholar] [CrossRef]
- Everette, J.D.; Bryant, Q.M.; Green, A.M.; Abbey, Y.A.; Wangila, G.W.; Walker, R.B. Thorough study of reactivity of various compound classes toward the folin-ciocalteu reagent. J. Agric. Food Chem. 2010, 58, 8139–8144. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiou, S.-Y.; Ha, C.-L.; Wu, P.-S.; Yeh, C.-L.; Su, Y.-S.; Li, M.-P.; Wu, M.-J. Antioxidant, Anti-Tyrosinase and Anti-Inflammatory Activities of Oil Production Residues from Camellia tenuifloria. Int. J. Mol. Sci. 2015, 16, 29522-29541. https://doi.org/10.3390/ijms161226184
Chiou S-Y, Ha C-L, Wu P-S, Yeh C-L, Su Y-S, Li M-P, Wu M-J. Antioxidant, Anti-Tyrosinase and Anti-Inflammatory Activities of Oil Production Residues from Camellia tenuifloria. International Journal of Molecular Sciences. 2015; 16(12):29522-29541. https://doi.org/10.3390/ijms161226184
Chicago/Turabian StyleChiou, Shu-Yuan, Choi-Lan Ha, Pei-Shan Wu, Chiu-Ling Yeh, Ying-Shan Su, Man-Po Li, and Ming-Jiuan Wu. 2015. "Antioxidant, Anti-Tyrosinase and Anti-Inflammatory Activities of Oil Production Residues from Camellia tenuifloria" International Journal of Molecular Sciences 16, no. 12: 29522-29541. https://doi.org/10.3390/ijms161226184