
Alternative RNA Structure-Coupled Gene Regulations in Tumorigenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
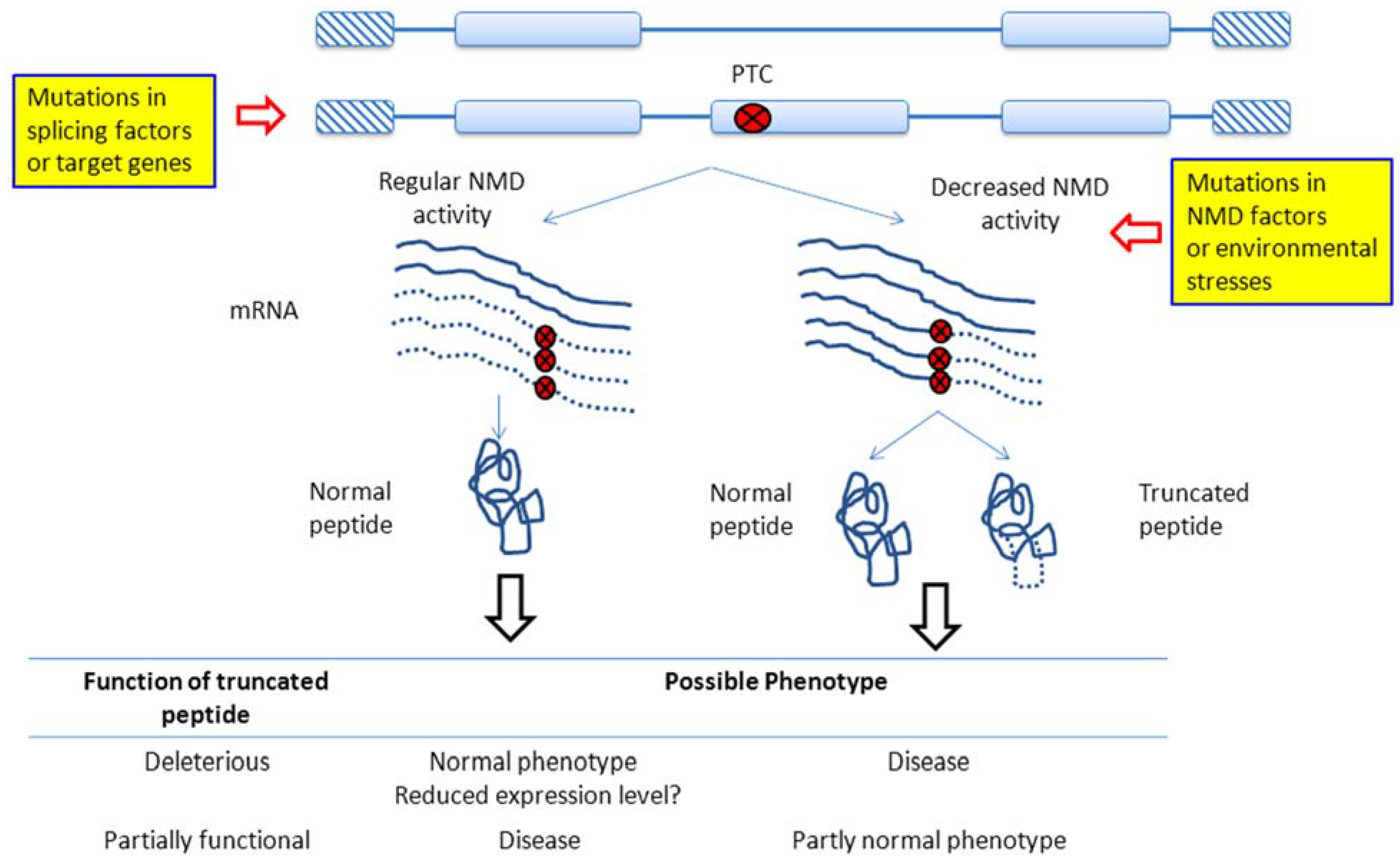
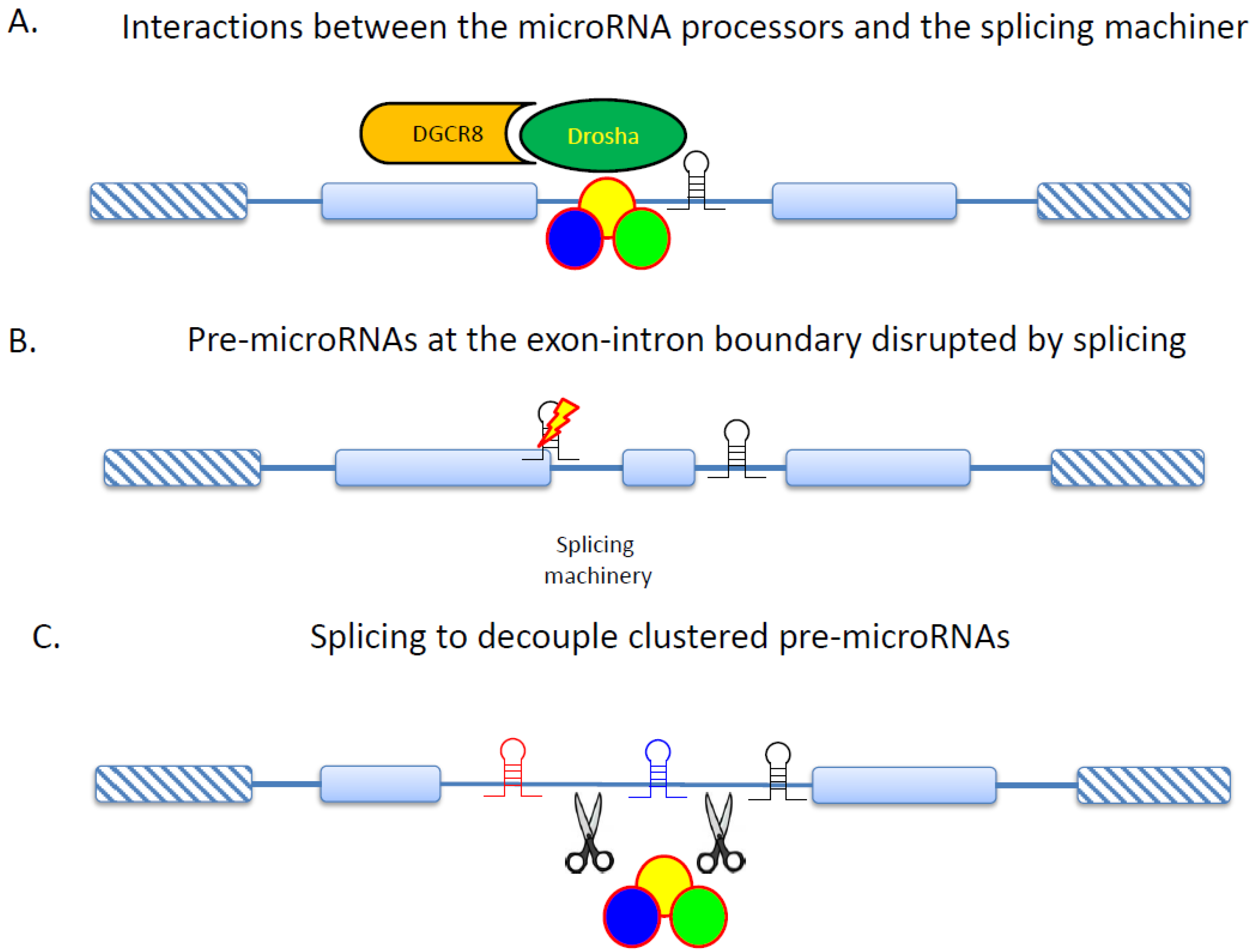
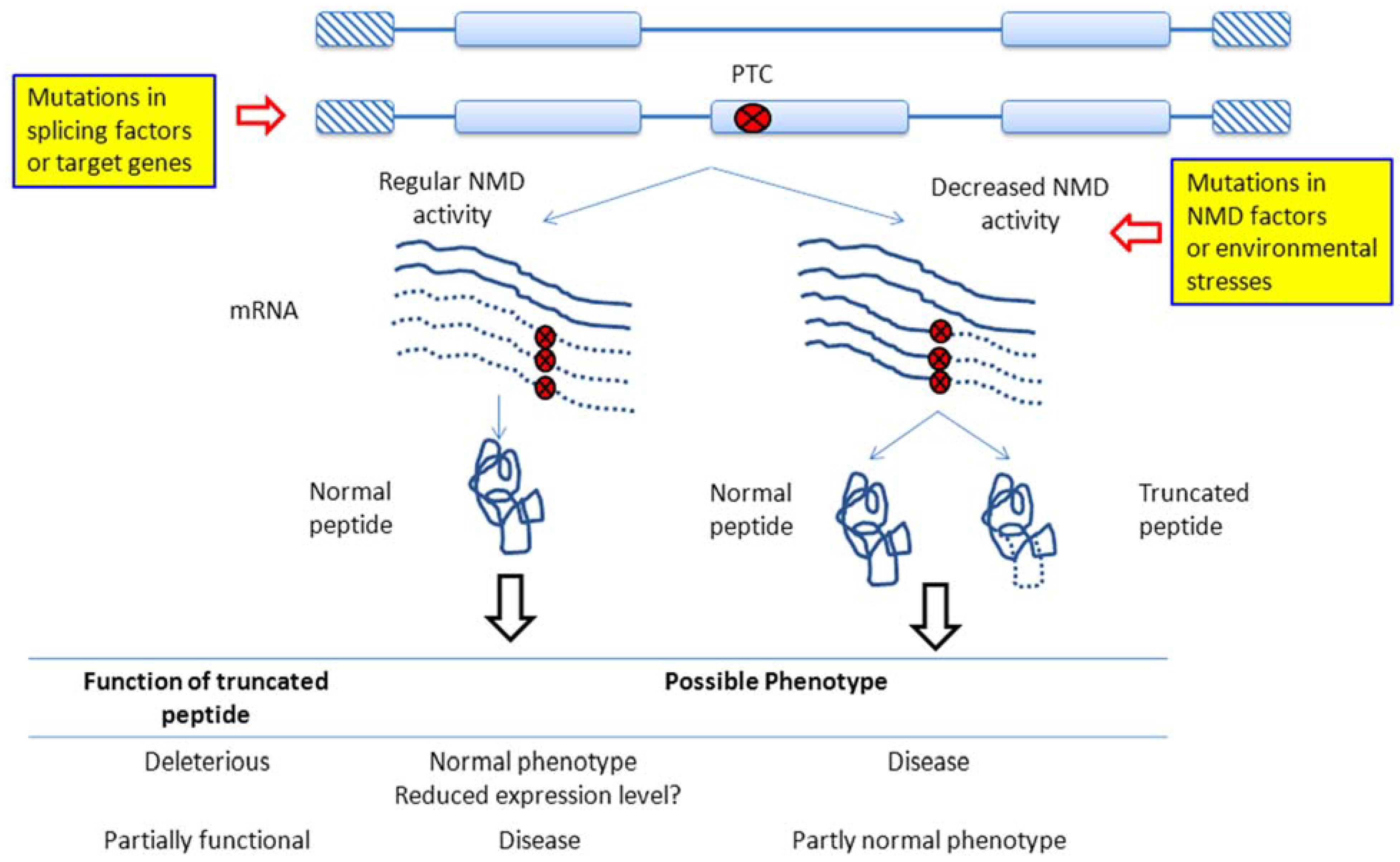
2. NMD Regulation in Cancer
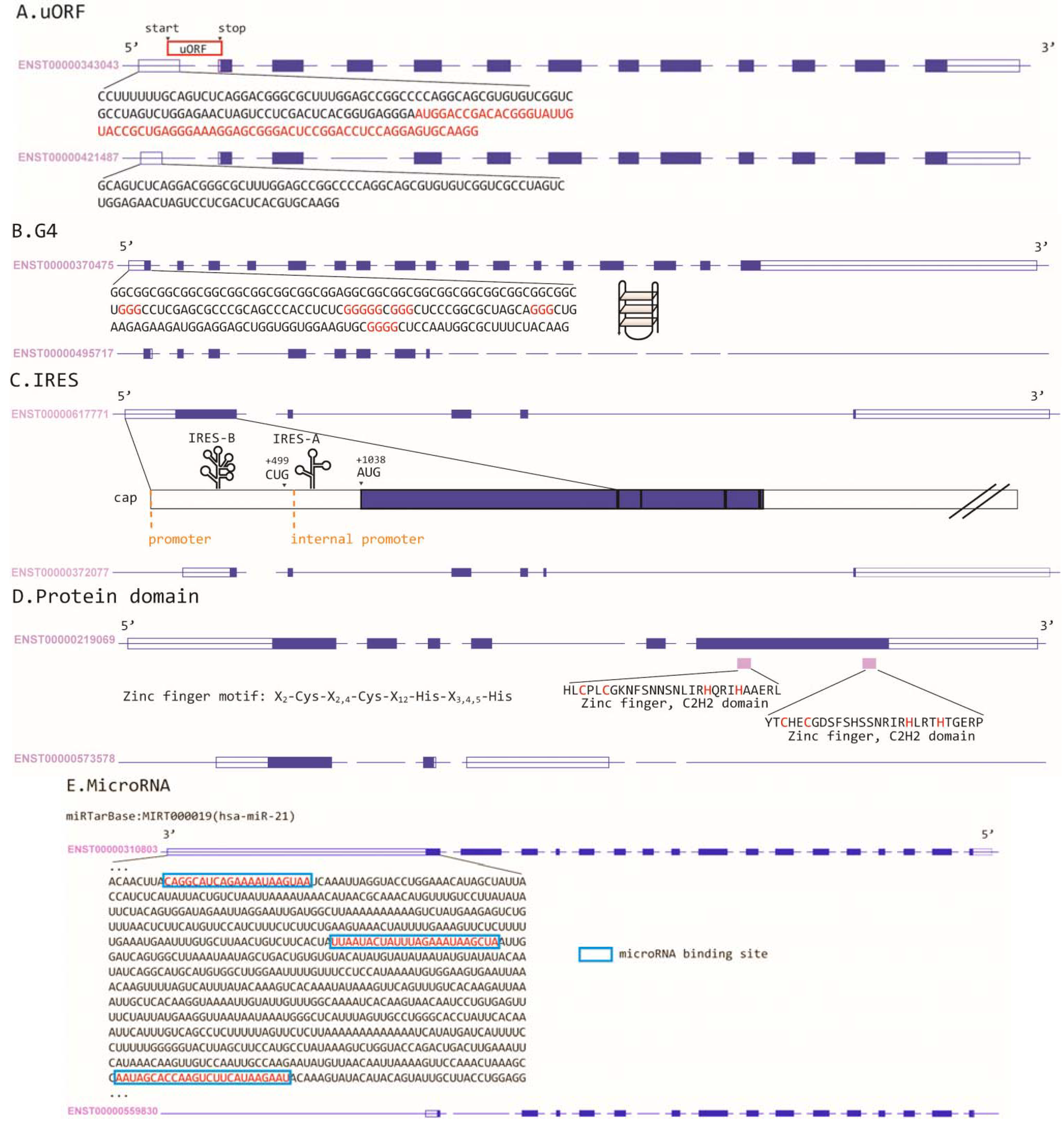
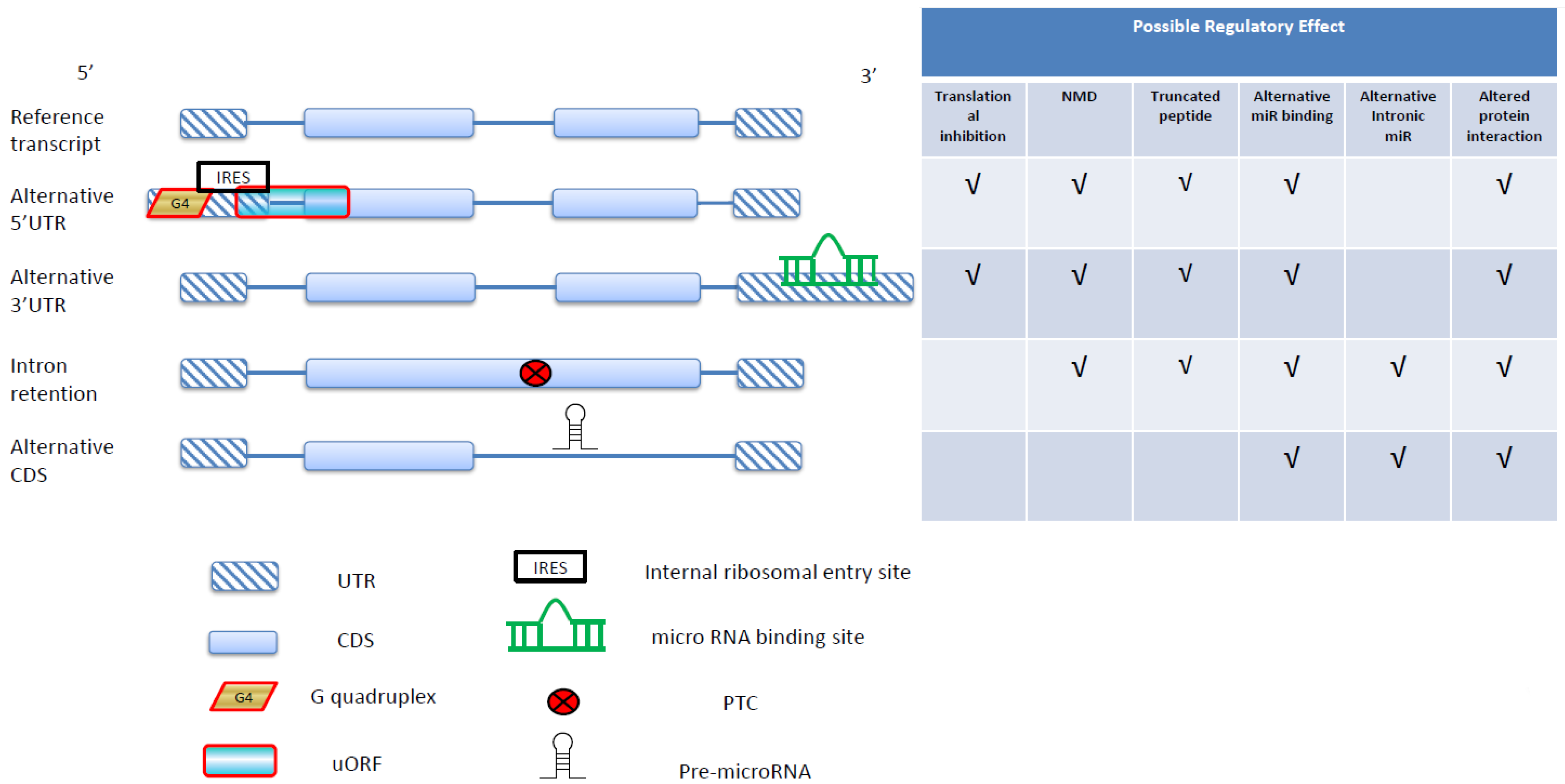
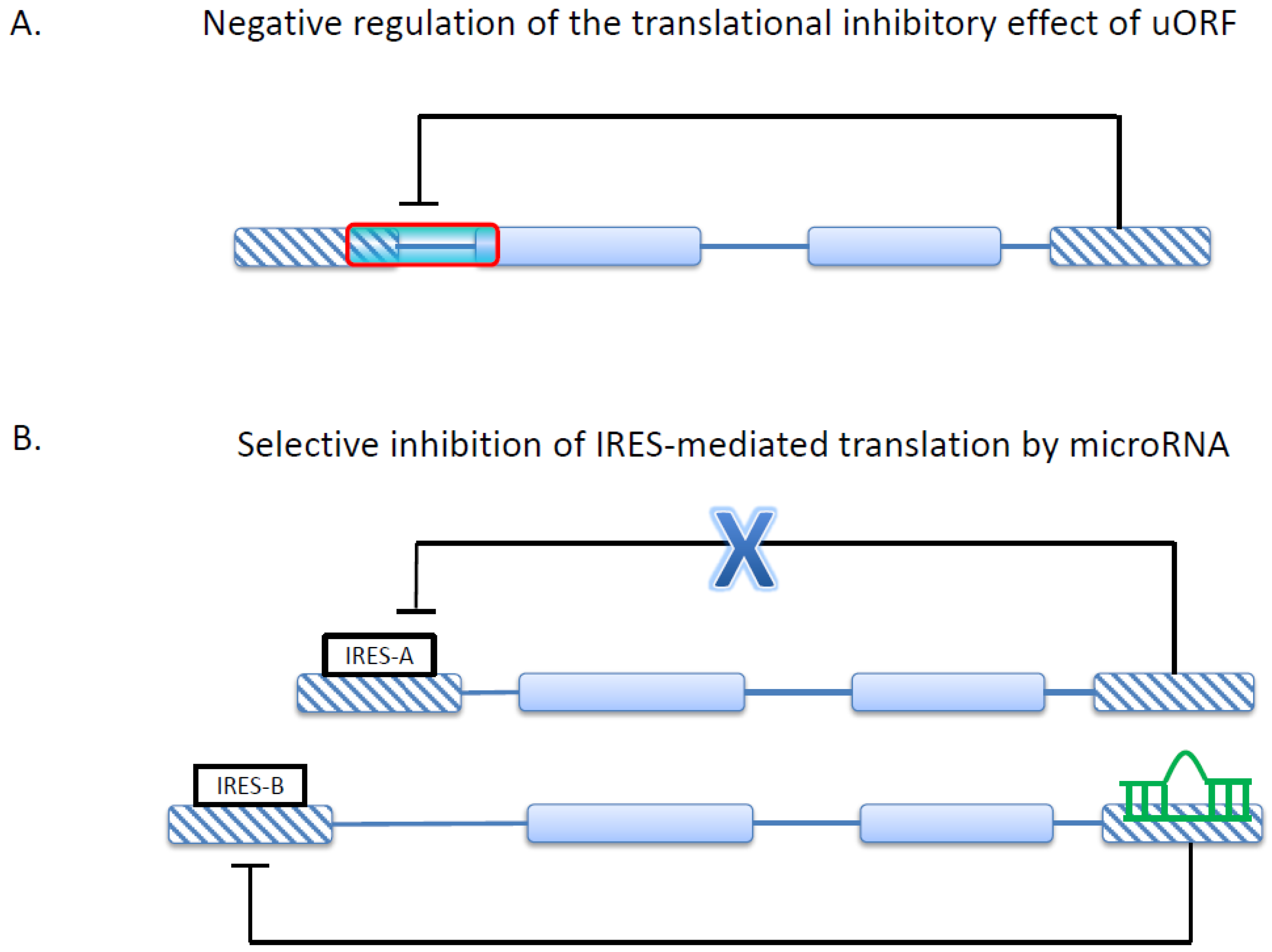
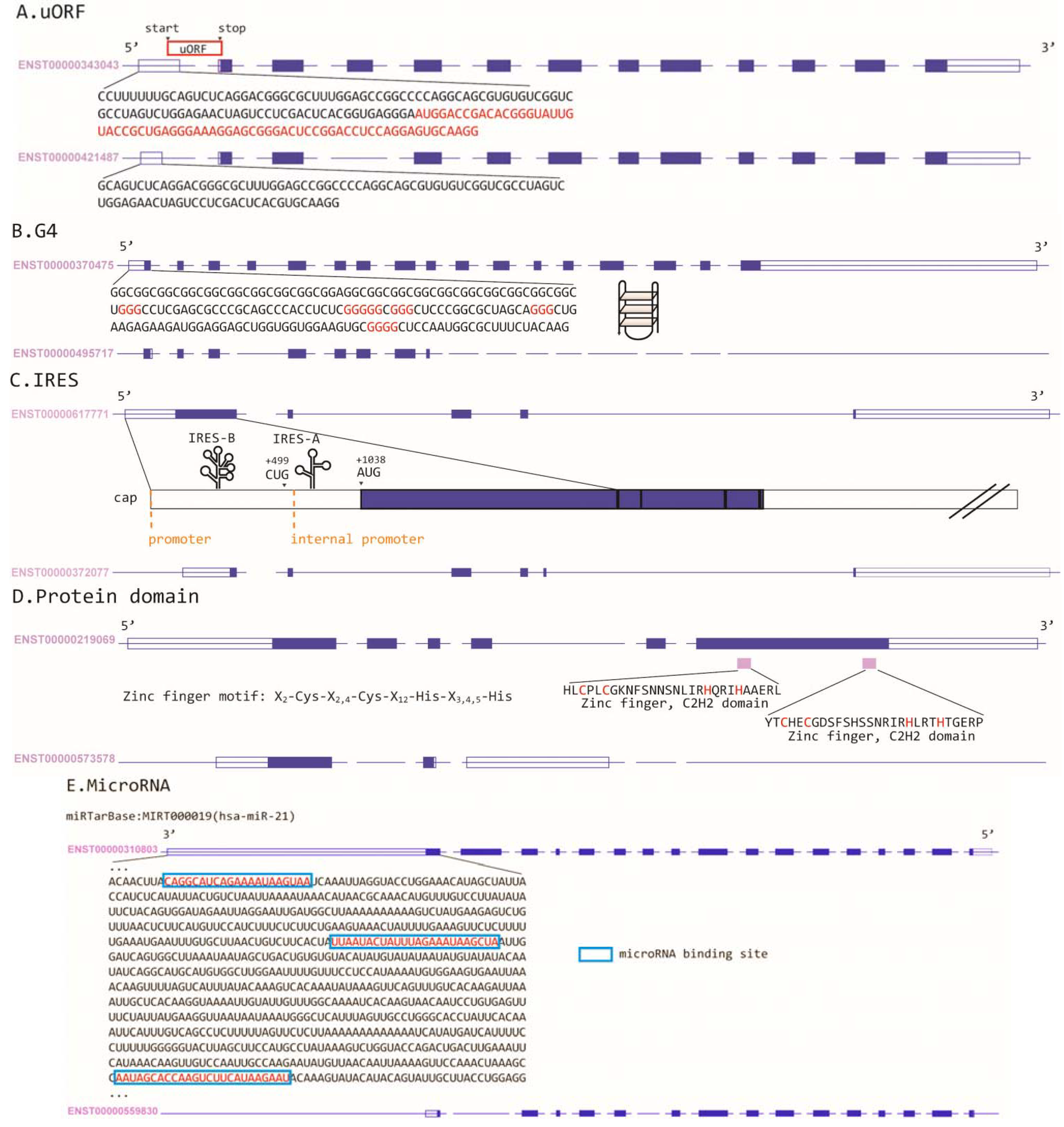
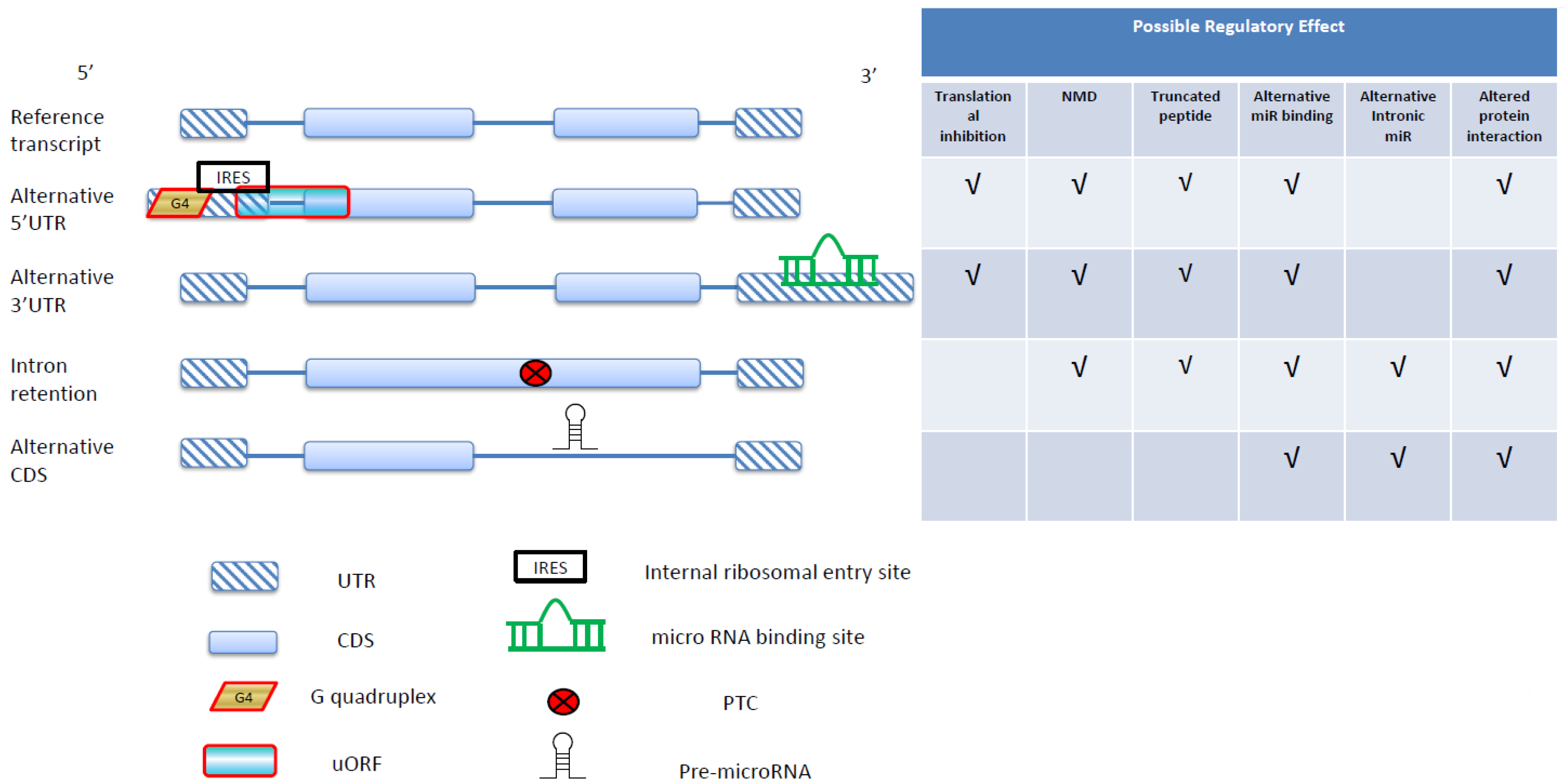
3. Alternative 5'UTR and Translational Regulations in Cancer
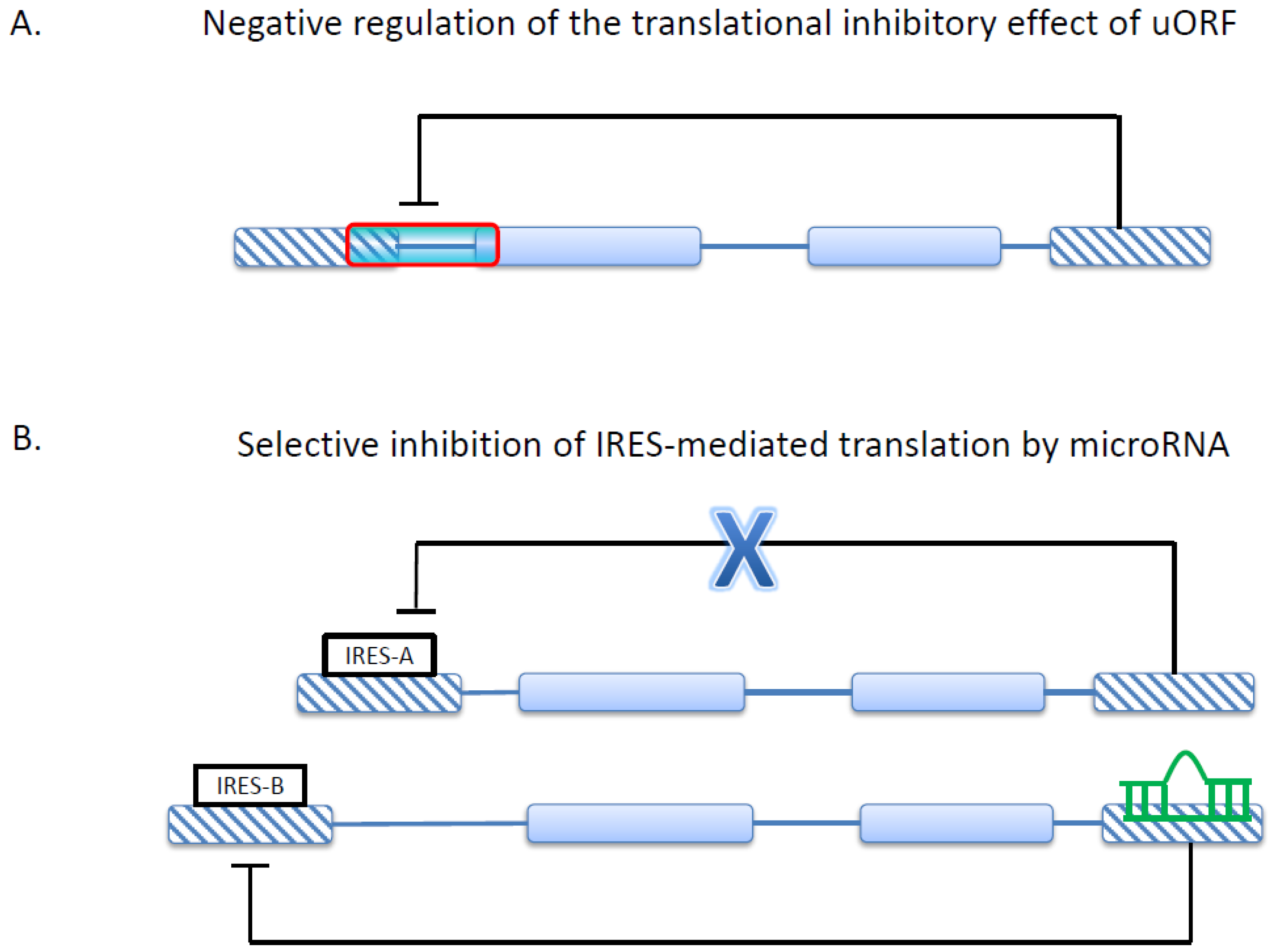
4. Alternative 3'UTR and MicroRNA-Mediated Gene Regulation in Cancer
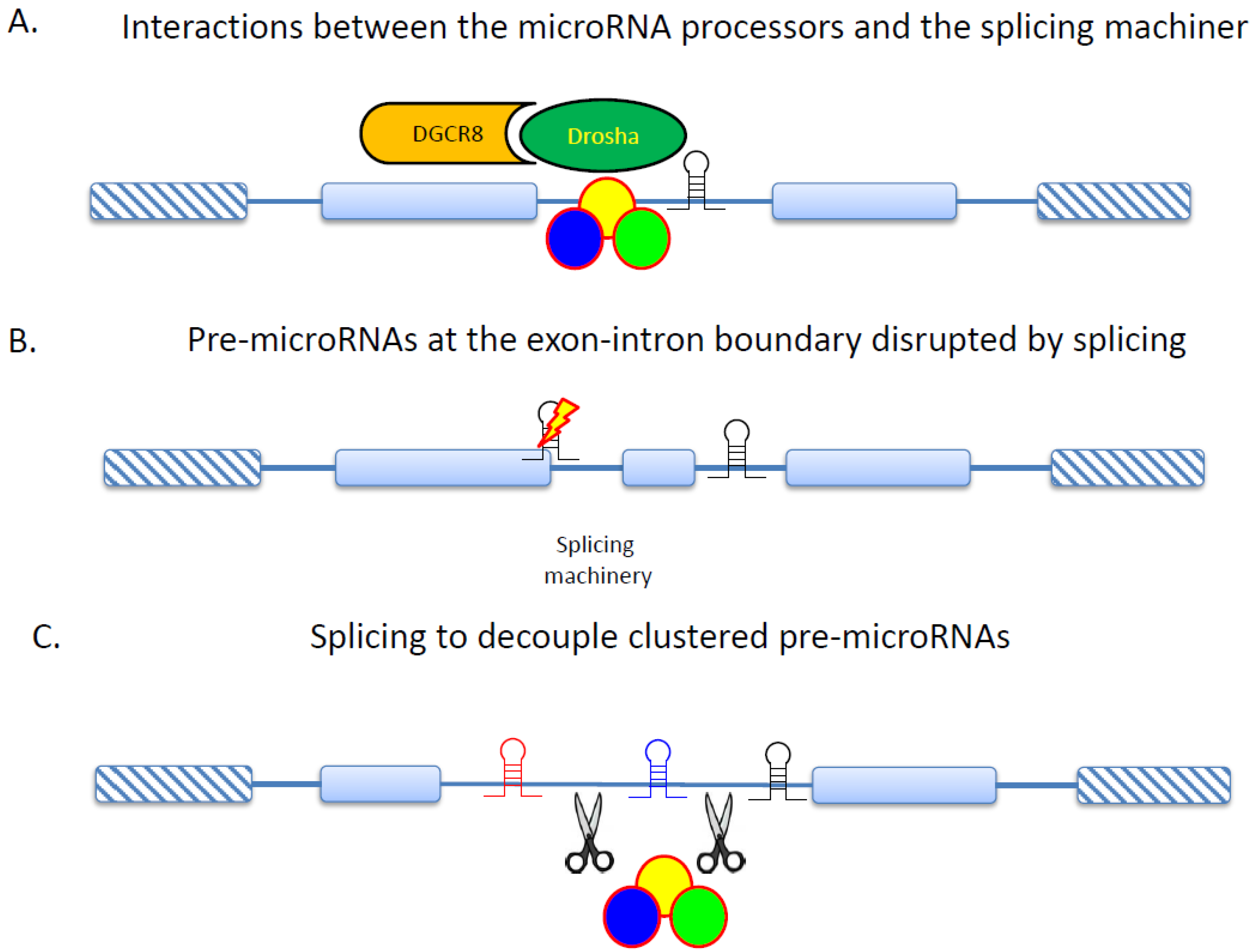
5. RNA Splicing and Protein Interactions in Cancer
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- </i>Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3154–3158. [Google Scholar] [CrossRef] [PubMed]
- Filipp, F.V. Cancer metabolism meets systems biology: Pyruvate kinase isoform PKM2 is a metabolic master regulator. J. Carcinog. 2013, 12. [Google Scholar] [CrossRef]
- Pimentel, H.; Parra, M.; Gee, S.; Ghanem, D.; An, X.; Li, J.; Mohandas, N.; Pachter, L.; Conboy, J.G. A dynamic alternative splicing program regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 2014, 42, 4031–4042. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cheng, Y.; Wu, W.; Liu, Y.; Wei, N.; Feng, X.; Xie, Z.; Feng, Y. SRSF10 regulates alternative splicing and is required for adipocyte differentiation. Mol. Cell. Biol. 2014, 34, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Loh, Y.H.; Li, H.; Cesana, M.; Ficarro, S.B.; Parikh, J.R.; Salomonis, N.; Toh, C.X.; Andreadis, S.T.; Luckey, C.J.; et al. Alternative splicing of MBD2 supports self-renewal in human pluripotent stem cells. Cell Stem Cell 2014, 15, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Martello, G. Let’s sp(l)ice up pluripotency! EMBO J. 2013, 32, 2903–2904. [Google Scholar] [CrossRef] [PubMed]
- Livyatan, I.; Meshorer, E. SON sheds light on RNA splicing and pluripotency. Nat. Cell Biol. 2013, 15, 1139–1140. [Google Scholar] [CrossRef] [PubMed]
- Kouro, H.; Kon, S.; Matsumoto, N.; Miyashita, T.; Kakuchi, A.; Ashitomi, D.; Saitoh, K.; Nakatsuru, T.; Togi, S.; Muromoto, R.; et al. The novel α4B murine α4 integrin protein splicing variant inhibits α4 protein-dependent cell adhesion. J. Biol. Chem. 2014, 289, 16389–16398. [Google Scholar] [CrossRef] [PubMed]
- Boucard, A.A.; Maxeiner, S.; Sudhof, T.C. Latrophilins function as heterophilic cell-adhesion molecules by binding to teneurins: Regulation by alternative splicing. J. Biol. Chem. 2014, 289, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Bechara, E.G.; Sebestyen, E.; Bernardis, I.; Eyras, E.; Valcarcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 2013, 52, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.; Roy, S.G.; Tsai, Y.S.; Tripathy, A.; Graves, L.M.; Wang, Z. The splicing activator DAZAP1 integrates splicing control into MEK/Erk-regulated cell proliferation and migration. Nat. Commun. 2014, 5, 3078. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Fujibuchi, W.; Unno, M. Splice variants in apoptotic pathway. Exp. Oncol. 2012, 34, 212–217. [Google Scholar] [PubMed]
- Akgul, C.; Moulding, D.A.; Edwards, S.W. Alternative splicing of Bcl-2-related genes: Functional consequences and potential therapeutic applications. Cell. Mol. Life Sci. 2004, 61, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Xie, J. Aberrant splicing in neurological diseases. Wiley Interdiscip. Rev. RNA 2013, 4, 631–649. [Google Scholar] [PubMed]
- Fan, X.; Tang, L. Aberrant and alternative splicing in skeletal system disease. Gene 2013, 528, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Lara-Pezzi, E.; Gomez-Salinero, J.; Gatto, A.; Garcia-Pavia, P. The alternative heart: Impact of alternative splicing in heart disease. J. Cardiovasc. Transl. Res. 2013, 6, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The alternative splicing side of cancer. Semin. Cell Dev. Biol. 2014, 32, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Stranger, B.E. Genomics of alternative splicing: evolution, development and pathophysiology. Hum. Genet. 2014, 133, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M. mRNA splicing variants: Exploiting modularity to outwit cancer therapy. Cancer Res. 2013, 73, 5309–5314. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2013, 33. [Google Scholar] [CrossRef]
- Pal, S.; Gupta, R.; Davuluri, R.V. Alternative transcription and alternative splicing in cancer. Pharmacol. Ther. 2012, 136, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Brugiolo, M.; Herzel, L.; Neugebauer, K.M. Counting on co-transcriptional splicing. F1000prime Rep. 2013, 5. [Google Scholar] [CrossRef]
- Kozak, M. The scanning model for translation: An update. J. Cell Biol. 1989, 108, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, J.D.; Perreault, J.P. 5'-UTR G-quadruplex structures acting as translational repressors. Nucleic Acids Res. 2010, 38, 7022–7036. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K.; Barbosa-Silva, A.; Andrade-Navarro, M.A.; Leutz, A. uORFdb—A comprehensive literature database on eukaryotic uORF biology. Nucleic Acids Res. 2013. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Wilkinson, M.F.; Gecz, J. Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neurosci. Biobehav. Rev. 2014, 46, 175–186. [Google Scholar] [CrossRef]
- Wu, C.T.; Chiou, C.Y.; Chiu, H.C.; Yang, U.C. Fine-tuning of microRNA-mediated repression of mRNA by splicing-regulated and highly repressive microRNA recognition element. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Buckley, P.T.; Khaladkar, M.; Kim, J.; Eberwine, J. Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. Wiley Interdiscip. Rev. RNA 2014, 5, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R. Regulation of gene expression through production of unstable mRNA isoforms. Biochem. Soc. Trans. 2014, 42, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Hamid, F.M.; Makeyev, E.V. Emerging functions of alternative splicing coupled with nonsense-mediated decay. Biochem. Soc. Trans. 2014, 42, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Haupt, L.M.; Griffiths, L.R. Review: Alternative Splicing (AS) of genes as an approach for generating protein complexity. Curr. Genomics 2013, 14, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Schor, I.E.; Allo, M.; Dujardin, G.; Petrillo, E.; Munoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Reviews. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef]
- Schwerk, C.; Schulze-Osthoff, K. Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell 2005, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Munaut, C.; Colige, A.C.; Lambert, C.A. Alternative splicing: A promising target for pharmaceutical inhibition of pathological angiogenesis? Curr. Pharm. Des. 2010, 16, 3864–3876. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rew. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef]
- Vegran, F.; Mary, R.; Gibeaud, A.; Mirjolet, C.; Collin, B.; Oudot, A.; Charon-Barra, C.; Arnould, L.; Lizard-Nacol, S.; Boidot, R. Survivin-3B potentiates immune escape in cancer but also inhibits the toxicity of cancer chemotherapy. Cancer Res. 2013, 73, 5391–5401. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Feng, A.; Chen, S.; Zhang, Y.; Xie, Q.; Yang, M.; Shao, Q.; Liu, J.; Yang, Q.; Kong, B.; et al. Osteopontin splice variants expressed by breast tumors regulate monocyte activation via MCP-1 and TGF-β1. Cell. Mol. Immunol. 2013, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, C.C.; Carstens, R.P. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin. Cancer Biol. 2012, 22, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Novikov, L.; Park, J.W.; Chen, H.; Klerman, H.; Jalloh, A.S.; Gamble, M.J. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol. Cell. Biol. 2011, 31, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F.; Maquat, L.E. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr. Opin. Cell Biol. 2005, 17, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Palacios, I.M. Nonsense-mediated mRNA decay: from mechanistic insights to impacts on human health. Brief. Funct. Genomics 2013, 12, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Jangi, M.; Boutz, P.L.; Paul, P.; Sharp, P.A. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014, 28, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.Z.; Grate, L.; Donohue, J.P.; Preston, C.; Nobida, N.; O’Brien, G.; Shiue, L.; Clark, T.A.; Blume, J.E.; Ares, M., Jr. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007, 21, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Kashima, I.; Higuchi, I.; Osame, M.; Ohno, S. Specific inhibition of nonsense-mediated mRNA decay components, SMG-1 or Upf1, rescues the phenotype of Ullrich disease fibroblasts. Mol. Ther. 2006, 14, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Shiraishi, T.; Shiga, A.; Onodera, O.; Higuchi, I.; Ohno, S. Inhibition of SMG-8, a subunit of SMG-1 kinase, ameliorates nonsense-mediated mRNA decay-exacerbated mutant phenotypes without cytotoxicity. Proc. Natl. Acad. Sci. USA 2013, 110, 15037–15042. [Google Scholar] [CrossRef] [PubMed]
- Sako, Y.; Usuki, F.; Suga, H. A novel therapeutic approach for genetic diseases by introduction of suppressor tRNA. Nucleic Acids Symp. Ser. 2006, 50, 239–240. [Google Scholar] [CrossRef]
- Gardner, L.B. Nonsense-mediated RNA decay regulation by cellular stress: Implications for tumorigenesis. Mol. Cancer Res. 2010, 8, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zavadil, J.; Martin, L.; Parisi, F.; Friedman, E.; Levy, D.; Harding, H.; Ron, D.; Gardner, L.B. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell. Biol. 2011, 31, 3670–3680. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Damgaard, I.; Bryder, D.; Theilgaard-Monch, K.; Thoren, L.A.; Nielsen, F.C.; Jacobsen, S.E.; Nerlov, C.; Porse, B.T. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008, 22, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Lingner, J. The double life of UPF1 in RNA and DNA stability pathways. Cell Cycle 2006, 5, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Varsally, W.; Brogna, S. UPF1 involvement in nuclear functions. Biochem. Soc. Trans. 2012, 40, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, M.; Zhang, B.; Jaeger, M.; Stamm, S.; Erbes, T.; Mayer, S.; Tong, X.; Stickeler, E. Hypoxia-dependent mRNA expression pattern of splicing factor YT521 and its impact on oncological important target gene expression. Mol. Carcinog. 2014, 53, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; zur Hausen, A.; Orlowska-Volk, M.; Jager, M.; Bettendorf, H.; Stamm, S.; Hirschfeld, M.; Yiqin, O.; Tong, X.; Gitsch, G.; et al. Alternative splicing-related factor YT521: An independent prognostic factor in endometrial cancer. Int. J. Gynecol. Cancer 2010, 20, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Miura, M.; Bergeron, L.; Zhu, H.; Yuan, J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 1994, 78, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Logette, E.; Desoche, L.; Solary, E.; Corcos, L. Nonsense-mediated mRNA decay among human caspases: The caspase-2S putative protein is encoded by an extremely short-lived mRNA. Cell Death Differ. 2005, 12, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.B.; Broz, S.D.; Levinson, A.D. Expression of the H-ras proto-oncogene is controlled by alternative splicing. Cell 1989, 58, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.B.; Levinson, A.D. A point mutation in the last intron responsible for increased expression and transforming activity of the c-Ha-ras oncogene. Nature 1988, 334, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Barbier, J.; Dutertre, M.; Bittencourt, D.; Sanchez, G.; Gratadou, L.; de la Grange, P.; Auboeuf, D. Regulation of H-ras splice variant expression by cross talk between the p53 and nonsense-mediated mRNA decay pathways. Mol. Cell. Biol. 2007, 27, 7315–7333. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; de La Iglesia, N.; Fernandez-Larrea, J.; Cifuentes, D.; Ferrer, J.C.; Guinovart, J.J.; Bach-Elias, M. Alternative splicing of the human proto-oncogene c-H-ras renders a new Ras family protein that trafficks to cytoplasm and nucleus. Cancer Res. 2003, 63, 5178–5187. [Google Scholar] [PubMed]
- Zhao, C.; Datta, S.; Mandal, P.; Xu, S.; Hamilton, T. Stress-sensitive regulation of IFRD1 mRNA decay is mediated by an upstream open reading frame. J. Biol. Chem. 2010, 285, 8552–8562. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.B. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol. Cell. Biol. 2008, 28, 3729–3741. [Google Scholar] [CrossRef] [PubMed]
- Camats, M.; Kokolo, M.; Heesom, K.J.; Ladomery, M.; Bach-Elias, M. P19 H-ras induces G1/S phase delay maintaining cells in a reversible quiescence state. PLoS One 2009, 4, e8513. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Karam, R.; Zhou, Y.; Su, F.; Ji, Y.; Li, G.; Xu, G.; Lu, L.; Wang, C.; Song, M.; et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014, 20, 596–598. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Svenson, K.B.; Longmate, W.M.; Gkirtzimanaki, K.; Sadej, R.; Wang, X.; Zhao, J.; Eliopoulos, A.G.; Berditchevski, F.; Dipersio, C.M. Suppression of integrin α3β1 in breast cancer cells reduces cyclooxygenase-2 gene expression and inhibits tumorigenesis, invasion, and cross-talk to endothelial cells. Cancer Res. 2010, 70, 6359–6367. [Google Scholar] [CrossRef] [PubMed]
- Subbaram, S.; Lyons, S.P.; Svenson, K.B.; Hammond, S.L.; McCabe, L.G.; Chittur, S.V.; DiPersio, C.M. Integrin α3β1 controls mRNA splicing that determines Cox-2 mRNA stability in breast cancer cells. J. Cell Sci. 2014, 127, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.C. Are all of the human exons alternatively spliced? Brief. Bioinform. 2013. [Google Scholar] [CrossRef]
- Hsu, M.K.; Chen, F.C. Selective constraint on the upstream open reading frames that overlap with coding sequences in animals. PLoS One 2012, 7, e48413. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Liao, B.Y.; Chen, F.C. Exploring the selective constraint on the sizes of insertions and deletions in 5' untranslated regions in mammals. BMC Evol. Biol. 2011, 11, 192. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, G.D.; Derecka, K.; Garner, T.P.; Hodgman, C.; Flint, A.P.; Searle, M.S. Repression of translation of human estrogen receptor α by G-quadruplex formation. Biochemistry 2009, 48, 11487–11495. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.A. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006, 22, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [PubMed]
- Somers, J.; Poyry, T.; Willis, A.E. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013, 45, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Yachie, N.; Okada, Y.; Saito, R.; Tomita, M. Bioinformatic analysis of post-transcriptional regulation by uORF in human and mouse. FEBS Lett. 2007, 581, 4184–4188. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K.; Smink, J.J.; Leutz, A. Upstream open reading frames: Molecular switches in (patho)physiology. BioEssays 2010, 32, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Le Quesne, J.P.; Spriggs, K.A.; Bushell, M.; Willis, A.E. Dysregulation of protein synthesis and disease. J. Pathol. 2010, 220, 140–151. [Google Scholar]
- Child, S.J.; Miller, M.K.; Geballe, A.P. Translational control by an upstream open reading frame in the HER-2/neu transcript. J. Biol. Chem. 1999, 274, 24335–24341. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Miller, M.K.; Geballe, A.P. Cell type-dependent and -independent control of HER-2/neu translation. Int. J. Biochem. Cell Biol. 1999, 31, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Trotta, C.R.; Peltz, S.W. Derepression of the HER-2 uORF is mediated by a novel post-transcriptional control mechanism in cancer cells. Genes Dev. 2006, 20, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Nerlov, C. The C/EBP family of transcription factors: A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Descombes, P.; Schibler, U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 1991, 67, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Calkhoven, C.F.; Muller, C.; Leutz, A. Translational control of C/EBPα and C/EBPβ isoform expression. Genes Dev. 2000, 14, 1920–1932. [Google Scholar] [PubMed]
- Jundt, F.; Raetzel, N.; Muller, C.; Calkhoven, C.F.; Kley, K.; Mathas, S.; Lietz, A.; Leutz, A.; Dorken, B. A rapamycin derivative (everolimus) controls proliferation through down-regulation of truncated CCAAT enhancer binding protein β and NF-κB activity in Hodgkin and anaplastic large cell lymphomas. Blood 2005, 106, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Zahnow, C.A. CCAAT/enhancer-binding protein β: Its role in breast cancer and associations with receptor tyrosine kinases. Expert Rev. Mol. Med. 2009, 11. [Google Scholar] [CrossRef]
- Nerlov, C. C/EBPα mutations in acute myeloid leukaemias. Nat. Rew. Cancer 2004, 4, 394–400. [Google Scholar] [CrossRef]
- Smith, M.L.; Cavenagh, J.D.; Lister, T.A.; Fitzgibbon, J. Mutation of CEBPA in familial acute myeloid leukemia. N. Engl. J. Med. 2004, 351, 2403–2407. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, K.; Krzyzosiak, W.J. Structural determinants of BRCA1 translational regulation. J. Biol. Chem. 2002, 277, 17349–17358. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Saji, S.; Eguchi, H.; Nakashima, S.; Hayashi, S. Distinct promoter usage of mdm2 gene in human breast cancer. Oncol. Rep. 2002, 9, 557–563. [Google Scholar] [PubMed]
- Brown, C.Y.; Mize, G.J.; Pineda, M.; George, D.L.; Morris, D.R. Role of two upstream open reading frames in the translational control of oncogene mdm2. Oncogene 1999, 18, 5631–5637. [Google Scholar] [CrossRef] [PubMed]
- Genolet, R.; Rahim, G.; Gubler-Jaquier, P.; Curran, J. The translational response of the human mdm2 gene in HEK293T cells exposed to rapamycin: A role for the 5'-UTRs. Nucleic Acids Res. 2011, 39, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Brannan, R.A.; Hanby, A.M.; Shaaban, A.M.; Verghese, E.T.; Peter, M.B.; Pollock, S.; Satheesha, S.; Szynkiewicz, M.; Speirs, V.; et al. Differential regulation of oestrogen receptor β isoforms by 5' untranslated regions in cancer. J. Cell. Mol. Med. 2010, 14, 2172–2184. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Brabletz, T.; Fearon, E.; Willis, A.L.; Hu, C.Y.; Li, X.Y.; Weiss, S.J. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11312–11317. [Google Scholar] [CrossRef] [PubMed]
- Hadjihannas, M.V.; Bruckner, M.; Jerchow, B.; Birchmeier, W.; Dietmaier, W.; Behrens, J. Aberrant Wnt/β-catenin signaling can induce chromosomal instability in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 10747–10752. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.A.; Brady, H.J. Regulation of axin2 expression at the levels of transcription, translation and protein stability in lung and colon cancer. Cancer Lett. 2006, 233, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Baranick, B.T.; Lemp, N.A.; Nagashima, J.; Hiraoka, K.; Kasahara, N.; Logg, C.R. Splicing mediates the activity of four putative cellular internal ribosome entry sites. Proc. Natl. Acad. Sci. USA 2008, 105, 4733–4738. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.A.; Hu, Y.; Inohara, N.; Nunez, G. Expression and functional analysis of Apaf-1 isoforms. Extra Wd-40 repeat is required for cytochrome c binding and regulated activation of procaspase-9. J. Biol. Chem. 2000, 275, 8461–8468. [Google Scholar] [CrossRef] [PubMed]
- Ensembl annotations on human Apaf1 (Version 77). Available online: http://www.ensembl.org (accessed on 29 October 2014).
- Bastide, A.; Karaa, Z.; Bornes, S.; Hieblot, C.; Lacazette, E.; Prats, H.; Touriol, C. An upstream open reading frame within an IRES controls expression of a specific VEGF-A isoform. Nucleic Acids Res. 2008, 36, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Martineau, Y.; le Bec, C.; Monbrun, L.; Allo, V.; Chiu, I.M.; Danos, O.; Moine, H.; Prats, H.; Prats, A.C. Internal ribosome entry site structural motifs conserved among mammalian fibroblast growth factor 1 alternatively spliced mRNAs. Mol. Cell. Biol. 2004, 24, 7622–7635. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, N.H.; Cloutier, M.; Lewis, S.M.; de Silva, N.; Blais, J.D.; Bell, J.C.; Holcik, M. Internal ribosome entry site-mediated translation of Apaf-1, but not XIAP, is regulated during UV-induced cell death. J. Biol. Chem. 2006, 281, 15155–15163. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.J.; Bruce, C.; Chioni, A.M.; Kocher, H.M.; Grose, R.P. The ins and outs of fibroblast growth factor receptor signalling. Clin. Sci. (Lond.) 2014, 127, 217–231. [Google Scholar] [CrossRef]
- Karaa, Z.S.; Iacovoni, J.S.; Bastide, A.; Lacazette, E.; Touriol, C.; Prats, H. The VEGF IRESes are differentially susceptible to translation inhibition by miR-16. RNA 2009, 15, 49–54. [Google Scholar] [CrossRef]
- Hamdollah Zadeh, M.A.; Amin, E.M.; Hoareau-Aveilla, C.; Domingo, E.; Symonds, K.E.; Ye, X.; Heesom, K.J.; Salmon, A.; D’Silva, O.; Betteridge, K.B.; et al. Alternative splicing of TIA-1 in human colon cancer regulates VEGF isoform expression, angiogenesis, tumour growth and bevacizumab resistance. Mol. Oncol. 2014. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Ladewig, E.; Okamura, K.; Flynt, A.S.; Westholm, J.O.; Lai, E.C. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res. 2012, 22, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Berillo, O.; Regnier, M.; Ivashchenko, A. Binding of intronic miRNAs to the mRNAs of host genes encoding intronic miRNAs and proteins that participate in tumourigenesis. Comp. Biol. Med. 2013, 43, 1374–1381. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Seow, Y.; Saayman, S.; Dijkstra, K.K.; el Andaloussi, S.; Weinberg, M.S.; Wood, M.J. The biogenesis and characterization of mammalian microRNAs of mirtron origin. Nucleic Acids Res. 2012, 40, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Schamberger, A.; Orban, T.I. Experimental validation of predicted mammalian microRNAs of mirtron origin. Methods Mol. Biol. 2014, 1182, 245–263. [Google Scholar] [PubMed]
- Agranat-Tamir, L.; Shomron, N.; Sperling, J.; Sperling, R. Interplay between pre-mRNA splicing and microRNA biogenesis within the supraspliceosome. Nucleic Acids Res. 2014, 42, 4640–4651. [Google Scholar] [CrossRef] [PubMed]
- Macias, S.; Cordiner, R.A.; Caceres, J.F. Cellular functions of the microprocessor. Biochem. Soc. Trans. 2013, 41, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Macias, S.; Plass, M.; Stajuda, A.; Michlewski, G.; Eyras, E.; Caceres, J.F. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol. 2012, 19, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Melamed, Z.; Levy, A.; Ashwal-Fluss, R.; Lev-Maor, G.; Mekahel, K.; Atias, N.; Gilad, S.; Sharan, R.; Levy, C.; Kadener, S.; Ast, G. Alternative splicing regulates biogenesis of miRNAs located across exon-intron junctions. Mol. Cell 2013, 50, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Reich, A.A.; Hastings, M.L. Drosha promotes splicing of a pre-microRNA-like alternative exon. PLoS Genet. 2014, 10, e1004312. [Google Scholar] [CrossRef] [PubMed]
- Bosia, C.; Osella, M.; Baroudi, M.E.; Cora, D.; Caselle, M. Gene autoregulation via intronic microRNAs and its functions. BMC Syst. Biol. 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. An intronic microRNA silences genes that are functionally antagonistic to its host gene. Nucleic Acids Res. 2008, 36, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Han, E.K.; McGonigal, T. Role of focal adhesion kinase in human cancer: A potential target for drug discovery. Anticancer Agents Med. Chem. 2007, 7, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sakabe, I.; Patel, S.; Zhang, Y.; Ahmad, I.; Gehan, E.A.; Whiteside, T.L.; Kasid, U. SCC-112, a novel cell cycle-regulated molecule, exhibits reduced expression in human renal carcinomas. Gene 2004, 328, 187–196. [Google Scholar] [CrossRef]
- Brancolini, C.; Bottega, S.; Schneider, C. Gas2, a growth arrest-specific protein, is a component of the microfilament network system. J. Cell Biol. 1992, 117, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, A.; Careccia, S.; Sagrestani, G.; Nanni, S.; Manni, I.; Schinzari, V.; Martens, J.H.; Farsetti, A.; Stunnenberg, H.G.; Gentileschi, M.P.; et al. Dual promoter usage as regulatory mechanism of let-7c expression in leukemic and solid tumors. Mol. Cancer Res. 2014, 12, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Relle, M.; Becker, M.; Meyer, R.G.; Stassen, M.; Schwarting, A. Intronic promoters and their noncoding transcripts: A new source of cancer-associated genes. Mol. Carcinog. 2014, 53, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P.; Palanichamy, J.K.; Singh, A.; Das, P.; Bhagat, M.; Kassab, M.A.; Sinha, S.; Chattopadhyay, P. Biogenesis of intronic miRNAs located in clusters by independent transcription and alternative splicing. RNA 2014, 20, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5'-UTR and 3'-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [PubMed]
- Meng, Y.; Shao, C.; Ma, X.; Wang, H. Introns targeted by plant microRNAs: A possible novel mechanism of gene regulation. Rice 2013, 6. [Google Scholar] [CrossRef]
- Fang, Z.; Rajewsky, N. The impact of miRNA target sites in coding sequences and in 3'UTRs. PLoS One 2011, 6, e18067. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Spies, N.; Burge, C.B.; Bartel, D.P. 3'UTR-isoform choice has limited influence on the stability and translational efficiency of most mRNAs in mouse fibroblasts. Genome Res. 2013, 23, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Maniar, J.M.; Fire, A.Z. “Inc-miRs”: Functional intron-interrupted miRNA genes. Genes Dev. 2011, 25, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharm. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Cheng, T.M.; Bates, P.A. Cancer networks and beyond: Interpreting mutations using the human interactome and protein structure. Semin. Cancer Biol. 2013, 23, 219–26. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmaros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: a novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [PubMed]
- Kotlyar, M.; Fortney, K.; Jurisica, I. Network-based characterization of drug-regulated genes, drug targets, and toxicity. Methods 2012, 57, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.R.; Zaidi, S.; Fang, Y.Y.; Uversky, V.N.; Radivojac, P.; Oldfield, C.J.; Cortese, M.S.; Sickmeier, M.; LeGall, T.; Obradovic, Z.; et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc. Natl. Acad. Sci. USA 2006, 103, 8390–8395. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Chalancon, G.; Dunker, A.K.; Bateman, A.; Balaji, S.; Fuxreiter, M.; Babu, M.M. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr. Opinion Struct. Biol. 2013, 23, 443–450. [Google Scholar] [CrossRef]
- Sinha, A.; Nagarajaram, H.A. Effect of alternative splicing on the degree centrality of nodes in protein-protein interaction networks of Homo sapiens. J. Proteome Res. 2013, 12, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Buljan, M.; Chalancon, G.; Eustermann, S.; Wagner, G.P.; Fuxreiter, M.; Bateman, A.; Babu, M.M. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 2012, 46, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.D.; Barrios-Rodiles, M.; Colak, R.; Irimia, M.; Kim, T.; Calarco, J.A.; Wang, X.; Pan, Q.; O’Hanlon, D.; Kim, P.M.; et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell 2012, 46, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J. An exon-centric perspective. Biochem. Cell Biol. 2012, 90, 603–612. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.-C. Alternative RNA Structure-Coupled Gene Regulations in Tumorigenesis. Int. J. Mol. Sci. 2015, 16, 452-475. https://doi.org/10.3390/ijms16010452
Chen F-C. Alternative RNA Structure-Coupled Gene Regulations in Tumorigenesis. International Journal of Molecular Sciences. 2015; 16(1):452-475. https://doi.org/10.3390/ijms16010452
Chicago/Turabian StyleChen, Feng-Chi. 2015. "Alternative RNA Structure-Coupled Gene Regulations in Tumorigenesis" International Journal of Molecular Sciences 16, no. 1: 452-475. https://doi.org/10.3390/ijms16010452