
De Novo Assembly and Characterization of Narrow-Ridged Finless Porpoise Renal Transcriptome and Identification of Candidate Genes Involved in Osmoregulation

Abstract
:1. Introduction
2. Results and Discussion
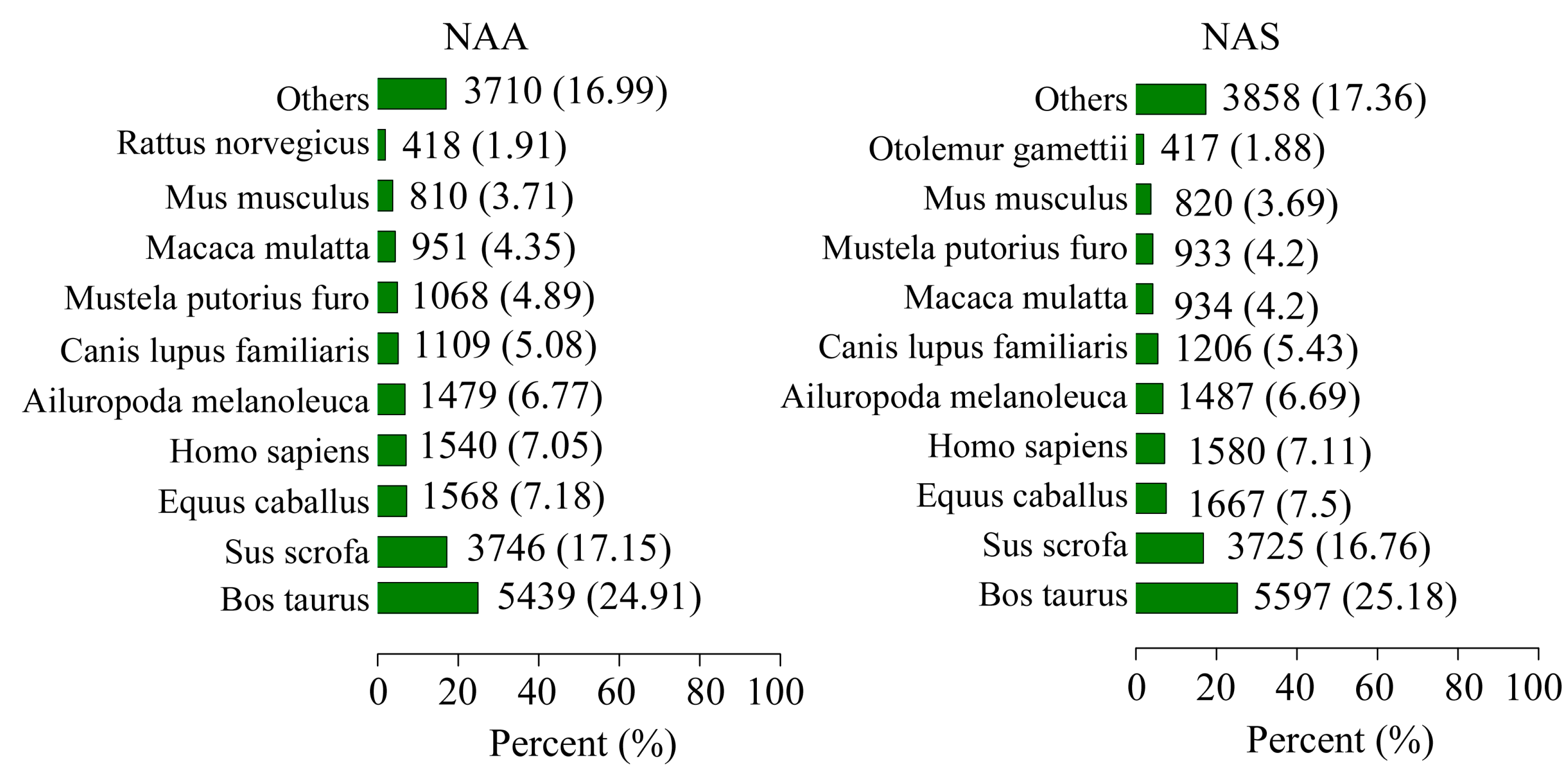
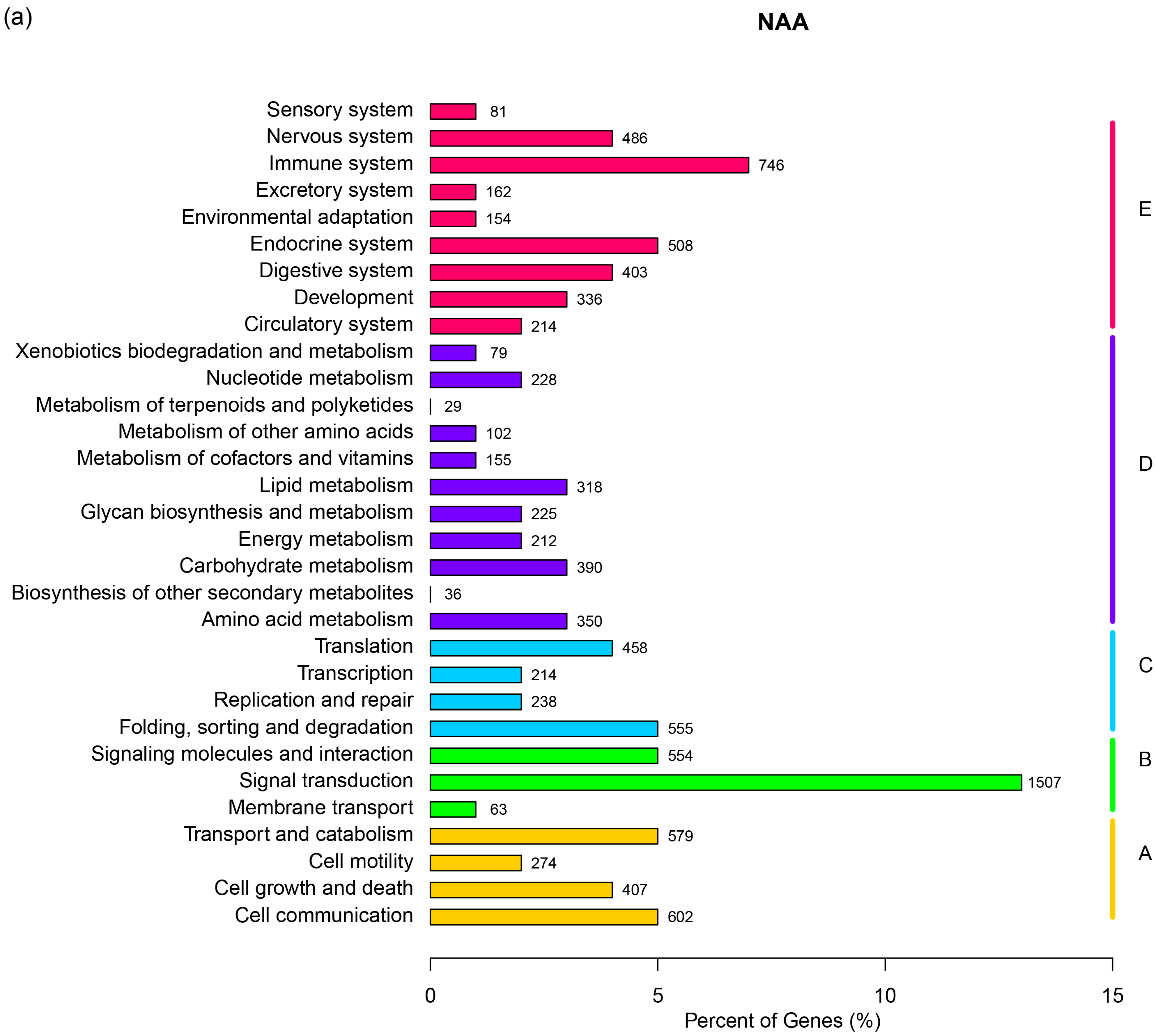
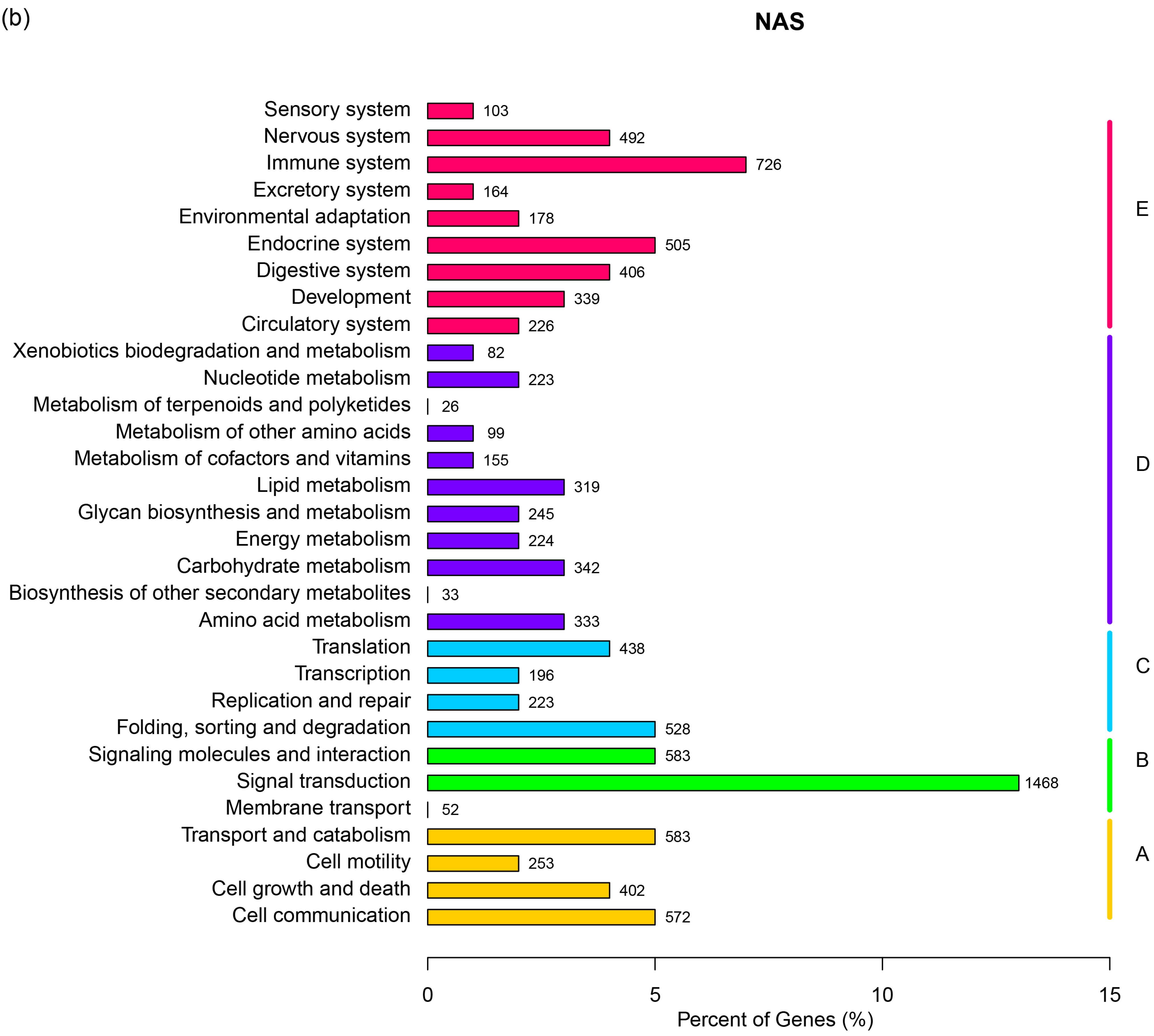
2.1. RNA Sequencing, de Novo Assembly and Functional Annotation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | NAA | NAS | Combined |
|---|---|---|---|
| Number of assembled transcripts | 105,813 | 114,057 | 165,547 |
| Number of unigenes | 68,073 | 73,449 | 103,077 |
| Mean unigene length (bp) | 735 | 740 | 685 |
| Maximum unigene length (bp) | 19,040 | 12,579 | 17,179 |
| Minimum unigene length (bp) | 201 | 201 | 201 |
| N50 (unigenes) (bp) | 1389 | 1447 | 1227 |
| N90 (unigenes) (bp) | 270 | 269 | 260 |
| Public Database | Number of Annotated Unigene | Percentage (%) of Assembled Unigenes | ||
|---|---|---|---|---|
| NAA | NAS | NAA (68,073) | NAS (73,449) | |
| NR | 21,849 | 22,231 | 32.10 | 30.27 |
| NT | 35,260 | 36,661 | 51.80 | 49.91 |
| SwissProt | 20,485 | 20,639 | 30.10 | 28.10 |
| GO | 17,550 | 18,163 | 25.78 | 24.73 |
| KOG | 11,555 | 11,867 | 16.97 | 16.16 |
| KEGG | 11,165 | 11,163 | 16.40 | 15.20 |

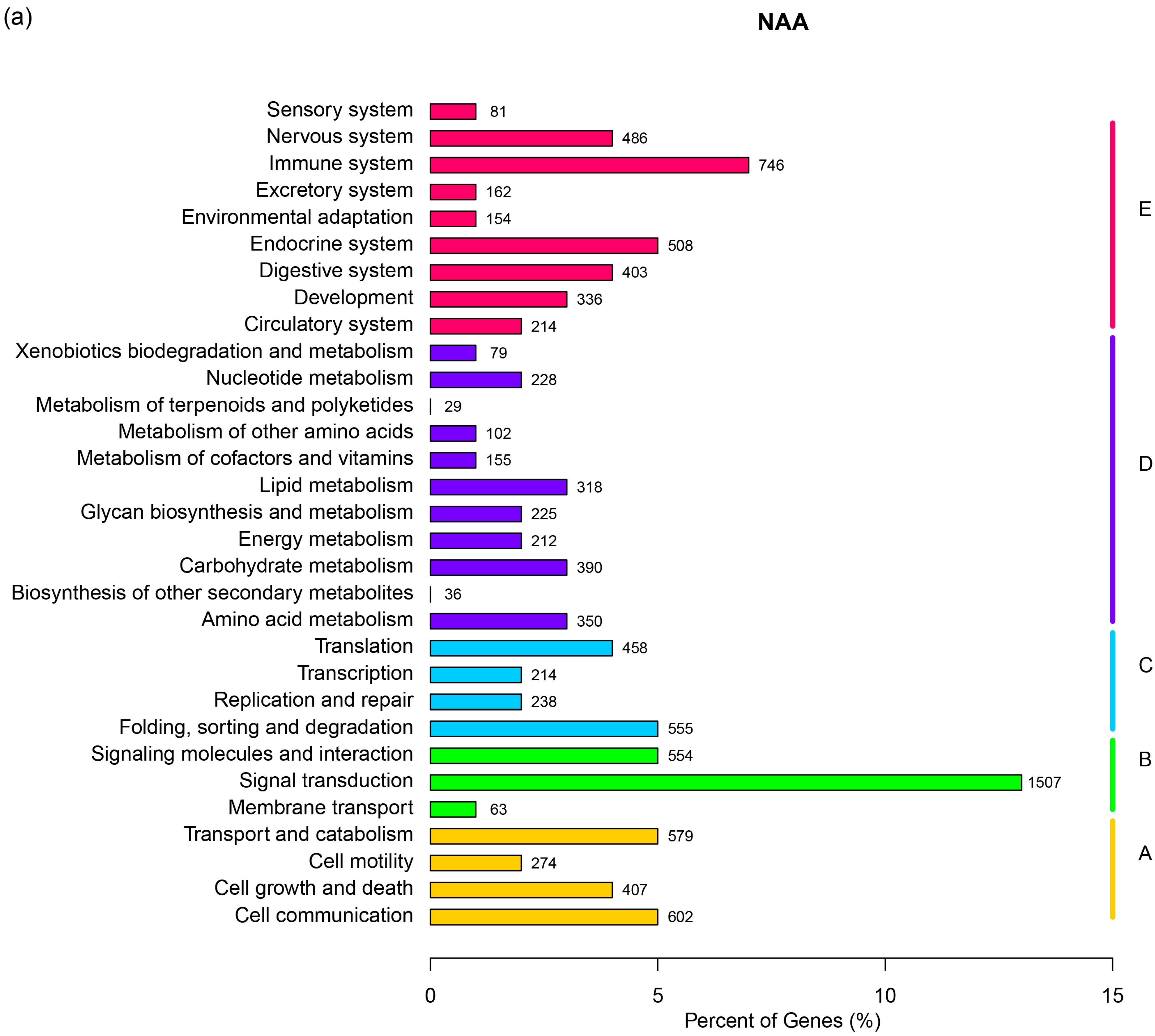

| KEGG Pathway | The Number of Unigenes in NAA | The Number of Unigenes in NAS |
|---|---|---|
| Renin-angiotensin system | 18 | 22 |
| Aldosterone-regulated sodium reabsorption | 41 | 35 |
| Proximal tubule bicarbonate reclamation | 18 | 19 |
| Vasopressin-regulated water reabsorption | 50 | 59 |
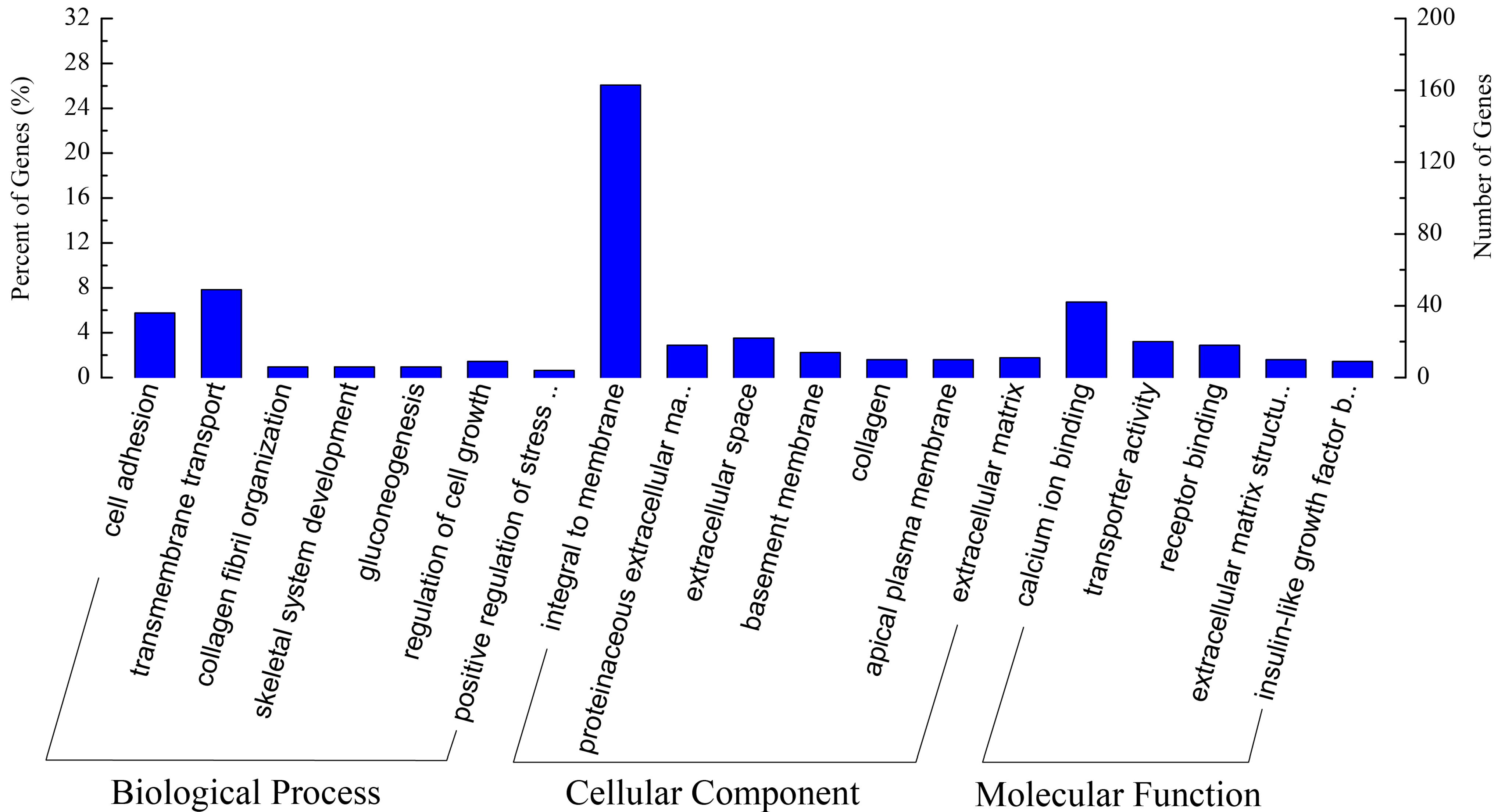
2.2. Preliminary Comparative Analysis of Transcriptomes between NAA and NAS

| Gene-ID | Gene Name | log2(Fold-Change) | Function in the Kidney | References |
|---|---|---|---|---|
| comp46908_c0 | angiotensin I converting enzyme precursor | −3.71 | Angiotensin I (ANG I) converting enzyme can convert angiotensin I to angiotensin II via the removal of two amino acids. | [19] |
| comp46499_c0 | type-1 angiotensin II receptor (ANGR II) | 3.32 | Angiotensin II (ANG II) has a greater effect on efferent arterioles than on afferent arterioles by binding to the type-1 angiotensin II receptor. Though it decreases blood flow, it raises systemic arterial blood pressure while increasing glomerular pressure and the filtration fraction and maintaining the glomerular filtration rate. Ultimately, decreased hydrostatic pressure and increased oncotic pressure in the peritubular capillaries will facilitate the reabsorption of tubular fluid. | [20,21,22] |
| comp45455_c0 | angiotensinogen | −2.52 | Angiotensinogen is the ultimate precursor of angiotensin II and is cleaved by renin to generate angiotensin I. | [19] |
| comp44526_c0 | sodium/hydrogen exchanger 3 (NHE3) precursor | −2.61 | Sodium/hydrogen exchanger 3 is responsible for Na+ reabsorption and H+ secretion and is distributed throughout the apical membrane of the proximal tubule and the thick ascending limb. | [23] |
| comp48685_c0 | aquaporin-2 (AQP2) | 2.30 | Aquaporin-2 is a water channel that localizes to the apical membrane and intracellular vesicles of the collecting duct. | [24] |
| comp16873_c0 | aquaporin-3 (AQP3) | 2.10 | Aquaporin-3 is a water channel that localizes to the basolateral membrane of the collecting duct. | [24] |
| comp46970_c0 | urea transporter 2 (UT2) | 2.42 | Urea transporter 2 mediates the diffusion of urea across the wall of the inner medullary collecting duct and thin descending limb of the loop of Henle and plays an important role in the maintenance of high osmotic pressure in the inner medulla. | [25,26] |
| comp47244_c0 | solute carrier family 12 member 1 | 1.02 | Solute carrier family 12 member 1 encodes a Na+–K+–Cl− cotransporter distributed throughout the luminal membrane of the thick ascending limb of the loop of Henle. | [27,28] |
| comp46740_c1 | atrial natriuretic peptide receptor 1 (ANPR1) precursor | 1.59 | Atrial natriuretic peptide binds the atrial natriuretic peptide receptor 1 and only inhibits the reabsorption of sodium in the inner medullary collecting duct and suppresses the secretion of renin, aldosterone and vasopressin but also directly affects the glomerular filtration rate. | [29,30] |
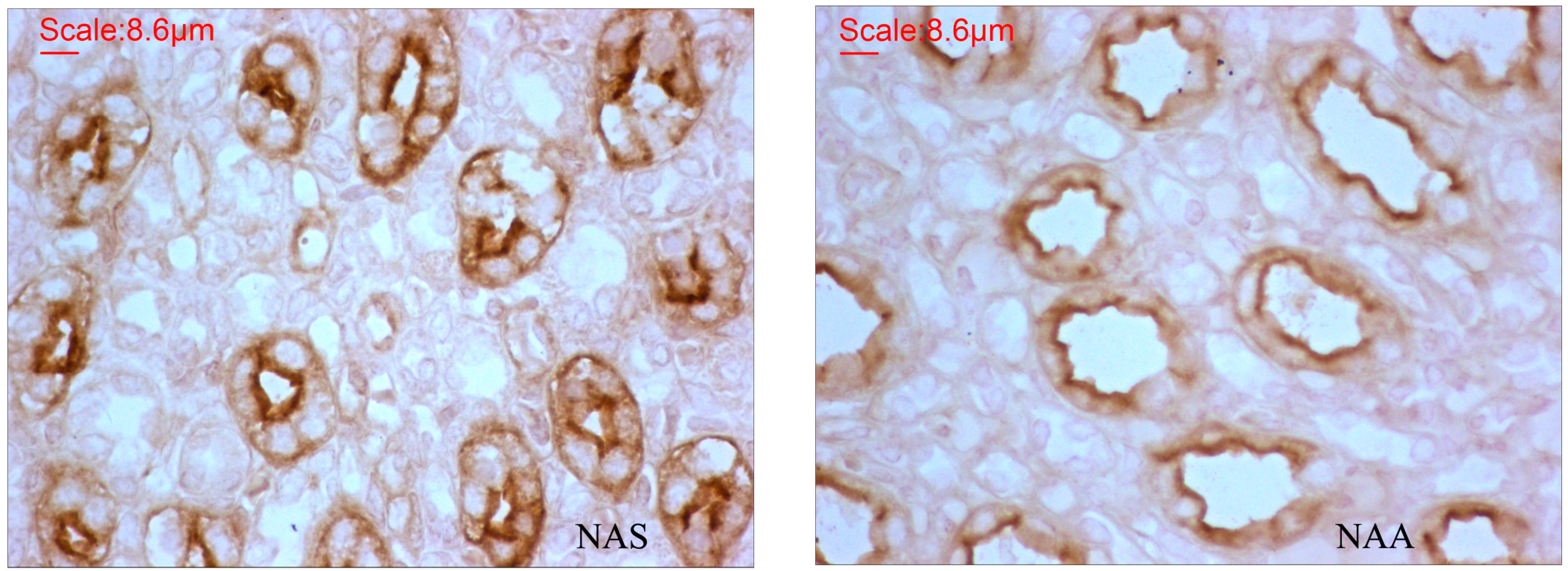
2.3. AQP2 Protein Expression
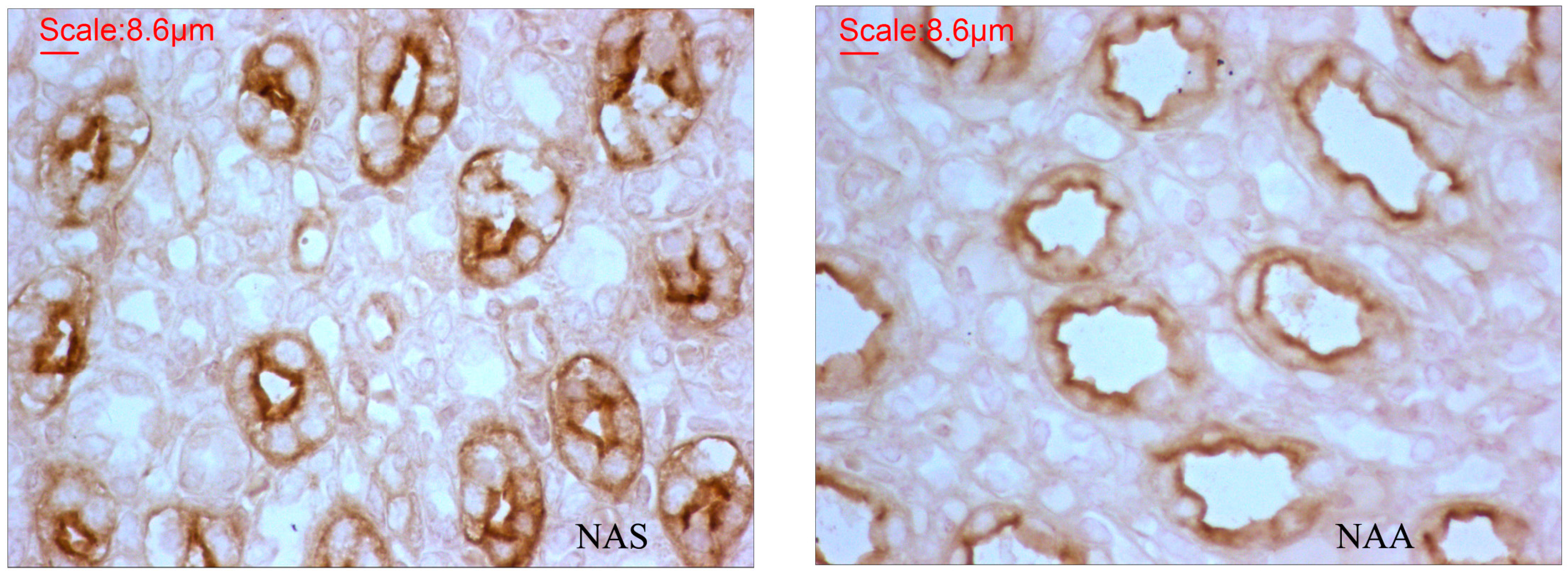
| IOD Mean ± SE | Density Mean ± SE | Area Sum Mean ± SE | |
|---|---|---|---|
| NAS | 84,122.2 ± 5388.5 | 26.4 ± 1.3 | 250,258.3 ± 15,482.7 |
| NAA | 36,534.0 ± 2752.8 | 17.0 ± 1.2 | 112,554.0 ± 8457.2 |
| Fold-change | 2.30 | 1.55 | 2.22 |
| p | <0.0001 | <0.0001 | <0.0001 |
3. Materials and Methods
3.1. Sample Preparation and RNA Sequencing
3.2. Basic Processing of RNA-Seq Data and Gene Functional Annotation
3.3. Comparative Analysis of Transcripts between NAA and NAS
3.4. AQP2 Protein Expression in the Kidney
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Appendix
Conflicts of Interest
References
- Gatesy, J.; O’Leary, M.A. Deciphering whale origins with molecules and fossils. Trends Ecol. Evol. 2001, 16, 562–570. [Google Scholar] [CrossRef]
- Thewissen, J.G.M.; Cooper, L.N.; Clementz, M.T.; Bajpai, S.; Tiwari, B.N. Whales originated from aquatic artiodactyls in the Eocene epoch of India. Nature 2007, 450, U1190–U1191. [Google Scholar] [CrossRef]
- Ortiz, R.M. Osmoregulation in marine mammals. J. Exp. Biol. 2001, 204, 1831–1844. [Google Scholar] [PubMed]
- Grunewald, R.W.; Kinne, R.K. Osmoregulation in the mammalian kidney: The role of organic osmolytes. J. Exp. Zool. 1999, 283, 708–724. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C. Renal cellular and tissue specializations in the bottlenose dolphin (Tursiops truncatus) and beluga whale (Delphinapterus leucas). Aquat. Mamm. 1997, 23, 75–84. [Google Scholar]
- Guo, A.; Hao, Y.; Wang, J.; Zhao, Q.; Wang, D. Concentrations of osmotically related constituents in plasma and urine of finless porpoise (Neophocaena asiaeorientalis): Implications for osmoregulatory strategies for marine mammals living in freshwater. Zool. Stud. 2014, 53, 10. [Google Scholar] [CrossRef]
- Xu, S.; Yang, Y.; Zhou, X.; Xu, J.; Zhou, K.; Yang, G. Adaptive evolution of the osmoregulation-related genes in cetaceans during secondary aquatic adaptation. BMC Evol. Biol. 2013, 13, 189. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Frasier, T.; Yang, S.; White, B. Detecting recent speciation events: The case of the finless porpoise (genus Neophocaena). Heredity 2008, 101, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Yang, S.C.; Wang, B.J.; Wang, L.S. Distinguishing between two species of finless porpoises (Neophocaena phocaenoides and N. asiaeorientalis) in areas of sympatry. Mammalia 2010, 74, 305–310. [Google Scholar]
- Yang, G.; Guo, L.; Bruford, M.W.; Wei, F.W.; Zhou, K.Y. Mitochondrial phylogeography and population history of finless porpoises in Sino-Japanese waters. Biol. J. Linn. Soc. 2008, 95, 193–204. [Google Scholar] [CrossRef]
- Janech, M.G.; Chen, R.; Klein, J.; Nowak, M.W.; McFee, W.; Paul, R.V.; Fitzgibbon, W.R.; Ploth, D.W. Molecular and functional characterization of a urea transporter from the kidney of a short-finned pilot whale. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2002, 282, R1490–R1500. [Google Scholar] [PubMed]
- Gingerich, P.D.; Ul Haq, M.; Zalmout, I.S.; Khan, I.H.; Malkani, M.S. Origin of whales from early artiodactyls: Hands and feet of Eocene Protocetidae from Pakistan. Science 2001, 293, 2239–2242. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.; Jia, K.; Xia, J.; Yang, L.; Chen, J.; Wu, Y.; Yi, M. De novo assembly of the indo-pacific humpback dolphin leucocyte transcriptome to identify putative genes involved in the aquatic adaptation and immune response. PLoS One 2013, 8, e72417. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, F.; Saijonmaa, O. Renin-angiotensin system revisited. J. Intern. Med. 2008, 264, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Pavey, S.A.; Collin, H.; Nosil, P.; Rogers, S.M. The role of gene expression in ecological speciation. Ann. N. Y. Acad. Sci. 2010, 1206, 110–129. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.S.; Cho, Y.S.; Guang, X.; Kang, S.G.; Jeong, J.-Y.; Cha, S.-S.; Oh, H.-M.; Lee, J.-H.; Yang, E.C.; Kwon, K.K. Minke whale genome and aquatic adaptation in cetaceans. Nat. Genet. 2014, 46, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Murphy, T.; Alexander, R.W. Molecular biology of the renin-angiotensin system. Circulation 1993, 87, 1816–1828. [Google Scholar] [CrossRef] [PubMed]
- Navar, L.; Rosivall, L. Contribution of the renin-angiotensin system to the control of intrarenal hemodynamics. Kidney Int. 1984, 25, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Navar, L.G.; Harrison-Bernard, L.M.; Nishiyama, A.; Kobori, H. Regulation of intrarenal angiotensin II in hypertension. Hypertension 2002, 39, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Mehr, A.P.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A.; Moe, O.W. Luminal Na+/H+ exchange in the proximal tubule. Pflug. Arch.-Eur. J. Physiol. 2009, 458, 5–21. [Google Scholar] [CrossRef]
- Nielsen, S.; Agre, P. The aquaporin family of water channels in kidney. Kidney Int. 1995, 48, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Sands, J.M. Regulation of renal urea transporters. J. Am. Soc. Nephrol. 1999, 10, 635–646. [Google Scholar] [PubMed]
- Bagnasco, S.M. Role and regulation of urea transporters. Pflug. Arch.-Eur. J. Physiol. 2005, 450, 217–226. [Google Scholar] [CrossRef]
- Quaggin, S.; Payne, J.; Forbush, B.; Igarashi, P. Localization of the renal Na–K–Cl cotransporter gene (Slc12a1) on mouse Chromosome 2. Mamm. Genome 1995, 6, 557–558. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Kaplan, M.; Plotkin, M.; Hebert, S. The Na–(K)–Cl cotransporter family in the mammalian kidney: Molecular identification and function(s). Nephrol. Dial. Transplant. 1996, 11, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Ballermann, B.; Brenner, B. Atrial natriuretic peptide and the kidney. Am. J. Kidney Dis. 1987, 10, 7. [Google Scholar] [PubMed]
- Zeidel, M.L. Renal actions of atrial natriuretic peptide: Regulation of collecting duct sodium and water transport. Annu. Rev. Physiol. 1990, 52, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Turvey, S.T.; Zhao, X.; Mei, Z. Neophocaena Asiaeorientalis ssp. Asiaeorientalis. IUCN 2013. IUCN Red List of Threatened Species, Version 2013.1; Available online: http://www.iucnredlist.org/details/43205774/0 (accessed on 2 September 2013).
- Suzuki, M.; Endo, N.; Nakano, Y.; Kato, H.; Kishiro, T.; Asahina, K. Localization of aquaporin-2, renal morphology and urine composition in the bottlenose dolphin and the Baird’s beaked whale. J. Comp. Physiol. B 2008, 178, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Frøkiaer, J.; Li, C.; Shi, Y.; Jensen, A.; Praetorius, H.; Hansen, H.; Topcu, O.; Sardeli, C.; Wang, W.; Kwon, T.-H. Renal aquaporins and sodium transporters with special focus on urinary tract obstruction. APMIS Suppl. 2002, 71–79. [Google Scholar]
- Noda, Y.; Sasaki, S. Regulation of aquaporin-2 trafficking and its binding protein complex. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1117–1125. [Google Scholar] [CrossRef]
- Wang, J.Y.; Reeves, R. Neophocaena Asiaeorientalis. IUCN 2013. IUCN Red List of Threatened Species, Version 2013.1; Available online: http://www.iucnredlist.org/details/41754/0 (accessed on 2 September 2013).
- Fleige, S.; Pfaffl, M.W. RNA integrity and the effect on the real-time qRT-PCR performance. Mol. Asp. Med. 2006, 27, 126–139. [Google Scholar] [CrossRef]
- Garbett, K.; Ebert, P.J.; Mitchell, A.; Lintas, C.; Manzi, B.; Mirnics, K.; Persico, A.M. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol. Dis. 2008, 30, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.S.; Moy, G.W.; Burton, R.S. Interpopulation patterns of divergence and selection across the transcriptome of the copepod Tigriopus californicus. Mol. Ecol. 2011, 20, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Elmer, K.R.; Fan, S.; Gunter, H.; Jones, J.; Boekhoff, S.; Kuraku, S.; Meyer, A. Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mol. Ecol. 2010, 19, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Luan, J.B.; Li, J.M.; Su, Y.L.; Xia, J.; Liu, S.S. Transcriptome analysis and comparison reveal divergence between two invasive whitefly cryptic species. BMC Genomics 2011, 12, 458. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Dessimoz, C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput. Biol. 2009, 5, e1000262. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In Proceedings of the Seventh International Conference on Intelligent Systems for Molecular Biology, 1999; AAAI Press: Menlo Park, CA, USA, 1999; pp. 138–158. [Google Scholar]
- Miyata, T.; Yasunaga, T. Molecular evolution of mRNA: A method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J. Mol. Evol. 1980, 16, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J. Educ. Behav. Stat. 2000, 25, 60–83. [Google Scholar] [CrossRef]
- Francisco, J.S.; Moraes, H.P.D.; Dias, E.P. Evaluation of the Image-Pro Plus 4.5 software for automatic counting of labeled nuclei by PCNA immunohistochemistry. Braz. Oral Res. 2004, 18, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Zhou, Z.-G.; Holmqvist, A.; Zhang, H.; Li, Y.; Adell, G.; Sun, X.-F. Survivin expression quantified by Image Pro-Plus compared with visual assessment. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 530–535. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, R.; Guo, A.-H.; Hao, Y.-J.; Zheng, J.-S.; Wang, D. De Novo Assembly and Characterization of Narrow-Ridged Finless Porpoise Renal Transcriptome and Identification of Candidate Genes Involved in Osmoregulation. Int. J. Mol. Sci. 2015, 16, 2220-2238. https://doi.org/10.3390/ijms16012220
Ruan R, Guo A-H, Hao Y-J, Zheng J-S, Wang D. De Novo Assembly and Characterization of Narrow-Ridged Finless Porpoise Renal Transcriptome and Identification of Candidate Genes Involved in Osmoregulation. International Journal of Molecular Sciences. 2015; 16(1):2220-2238. https://doi.org/10.3390/ijms16012220
Chicago/Turabian StyleRuan, Rui, Ai-Huan Guo, Yu-Jiang Hao, Jin-Song Zheng, and Ding Wang. 2015. "De Novo Assembly and Characterization of Narrow-Ridged Finless Porpoise Renal Transcriptome and Identification of Candidate Genes Involved in Osmoregulation" International Journal of Molecular Sciences 16, no. 1: 2220-2238. https://doi.org/10.3390/ijms16012220