
Structure and Function of SET and MYND Domain-Containing Proteins

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
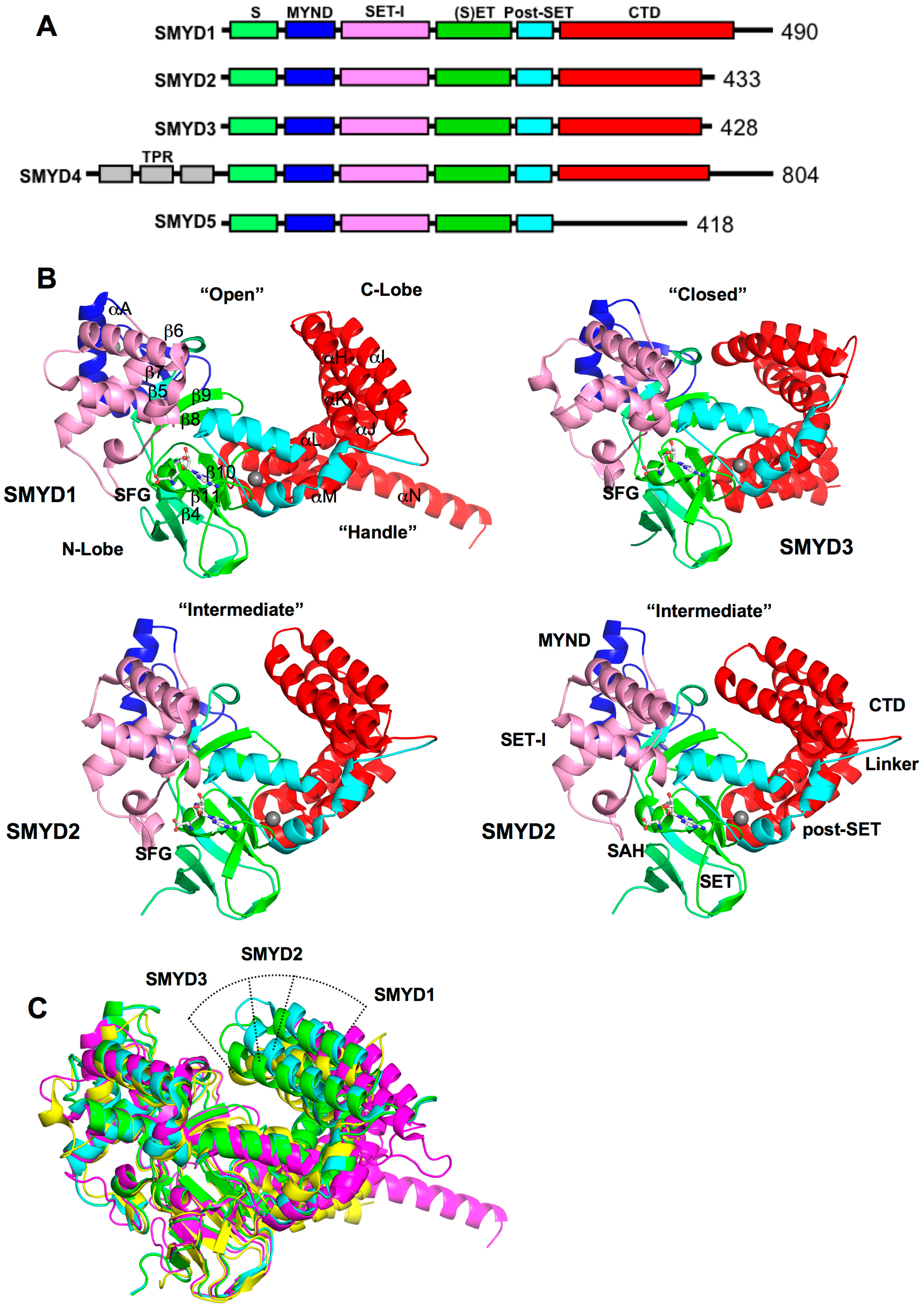
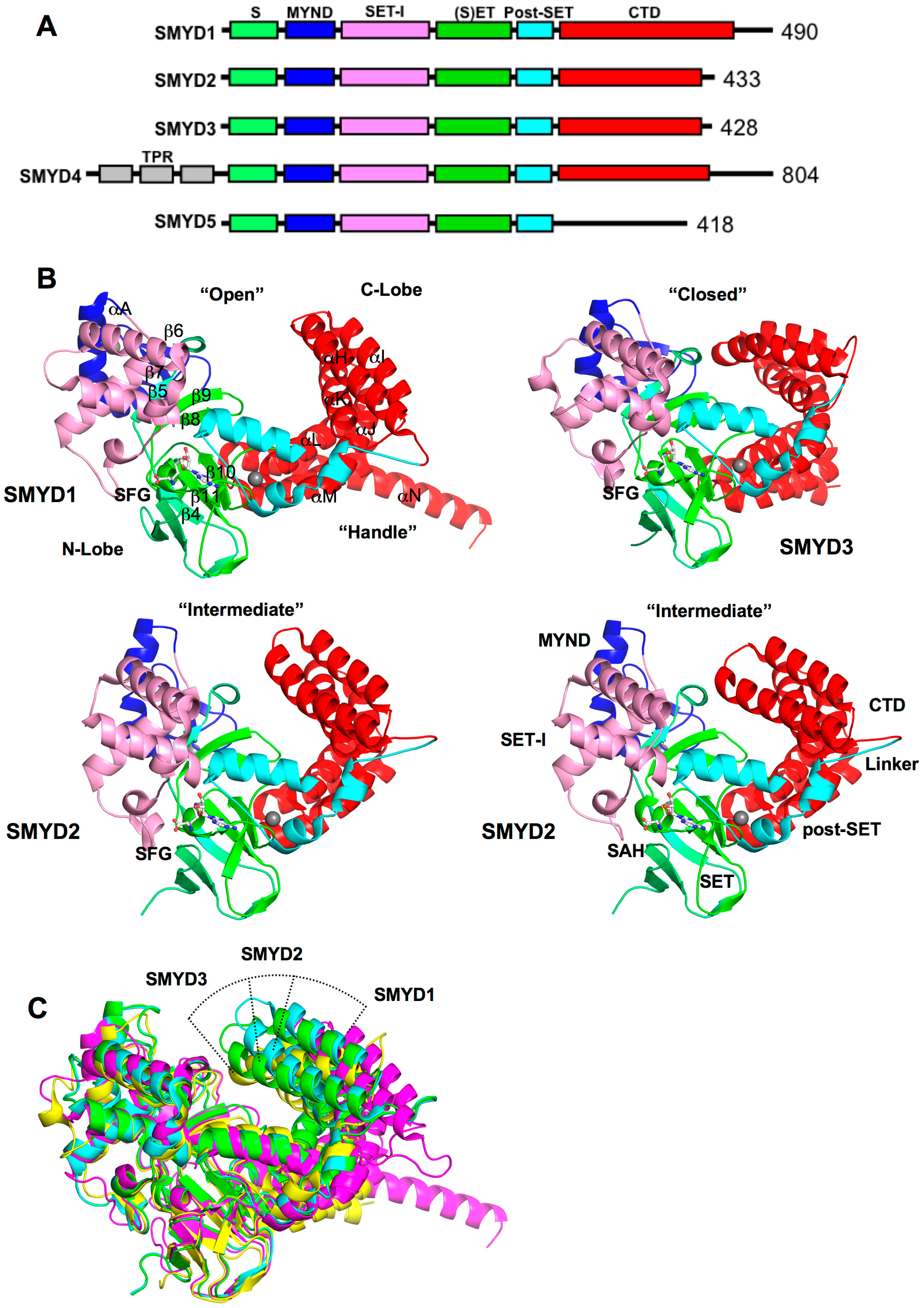
2. SMYD Structure and Function
2.1. Overall SMYD Structure
2.2. SET, the Evolutionary Conserved Methyltransferase Domain
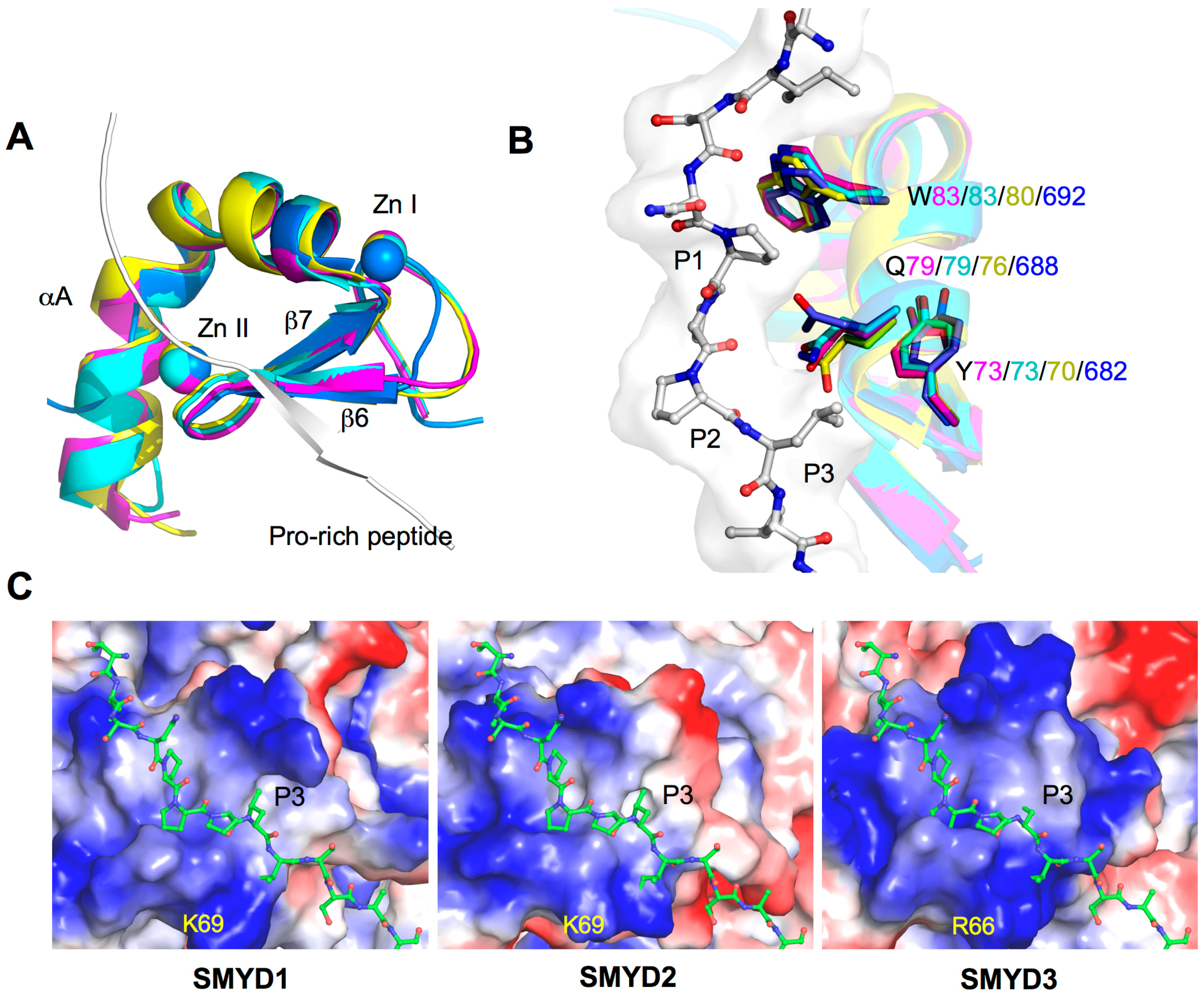
2.3. MYND, the Zinc Finger Motif

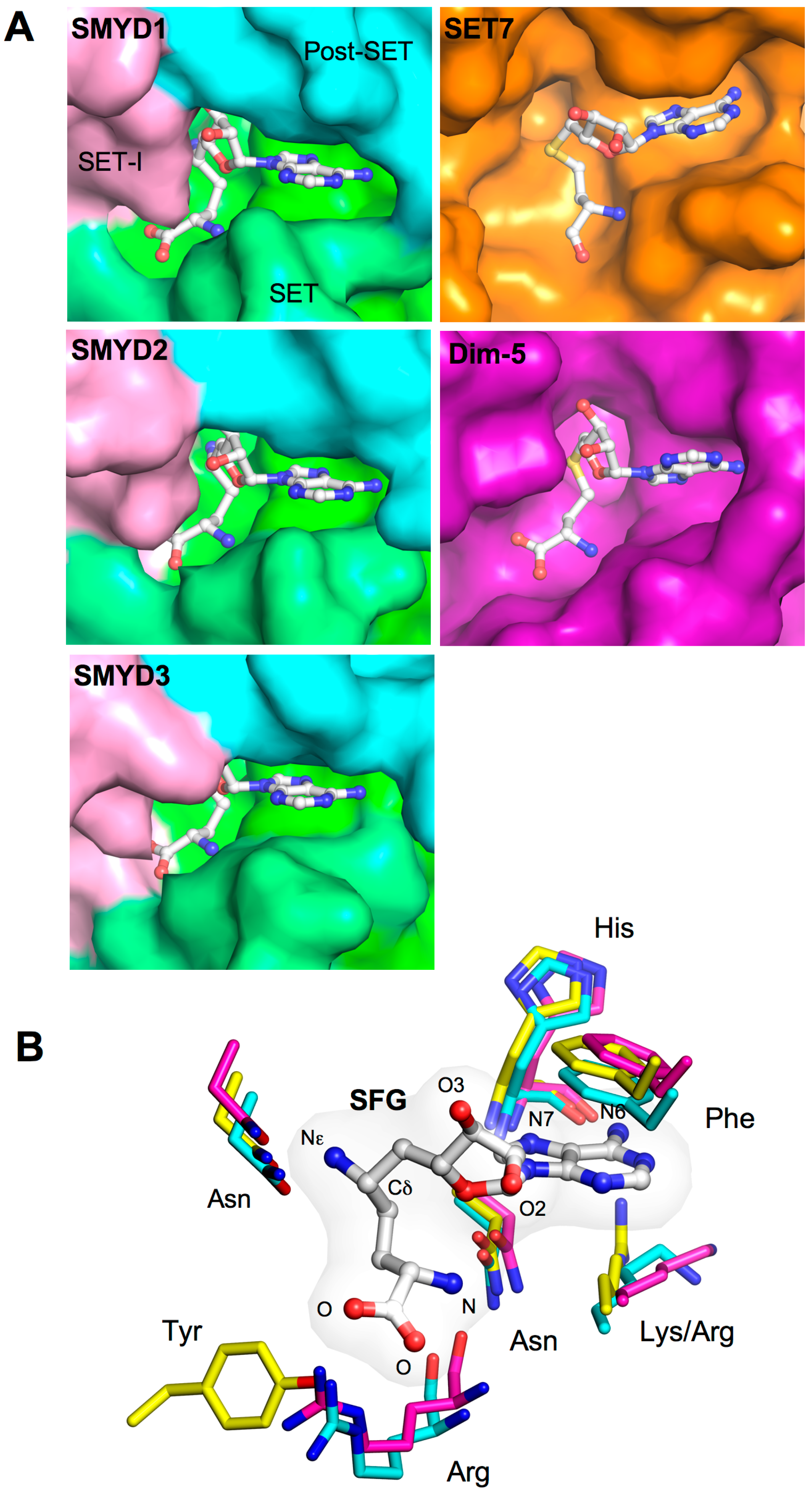
2.4. Cofactor Binding Pocket
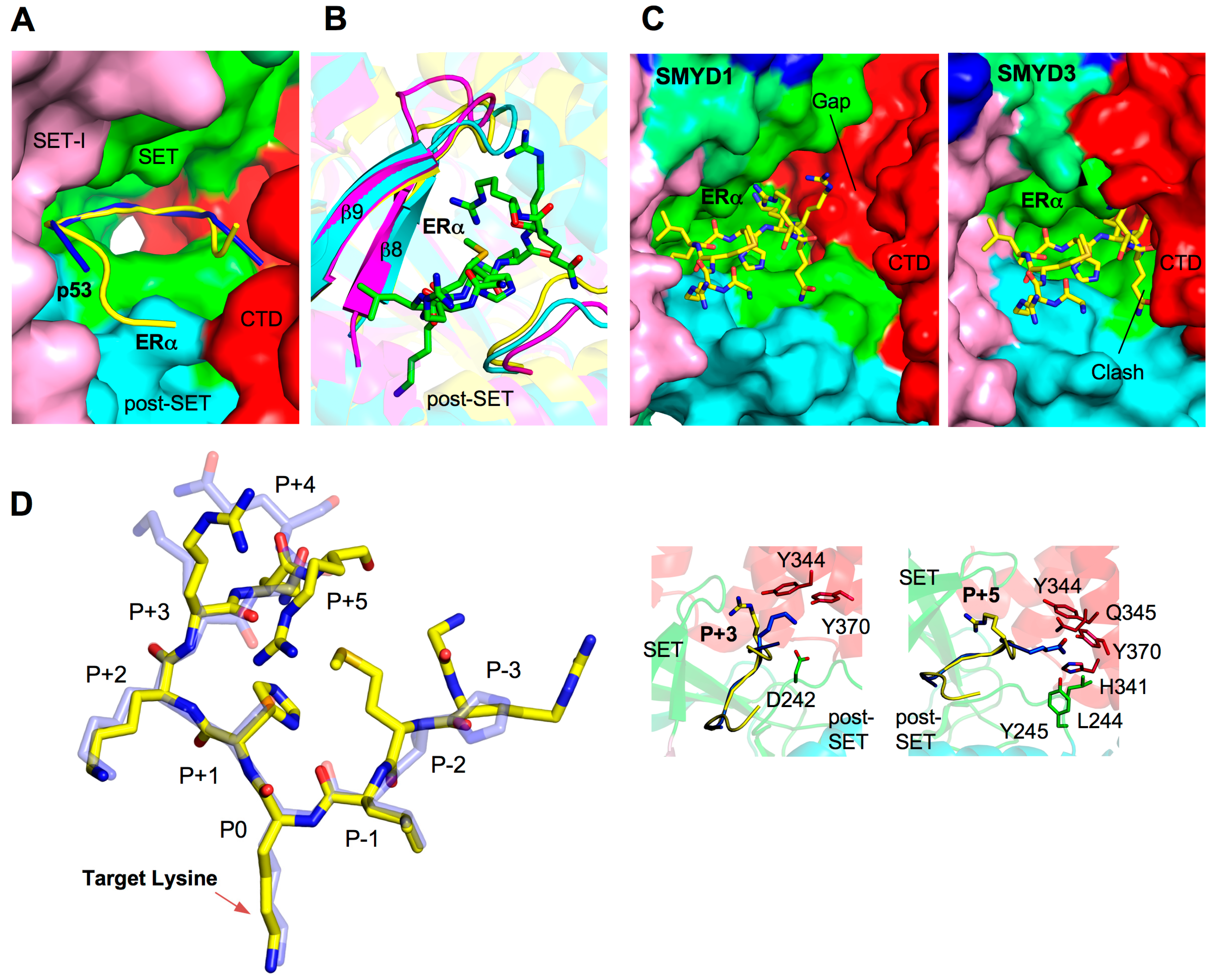
2.5. Substrate Peptide Binding Site
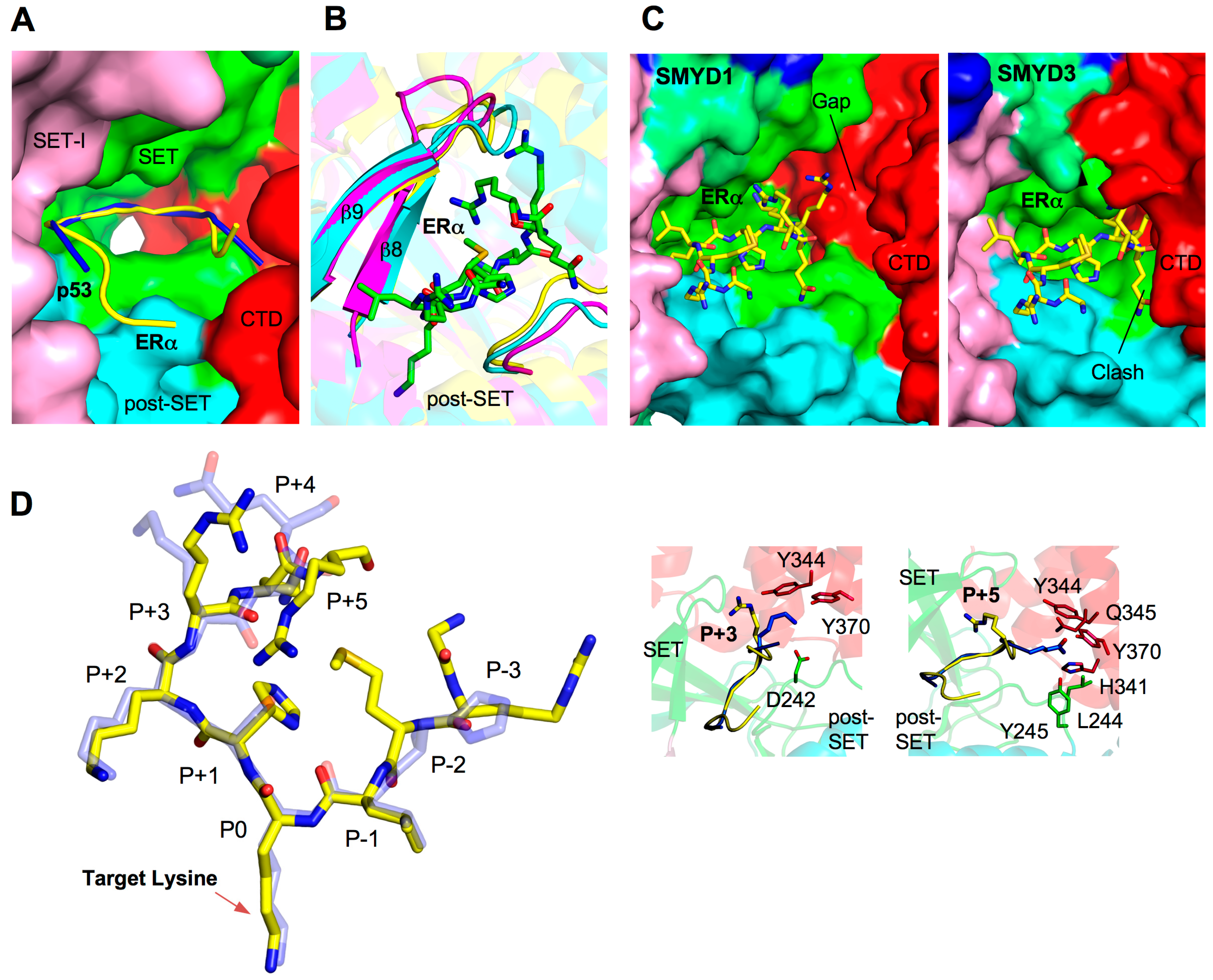
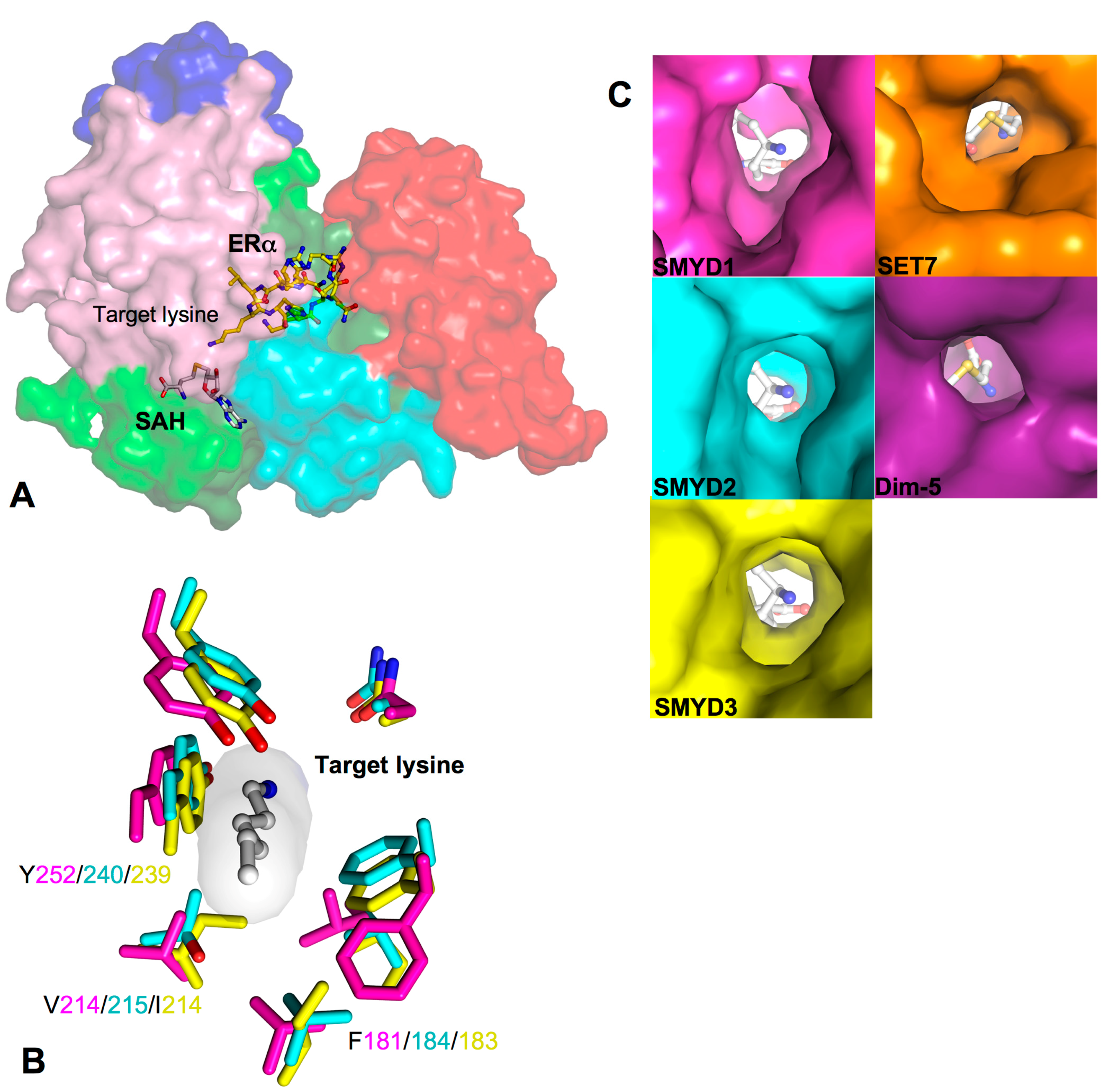
2.6. Target Lysine Access Channel
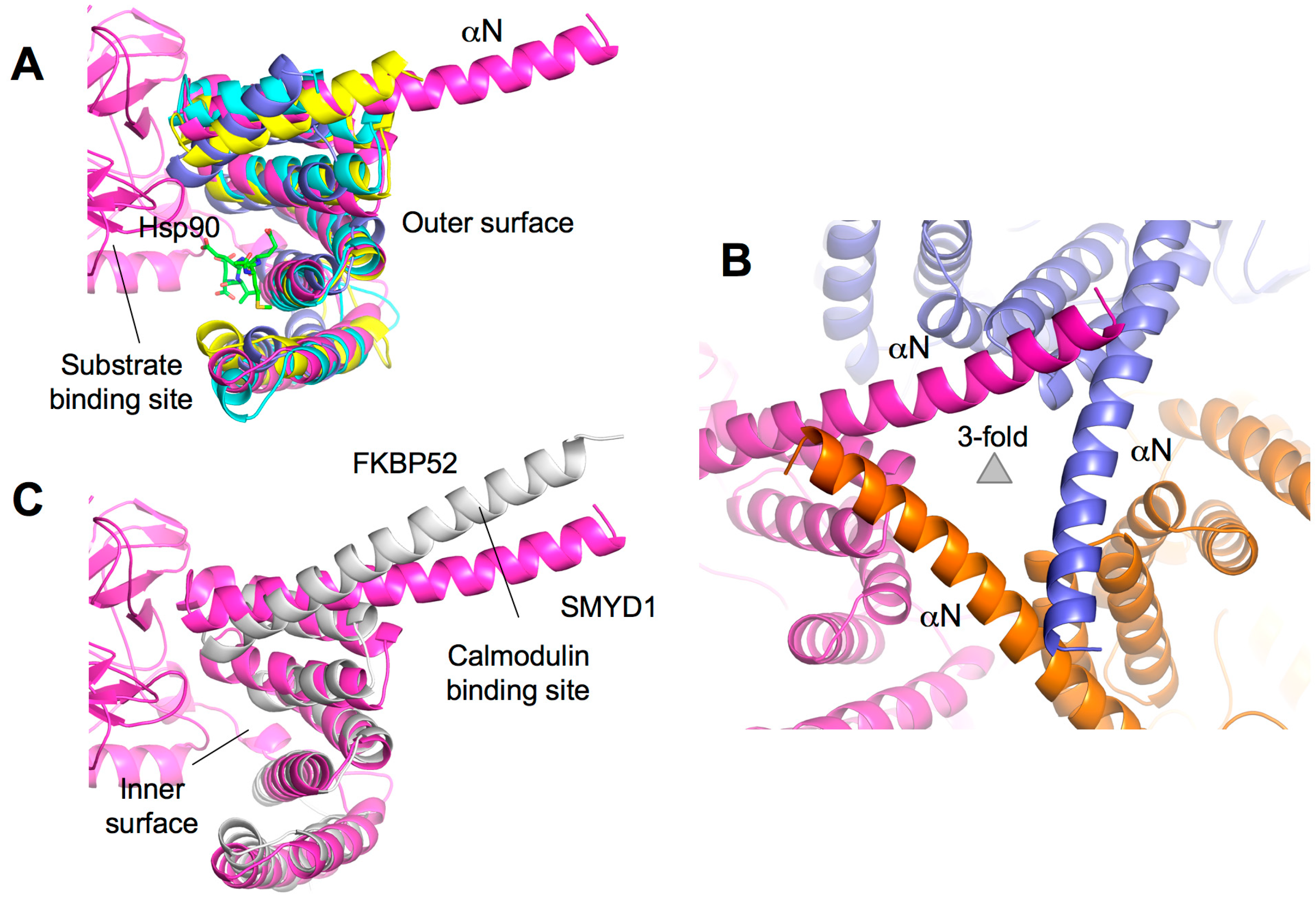
2.7. TPR-Like C-Terminal Domain
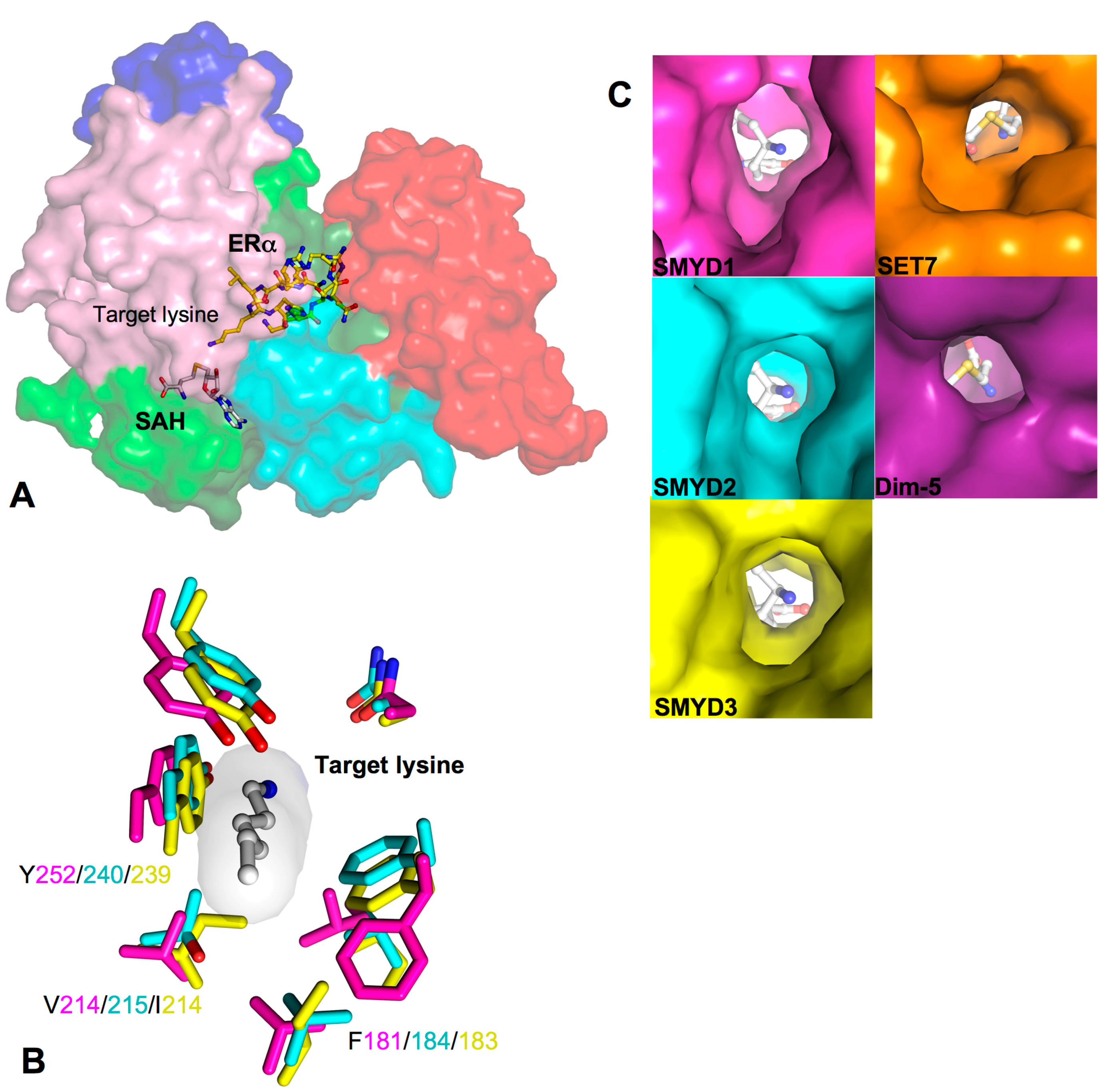
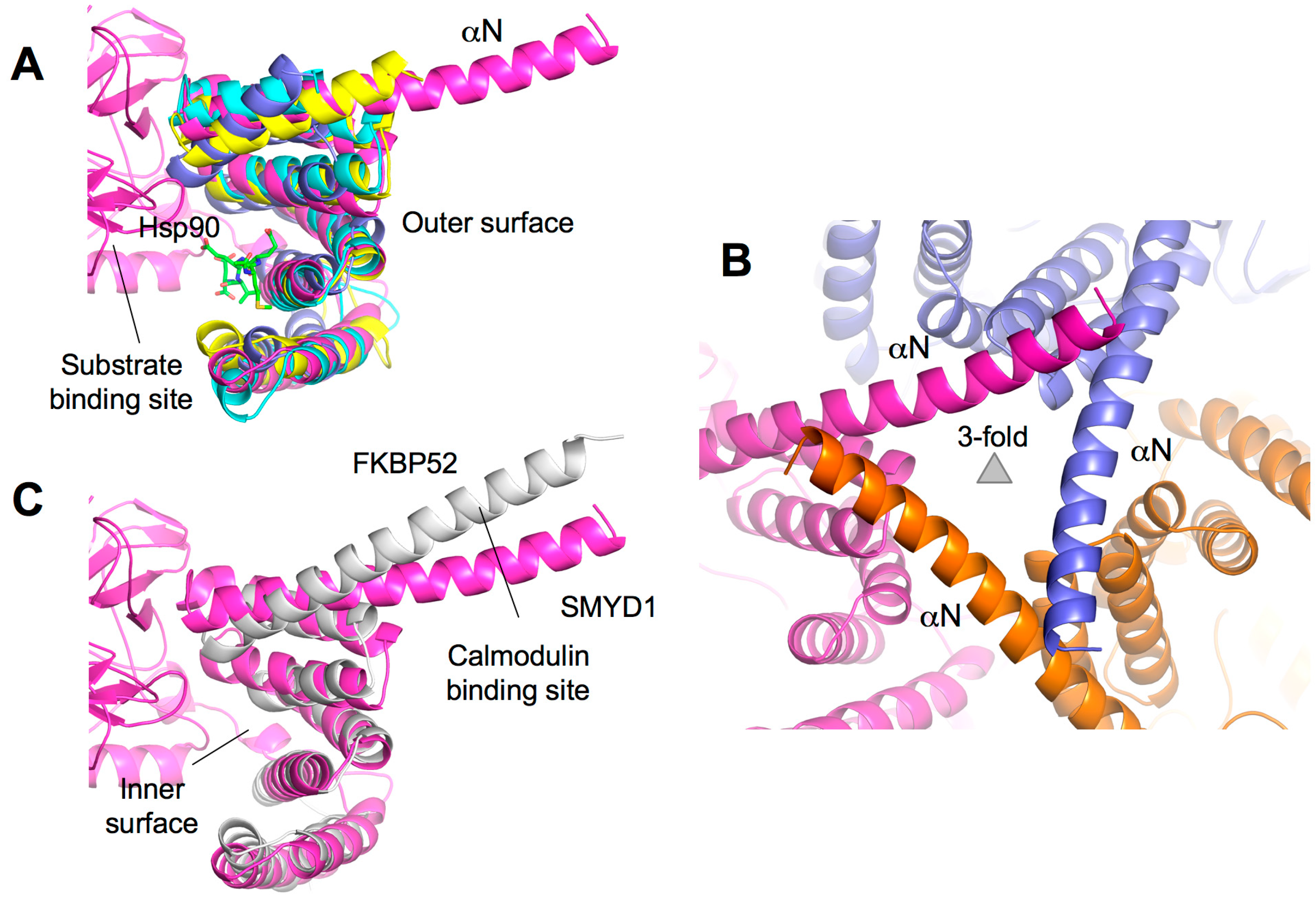
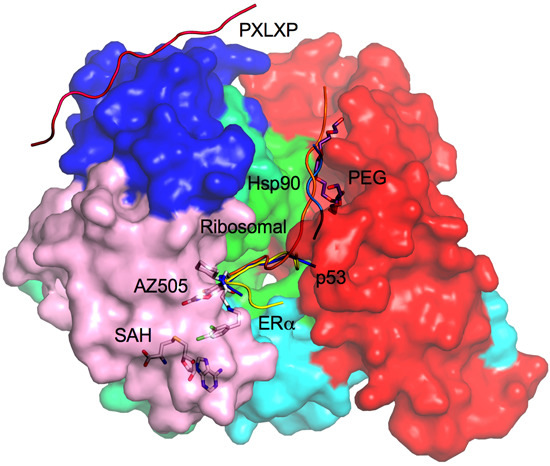
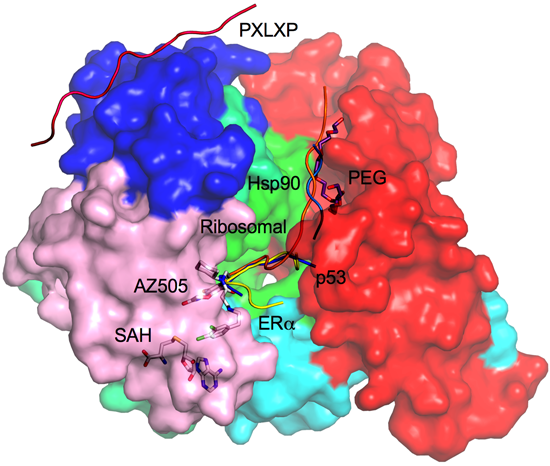
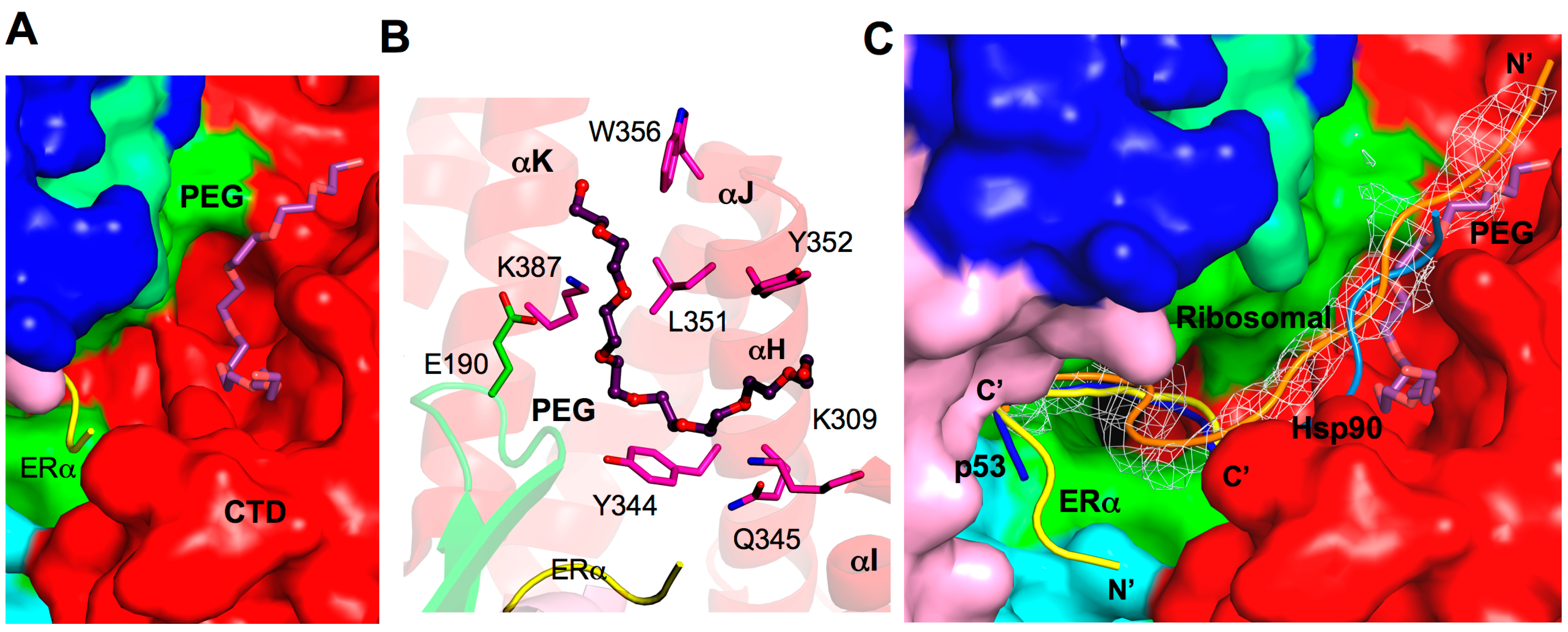
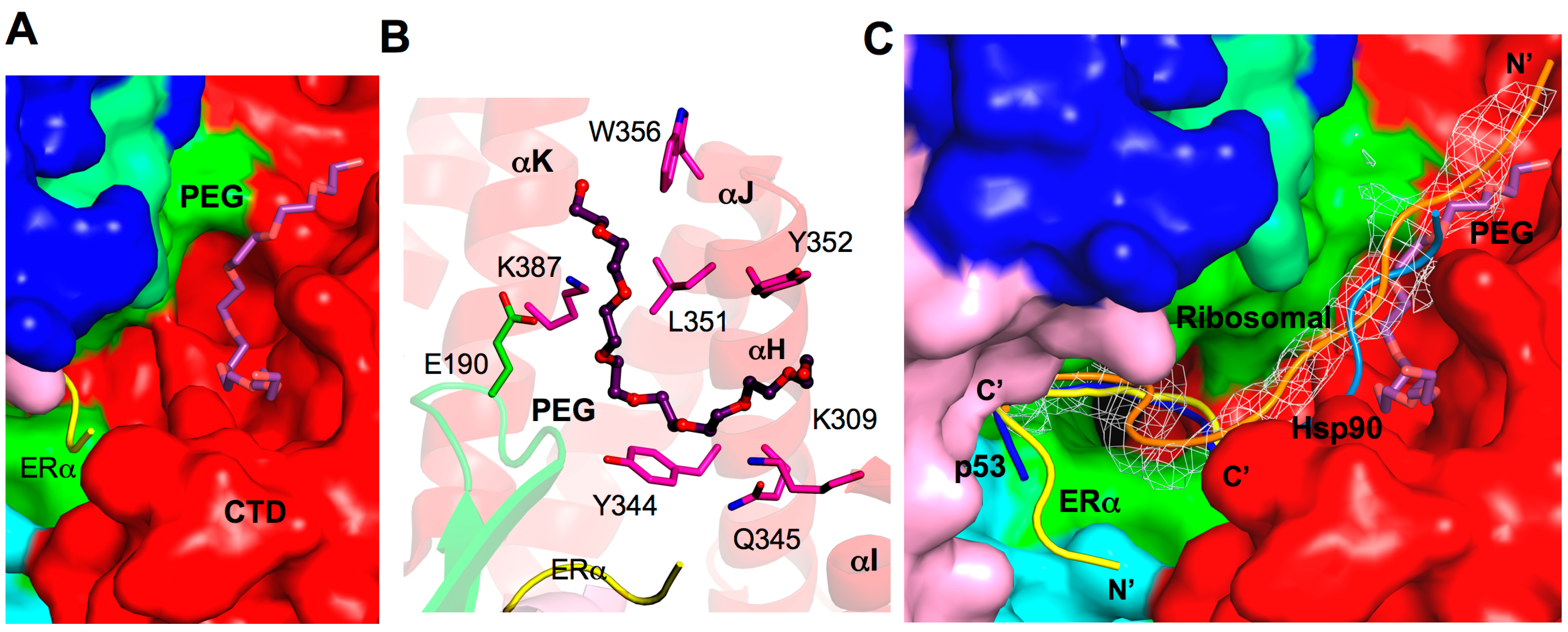
2.8. Additional Substrate Binding Site?
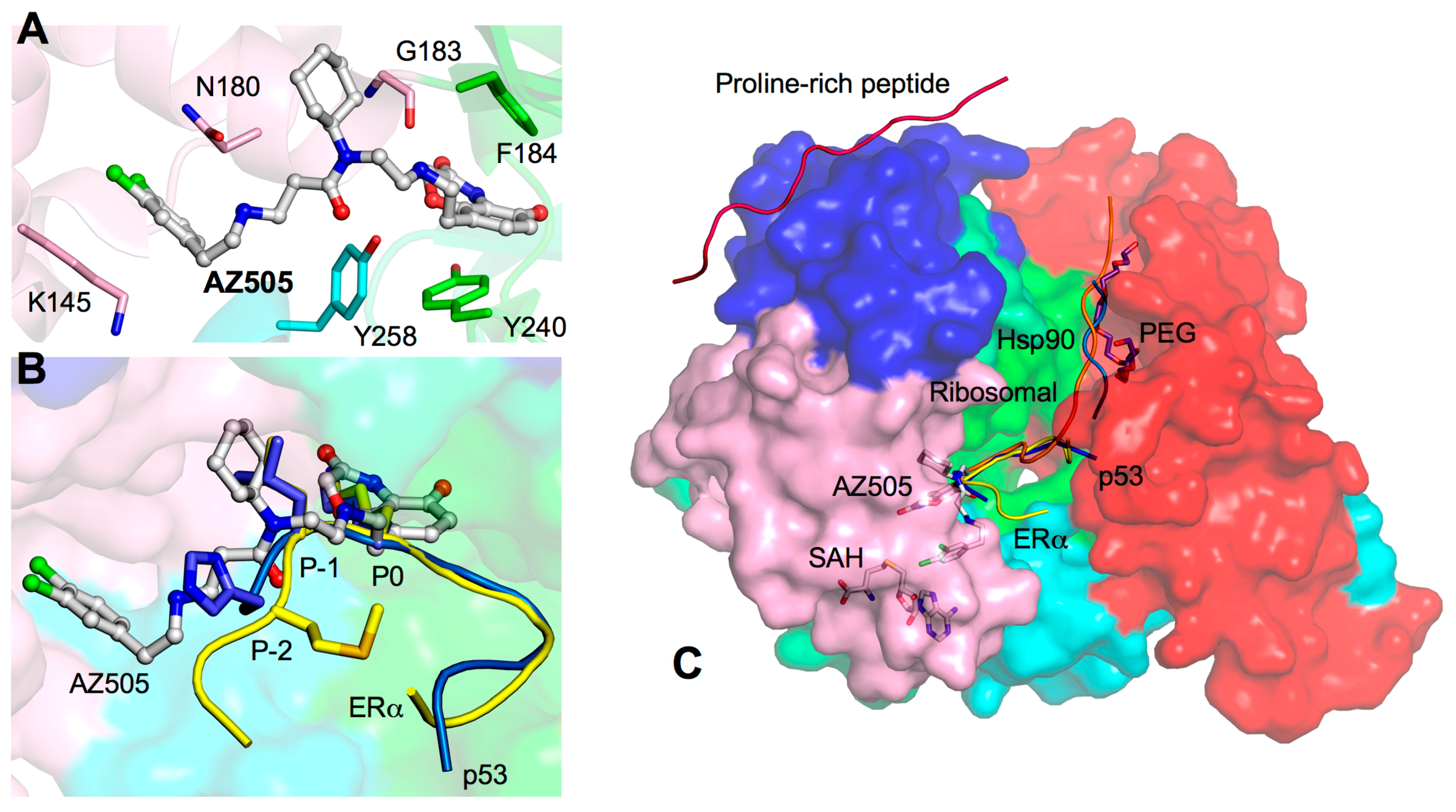
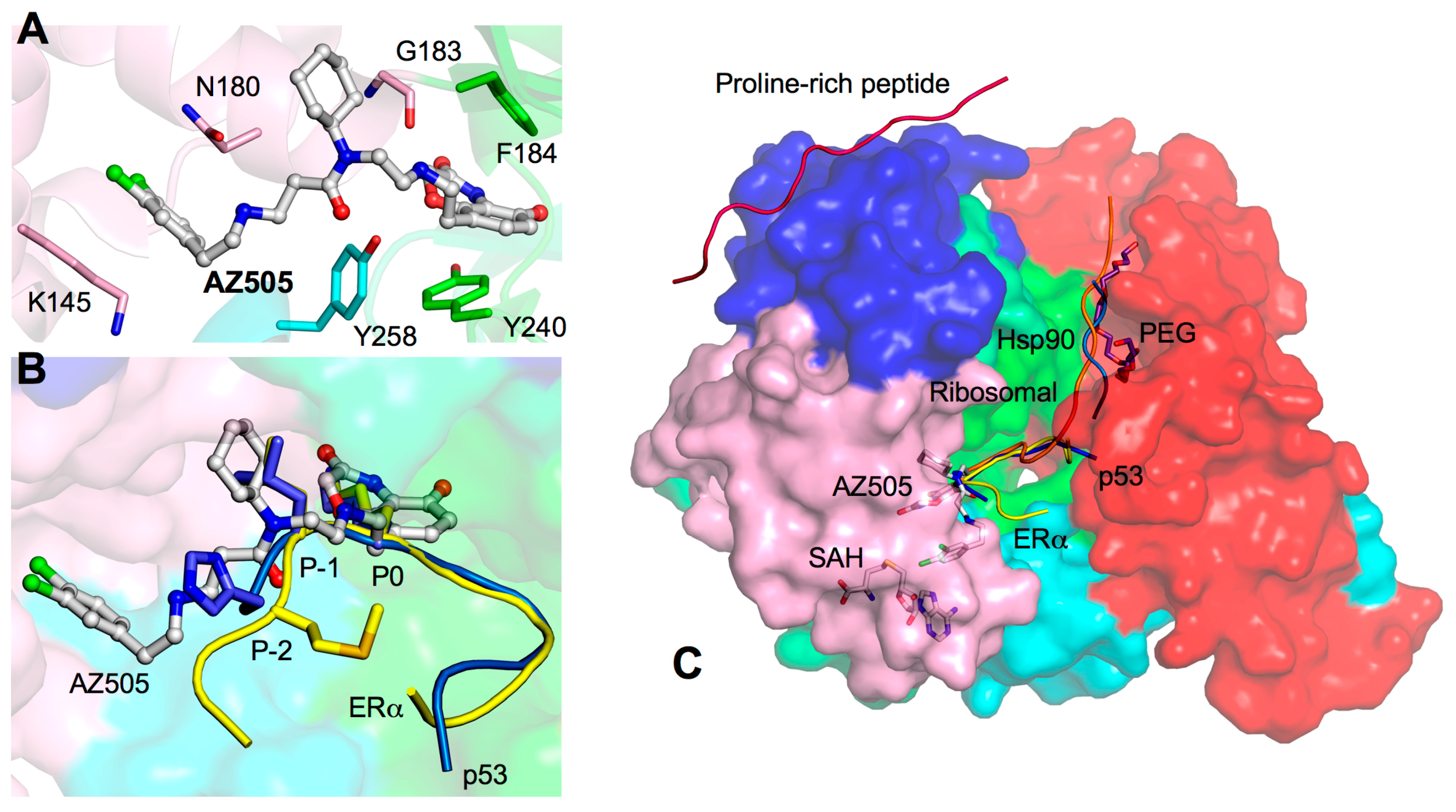
3. Drug Design Perspective
4. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Gottlieb, P.D.; Pierce, S.A.; Sims, R.J.; Yamagishi, H.; Weihe, E.K.; Harriss, J.V.; Maika, S.D.; Kuziel, W.A.; King, H.L.; Olson, E.N.; et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat. Genet. 2002, 31, 25–32. [Google Scholar] [PubMed]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Sims, R.J., 3rd; Gottlieb, P.D.; Tucker, P.W. Identification and characterization of Smyd2: A split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 2006, 5. [Google Scholar] [CrossRef]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Reynoird, N.; Khatri, P.; Jansen, P.W.; Wilkinson, A.W.; Liu, S.; Barbash, O.; van Aller, G.S.; Huddleston, M.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Du, S.J.; Tan, X.; Zhang, J. SMYD proteins: Key regulators in skeletal and cardiac muscle development and function. Anat. Rec. 2014, 297, 1650–1662. [Google Scholar] [CrossRef]
- Sirinupong, N.; Brunzelle, J.; Ye, J.; Pirzada, A.; Nico, L.; Yang, Z. Crystal structure of cardiac-specific histone methyltransferase smyd1 reveals unusual active site architecture. J. Biol. Chem. 2010, 285, 40635–40644. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, W.; Gaudet, J.; Cheney, M.D.; Roudaia, L.; Cierpicki, T.; Klet, R.C.; Hartman, K.; Laue, T.M.; Speck, N.A.; et al. Structural basis for recognition of SMRT/N-CoR by the MYND domain and its contribution to AML1/ETO’s activity. Cancer Cell 2007, 11, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Spadaccini, R.; Perrin, H.; Bottomley, M.J.; Ansieau, S.; Sattler, M. Structure and functional analysis of the MYND domain. J. Mol. Biol. 2006, 358, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Sirinupong, N.; Brunzelle, J.; Doko, E.; Yang, Z. Structural insights into the autoinhibition and posttranslational activation of histone methyltransferase SMYD3. J. Mol. Biol. 2011, 406, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sirinupong, N.; Brunzelle, J.; Yang, Z. Crystal structures of histone and p53 methyltransferase SMYD2 reveal a conformational flexibility of the autoinhibitory C-terminal domain. PLoS One 2011, 6, e21640. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.K.; Ratajczak, T. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones 2011, 16, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Abu-Farha, M.; Lambert, J.P.; Al-Madhoun, A.S.; Elisma, F.; Skerjanc, I.S.; Figeys, D. The tale of two domains: Proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell. Proteomics 2008, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Rotllant, J.; Li, H.; de Deyne, P.; Du, S.J. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc. Natl. Acad. Sci. USA 2006, 103, 2713–2718. [Google Scholar] [CrossRef] [PubMed]
- Medjkane, S.; Cock-Rada, A.; Weitzman, J.B. Role of the SMYD3 histone methyltransferase in tumorigenesis: Local or global effects? Cell Cycle 2012, 11, 810–820. [Google Scholar] [CrossRef]
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 2012, 72, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Furukawa, Y.; Tsunoda, T.; Yue, C.T.; Yang, K.C.; Nakamura, Y. Molecular diagnosis of colorectal tumors by expression profiles of 50 genes expressed differentially in adenomas and carcinomas. Oncogene 2002, 21, 4120–4128. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Satoh, S.; Kato, T.; Kitahara, O.; Yanagawa, R.; Yamaoka, Y.; Tsunoda, T.; Furukawa, Y.; Nakamura, Y. Genome-wide analysis of gene expression in human hepatocellular carcinomas using cdna microarray: Identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 2001, 61, 2129–2137. [Google Scholar] [PubMed]
- Hu, L.; Zhu, Y.T.; Qi, C.; Zhu, Y.J. Identification of SMYD4 as a potential tumor suppressor gene involved in breast cancer development. Cancer Res. 2009, 69, 4067–4072. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.C.; Travers, A.A. A drosophila SMYD4 homologue is a muscle-specific transcriptional modulator involved in development. PLoS One 2008, 3, e3008. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Pascual, G.; Liu, W.; Kaikkonen, M.U.; Do, K.; Spann, N.J.; Boutros, M.; Perrimon, N.; Rosenfeld, M.G.; Glass, C.K. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone h4k20. Mol. Cell 2012, 48, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Saddic, L.A.; West, L.E.; Aslanian, A.; Yates, J.R., 3rd; Rubin, S.M.; Gozani, O.; Sage, J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Hayami, S.; Toyokawa, G.; Maejima, K.; Yamane, Y.; Suzuki, T.; Dohmae, N.; Kogure, M.; Kang, D.; Neal, D.E.; et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia 2012, 14, 476–486. [Google Scholar] [PubMed]
- Komatsu, S.; Imoto, I.; Tsuda, H.; Kozaki, K.I.; Muramatsu, T.; Shimada, Y.; Aiko, S.; Yoshizumi, Y.; Ichikawa, D.; Otsuji, E.; et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009, 30, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Sajjad, A.; Novoyatleva, T.; Vergarajauregui, S.; Troidl, C.; Schermuly, R.T.; Tucker, H.O.; Engel, F.B. Lysine methyltransferase SMYD2 suppresses p53-dependent cardiomyocyte apoptosis. Biochim. Biophys. Acta 2014, 1843, 2556–2562. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Kang, D.; Suzuki, T.; Masuda, A.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. The histone methyltransferase SMYD2 methylates PARP1 and promotes poly(ADP-ribosyl)ation activity in cancer cells. Neoplasia 2014, 16, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Kunizaki, M.; Hamamoto, R.; Silva, F.P.; Yamaguchi, K.; Nagayasu, T.; Shibuya, M.; Nakamura, Y.; Furukawa, Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007, 67, 10759–10765. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Larsen, N.A.; Howard, T.; Pollard, H.; Green, I.; Grande, C.; Cheung, T.; Garcia-Arenas, R.; Cowen, S.; Wu, J.; et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.W.; Brown, M.; Park, F.; Emtage, S.; Harriss, J.; Das, C.; Zhu, L.; Crew, A.; Arnold, L.; Shaaban, S.; et al. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS One 2011, 6, e22290. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhong, C.; Zhang, T.; Ding, J. Structure of human lysine methyltransferase smyd2 reveals insights into the substrate divergence in smyd proteins. J. Mol. Cell Biol. 2011, 3, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Trescott, L.; Holcomb, J.; Zhang, X.; Brunzelle, J.; Sirinupong, N.; Shi, X.; Yang, Z. Structural insights into estrogen receptor alpha methylation by histone methyltransferase SMYD2, a cellular event implicated in estrogen signaling regulation. J. Mol. Biol. 2014, 426, 3413–3425. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.; Zhang, H.; Luo, X.; Dai, J.; Zhou, S.; Gu, J.; Zhu, J.; Atadja, P.; Lu, C.; et al. Structure of human SMYD2 protein reveals the basis of p53 tumor suppressor methylation. J. Biol. Chem. 2011, 286, 38725–38737. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J., 3rd; Weihe, E.K.; Zhu, L.; O’Malley, S.; Harriss, J.V.; Gottlieb, P.D. M-bop, a repressor protein essential for cardiogenesis, interacts with sknac, a heart- and muscle-specific transcription factor. J. Biol. Chem. 2002, 277, 26524–26529. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.F.; Collazo, E.; Brunzelle, J.S.; Trievel, R.C. Structural and functional analysis of set8, a histone h4 lys-20 methyltransferase. Genes Dev. 2005, 19, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; Jing, C.; Walker, P.A.; Martin, S.R.; Howell, S.A.; Blackburn, G.M.; Gamblin, S.J.; Xiao, B. Crystal structure and functional analysis of the histone methyltransferase SET7/9. Cell 2002, 111, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cheung, T.; Grande, C.; Ferguson, A.D.; Zhu, X.; Theriault, K.; Code, E.; Birr, C.; Keen, N.; Chen, H. Biochemical characterization of human SET and MYND domain-containing protein 2 methyltransferase. Biochemistry 2011, 50, 6488–6497. [Google Scholar] [CrossRef] [PubMed]
- Kateb, F.; Perrin, H.; Tripsianes, K.; Zou, P.; Spadaccini, R.; Bottomley, M.; Franzmann, T.M.; Buchner, J.; Ansieau, S.; Sattler, M. Structural and functional analysis of the DEAF-1 and BS69 MYND domains. PLoS One 2013, 8, e54715. [Google Scholar] [CrossRef] [PubMed]
- Abu-Farha, M.; Lanouette, S.; Elisma, F.; Tremblay, V.; Butson, J.; Figeys, D.; Couture, J.F. Proteomic analyses of the SMYD family interactomes identify HSP90 as a novel target for SMYD2. J. Mol. Cell Biol. 2011, 3, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wu, J.; Sun, B.; Zhong, C.; Ding, J. Structural and biochemical studies of human lysine methyltransferase Smyd3 reveal the important functional roles of its post-SET and TPR domains and the regulation of its activity by DNA binding. Nucleic Acids Res. 2011, 39, 4438–4449. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.G.; Zhang, C.L.; Zhao, W.W.; Liu, Z.P.; Liu, L.; Mu, A.; Guo, S.; Wang, N.; Zhou, H.; Zhang, T.C. Histone methyltransferase SMYD3 promotes MRTF-A-mediated transactivation of MYL9 and migration of MCF-7 breast cancer cells. Cancer Lett. 2014, 344, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, D.; Han, H.; Fan, Y.; Schain, F.; Xu, Z.; Claesson, H.E.; Bjorkholm, M.; Sjoberg, J. Transcriptional regulation of 15-lipoxygenase expression by histone h3 lysine 4 methylation/demethylation. PLoS One 2012, 7, e52703. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Heo, K.; Kim, J.H.; Kim, K.; Choi, J.; An, W. Requirement of histone methyltransferase SMYD3 for estrogen receptor-mediated transcription. J. Biol. Chem. 2009, 284, 19867–19877. [Google Scholar] [CrossRef] [PubMed]
- Van Aller, G.S.; Reynoird, N.; Barbash, O.; Huddleston, M.; Liu, S.; Zmoos, A.F.; McDevitt, P.; Sinnamon, R.; Le, B.; Mas, G.; et al. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 2012, 7, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Z.; Khan, S.I.; Horton, J.R.; Tamaru, H.; Selker, E.U.; Cheng, X. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell 2003, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, G.M.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tanaka, K.; Yan, J.; Li, J.; Peng, D.; Jiang, Y.; Yang, Z.; Barton, M.C.; Wen, H.; Shi, X. Regulation of estrogen receptor alpha by histone methyltransferase smyd2-mediated protein methylation. Proc. Natl. Acad. Sci. USA 2013, 110, 17284–17289. [Google Scholar] [CrossRef] [PubMed]
- Onuoha, S.C.; Coulstock, E.T.; Grossmann, J.G.; Jackson, S.E. Structural studies on the co-chaperone Hop and its complexes with Hsp90. J. Mol. Biol. 2008, 379, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Masison, D.C. Independent regulation of Hsp70 and Hsp90 chaperones by Hsp70/Hsp90-organizing protein sti1 (Hop1). J. Biol. Chem. 2005, 280, 34178–34185. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Li, P.; Liu, Y.; Lou, Z.; Ding, Y.; Shu, C.; Ye, S.; Bartlam, M.; Shen, B.; Rao, Z. 3D structure of human FK506-binding protein 52: Implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc. Natl. Acad. Sci. USA 2004, 101, 8348–8353. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.H.; Sanchez, E.R. FKBP52. Int. J. Biochem. Cell Bsiol. 2005, 37, 42–47. [Google Scholar] [CrossRef]
- Deshpande, C.N.; Harrop, S.J.; Boucher, Y.; Hassan, K.A.; Di Leo, R.; Xu, X.; Cui, H.; Savchenko, A.; Chang, C.; Labbate, M.; et al. Crystal structure of an integron gene cassette-associated protein from vibrio cholerae identifies a cationic drug-binding module. PLoS One 2011, 6, e16934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Hemel, D.; Brige, A.; Savvides, S.N.; van Beeumen, J. Ligand-induced conformational changes in the capping subdomain of a bacterial old yellow enzyme homologue and conserved sequence fingerprints provide new insights into substrate binding. J. Biol. Chem. 2006, 281, 28152–28161. [Google Scholar] [CrossRef] [PubMed]
- Aufhammer, S.W.; Warkentin, E.; Ermler, U.; Hagemeier, C.H.; Thauer, R.K.; Shima, S. Crystal structure of methylenetetrahydromethanopterin reductase (Mer) in complex with coenzyme F420: Architecture of the F420/FMN binding site of enzymes within the nonprolyl cis-peptide containing bacterial luciferase family. Protein Sci. 2005, 14, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, L.H.; Andrade, R.V.; Felipe, M.S.; Motoyama, A.B.; Pittella Silva, F. Smyd2 is highly expressed in pediatric acute lymphoblastic leukemia and constitutes a bad prognostic factor. Leuk. Res. 2014, 38, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Thum, T. Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J. 2003, 17, 1592–1608. [Google Scholar] [CrossRef] [PubMed]
- Gambetta, K.; Al-Ahdab, M.K.; Ilbawi, M.N.; Hassaniya, N.; Gupta, M. Transcription repression and blocks in cell cycle progression in hypoplastic left heart syndrome. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2268–H2275. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Ichikawa, D.; Hirajima, S.; Nagata, H.; Nishimura, Y.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Konishi, H.; et al. Overexpression of Smyd2 contributes to malignant outcome in gastric cancer. Br. J. Cancer 2014, 16. [Google Scholar] [CrossRef]
- Liu, C.; Wang, C.; Wang, K.; Liu, L.; Shen, Q.; Yan, K.; Sun, X.; Chen, J.; Liu, J.; Ren, H.; et al. Smyd3 as an oncogenic driver in prostate cancer by stimulation of androgen receptor transcription. J. Natl. Cancer Inst. 2013, 105, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Z.; Luo, X.G.; Shen, J.; Zou, J.N.; Lu, Y.H.; Xi, T. Knockdown of Smyd3 by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep. 2008, 41, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Silva, F.P.; Tsuge, M.; Nishidate, T.; Katagiri, T.; Nakamura, Y.; Furukawa, Y. Enhanced Smyd3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006, 97, 113–118. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, K.J.; Baird, A.M.; Kilmartin, L.; Leonard, J.; Sacevich, C.; Gray, S.G. Epigenetic regulation of glucose transporters in non-small cell lung cancer. Cancers 2011, 3, 1550–1565. [Google Scholar] [CrossRef] [PubMed]
- Vieira, F.Q.; Costa-Pinheiro, P.; Ramalho-Carvalho, J.; Pereira, A.; Menezes, F.D.; Antunes, L.; Carneiro, I.; Oliveira, J.; Henrique, R.; Jeronimo, C. Deregulated expression of selected histone methylases and demethylases in prostate carcinoma. Endocr. Relat. Cancer 2014, 21, 51–61. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Xu, J.; Zhang, J.; Xie, D.; Ye, H.; Xiao, Z.; Cai, M.; Xu, K.; Zeng, Y.; Li, H.; et al. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum. Pathol. 2012, 43, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.G.; Xi, T.; Guo, S.; Liu, Z.P.; Wang, N.; Jiang, Y.; Zhang, T.C. Effects of Smyd3 overexpression on transformation, serum dependence, and apoptosis sensitivity in NIH3T3 cells. IUBMB Life 2009, 61, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.G.; Zou, J.N.; Wang, S.Z.; Zhang, T.C.; Xi, T. Novobiocin decreases SMYD3 expression and inhibits the migration of MDA-MB-231 human breast cancer cells. IUBMB Life 2010, 62, 194–199. [Google Scholar] [PubMed]
- Sponziello, M.; Durante, C.; Boichard, A.; Dima, M.; Puppin, C.; Verrienti, A.; Tamburrano, G.; di Rocco, G.; Redler, A.; Lacroix, L.; et al. Epigenetic-related gene expression profile in medullary thyroid cancer revealed the overexpression of the histone methyltransferases EZH2 and SMYD3 in aggressive tumours. Mol. Cell. Endocrinol. 2014, 392, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.W.; Zhang, H.; Wang, B.L.; Sun, P.; Wang, Y.G.; Zhang, P. Effect of the downregulation of SMYD3 expression by RNAi on RIZ1 expression and proliferation of esophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 1064–1072. [Google Scholar] [PubMed]
- Josse, R.; Dumont, J.; Fautrel, A.; Robin, M.A.; Guillouzo, A. Identification of early target genes of aflatoxin B1 in human hepatocytes, inter-individual variability and comparison with other genotoxic compounds. Toxicol. Appl. Pharmacol. 2012, 258, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Donahoe, J.; Perry, A.; Lemke, N.; Gorse, K.; Kittiniyom, K.; Rempel, S.A.; Gutierrez, J.A.; Newsham, I.F. Loss of DAL-1, a protein 4.1-related tumor suppressor, is an important early event in the pathogenesis of meningiomas. Hum. Mol. Genet. 2000, 9, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Tran, Y.K.; Bogler, O.; Gorse, K.M.; Wieland, I.; Green, M.R.; Newsham, I.F. A novel member of the NF2/ERM/4.1 superfamily with growth suppressing properties in lung cancer. Cancer Res. 1999, 59, 35–43. [Google Scholar] [PubMed]
- Wiesner, S.; Wybenga-Groot, L.E.; Warner, N.; Lin, H.; Pawson, T.; Forman-Kay, J.D.; Sicheri, F. A change in conformational dynamics underlies the activation of eph receptor tyrosine kinases. EMBO J. 2006, 25, 4686–4696. [Google Scholar] [CrossRef] [PubMed]
- Gosu, V.; Choi, S. Structural dynamic analysis of apo and ATP-bound IRAK4 kinase. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spellmon, N.; Holcomb, J.; Trescott, L.; Sirinupong, N.; Yang, Z. Structure and Function of SET and MYND Domain-Containing Proteins. Int. J. Mol. Sci. 2015, 16, 1406-1428. https://doi.org/10.3390/ijms16011406
Spellmon N, Holcomb J, Trescott L, Sirinupong N, Yang Z. Structure and Function of SET and MYND Domain-Containing Proteins. International Journal of Molecular Sciences. 2015; 16(1):1406-1428. https://doi.org/10.3390/ijms16011406
Chicago/Turabian StyleSpellmon, Nicholas, Joshua Holcomb, Laura Trescott, Nualpun Sirinupong, and Zhe Yang. 2015. "Structure and Function of SET and MYND Domain-Containing Proteins" International Journal of Molecular Sciences 16, no. 1: 1406-1428. https://doi.org/10.3390/ijms16011406