Radiosensitization of Human Leukemic HL-60 Cells by ATR Kinase Inhibitor (VE-821): Phosphoproteomic Analysis

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Overall Phosphoproteomic Analysis Reveals Thousands of Phosphorylation Sites
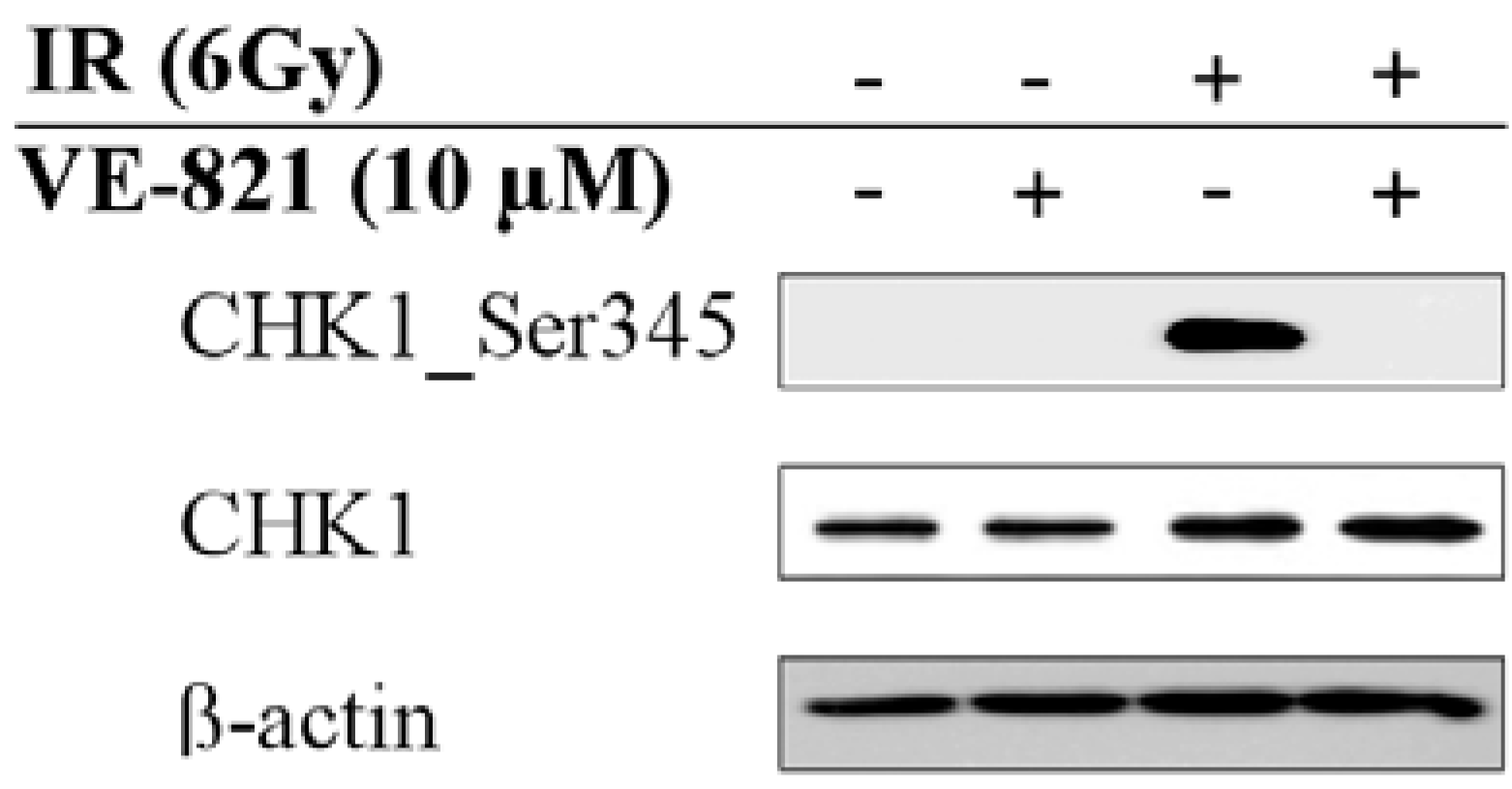
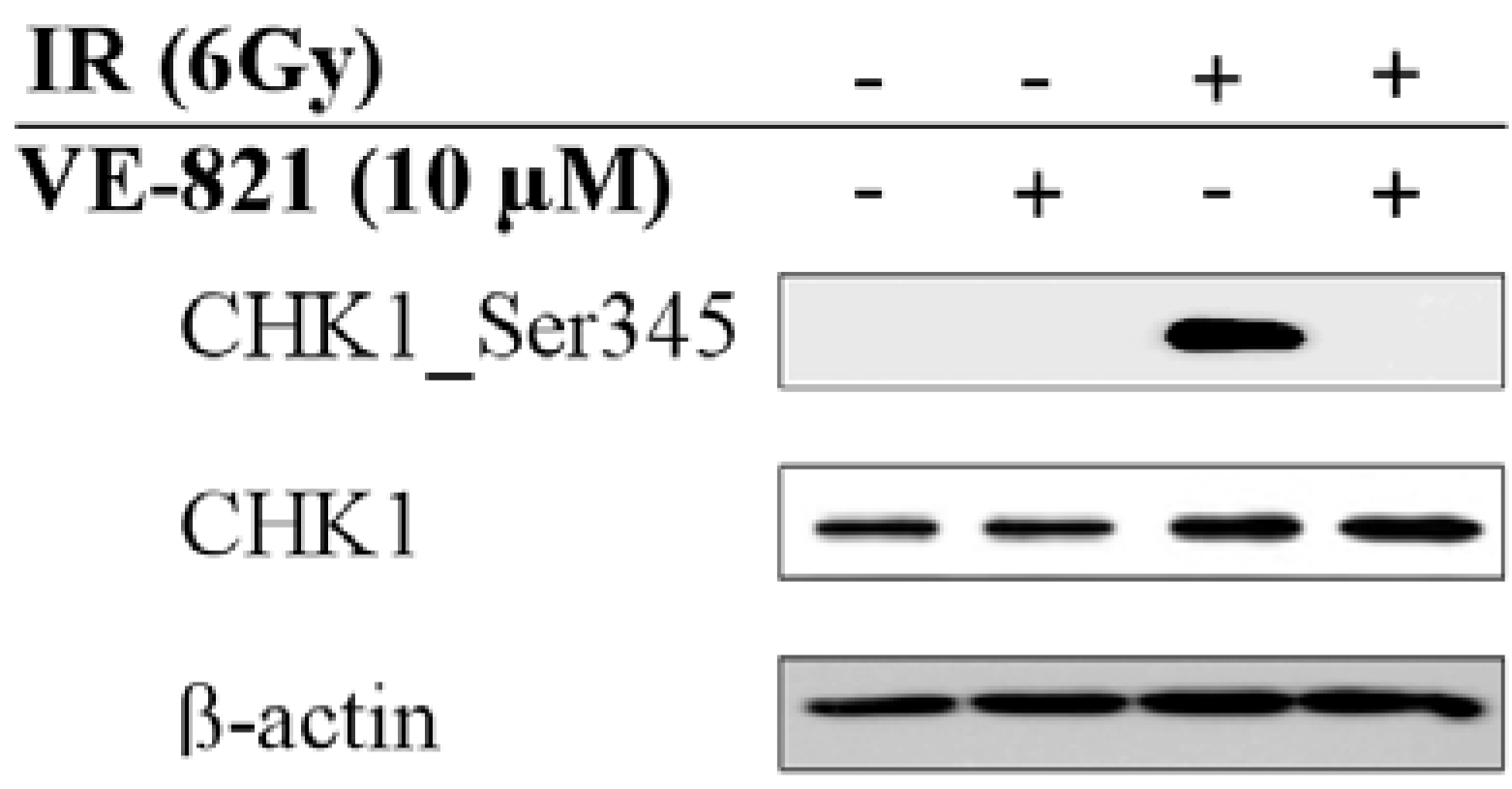
2.2. Combination of Ionizing Radiation and VE-821 Treatment Abrogated Phosphorylation of Checkpoint Kinase-1 on Ser345
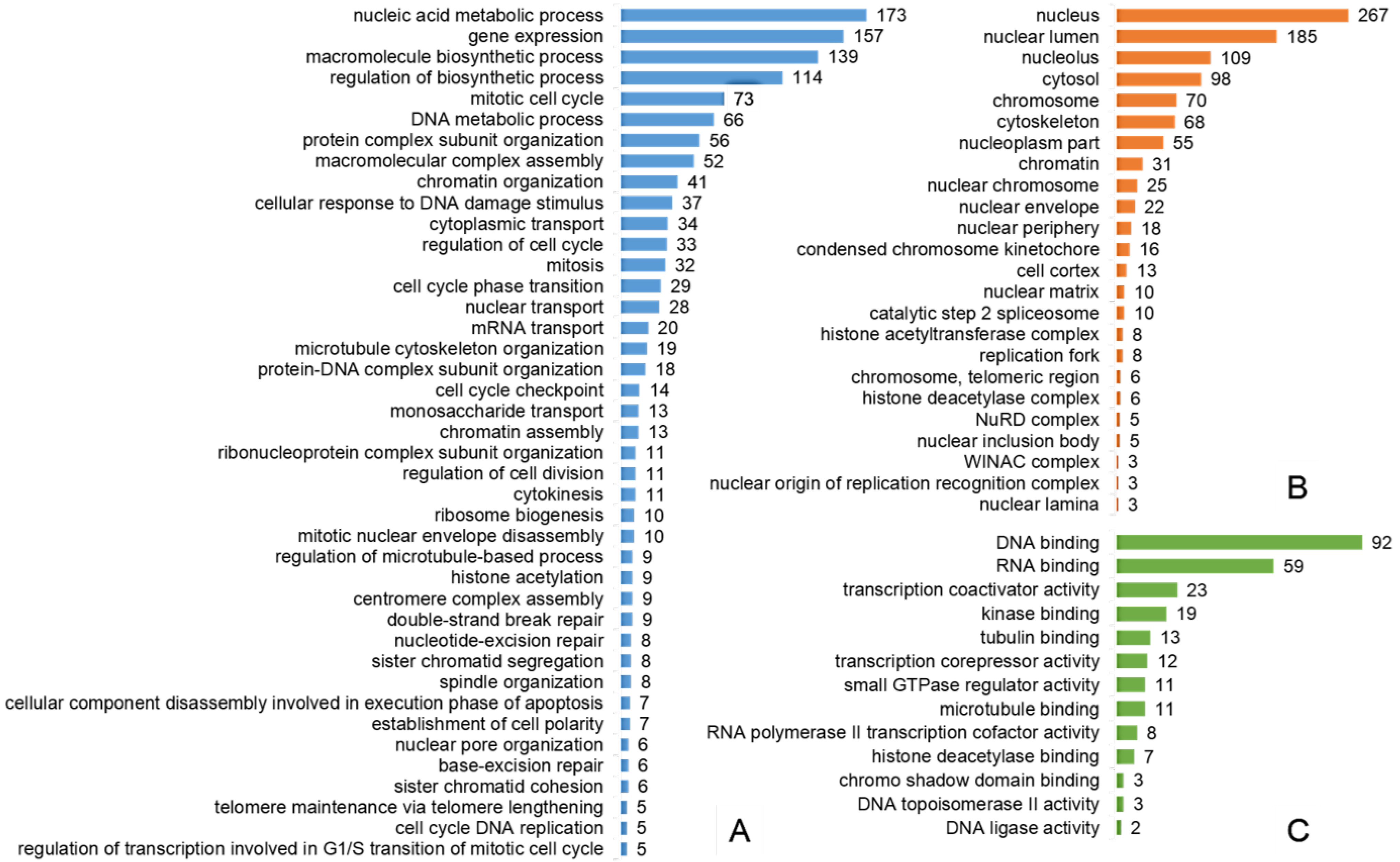
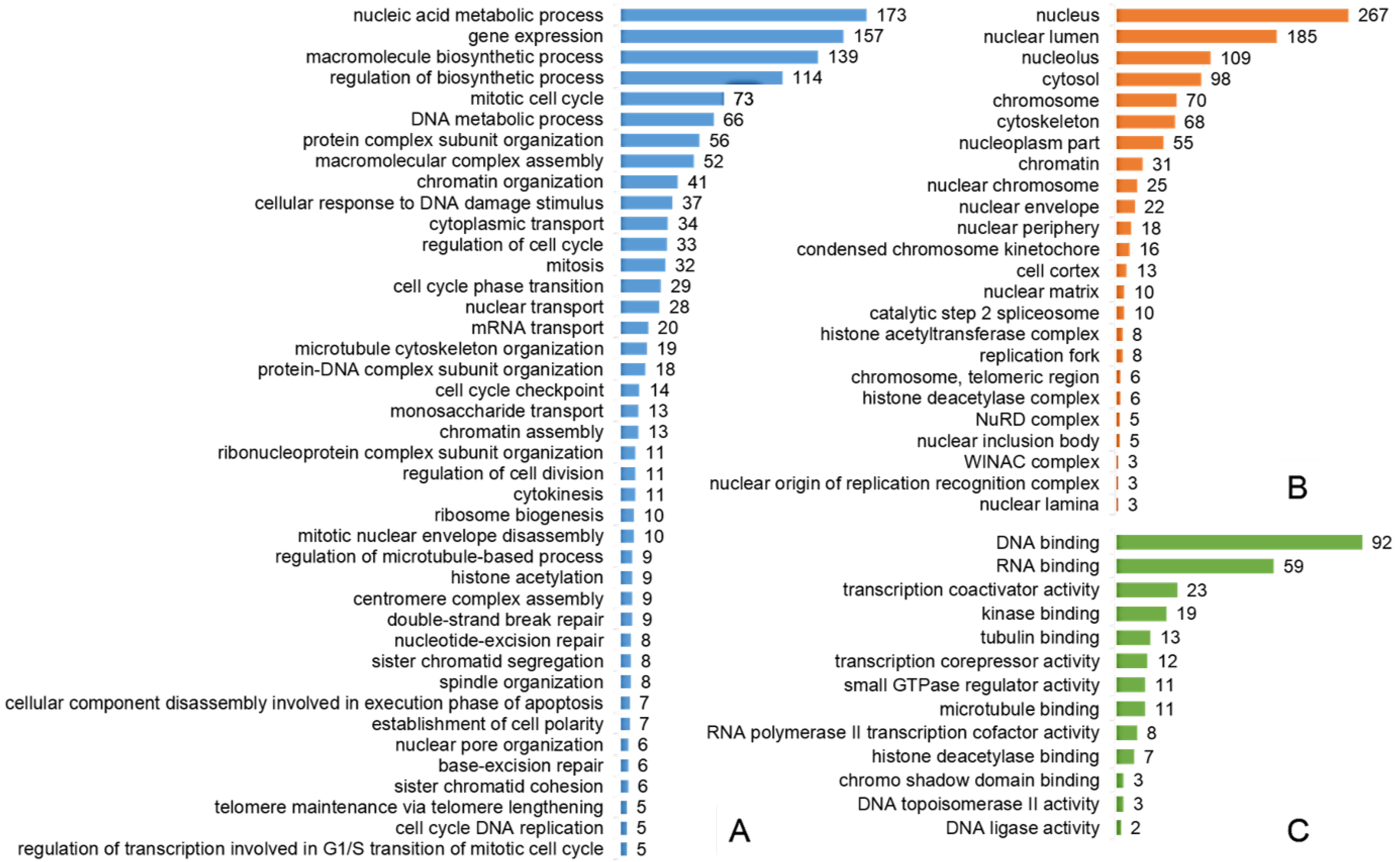
2.3. Most of the Regulated Phosphoproteins Were Localized in the Nucleus and Related to Mitosis, Cell Cycle Regulation, DNA Damage Response and Gene Expression
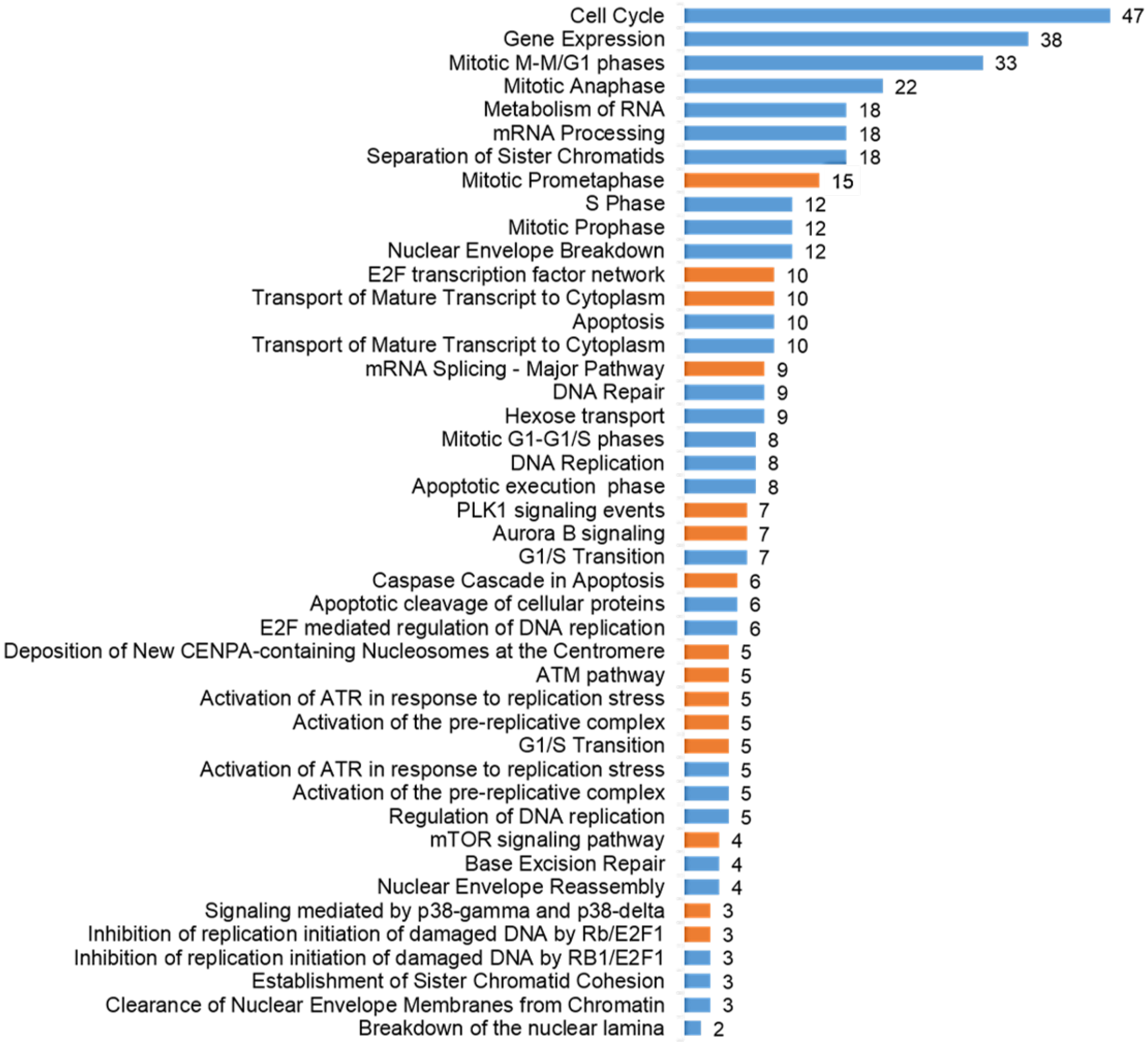
2.4. The Pathways Affected by Co-Treatment Were Involved in Cell Cycle Progression, Stimulus-Based Changes in Gene Expression, DDR and Apoptosis
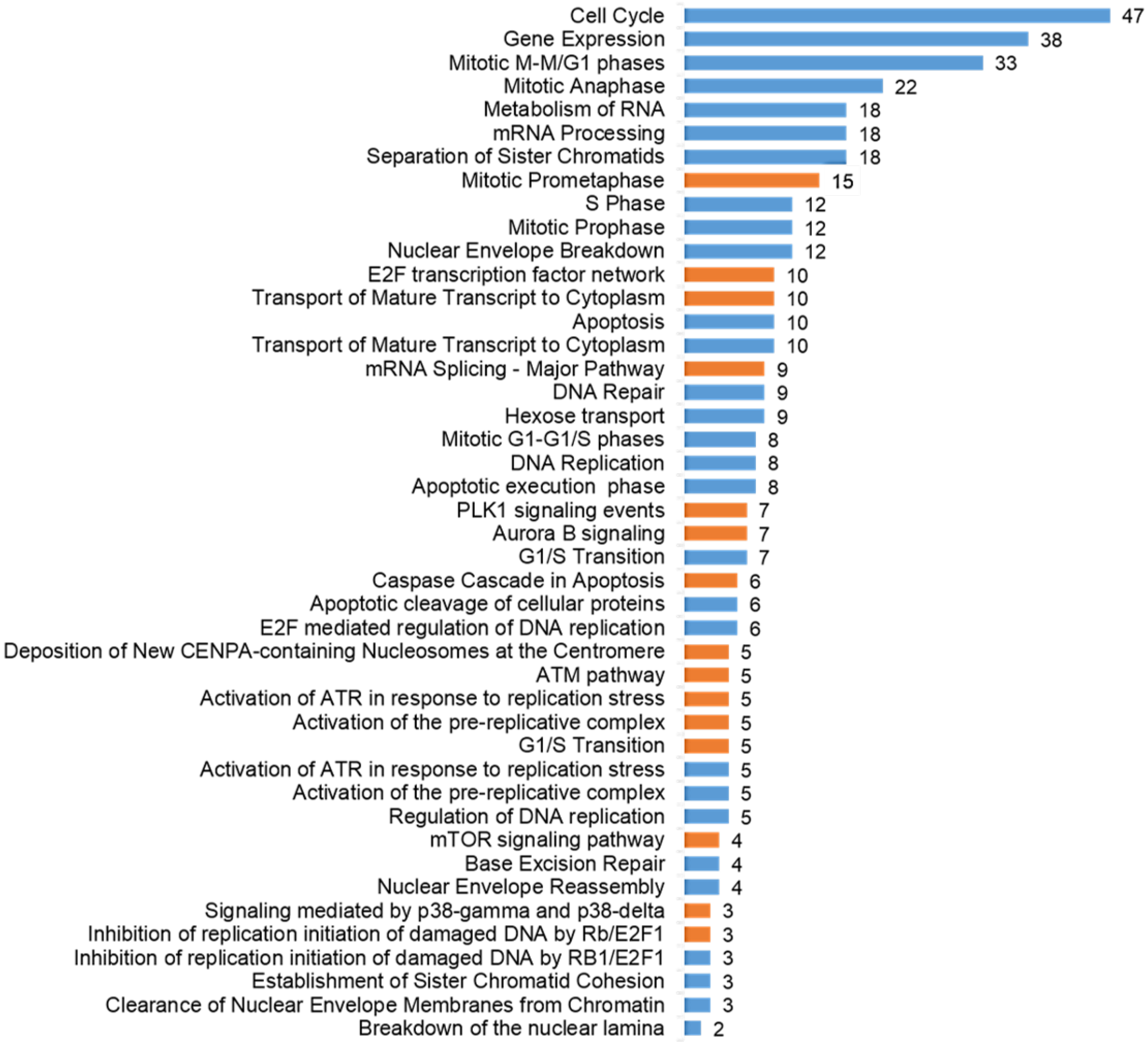
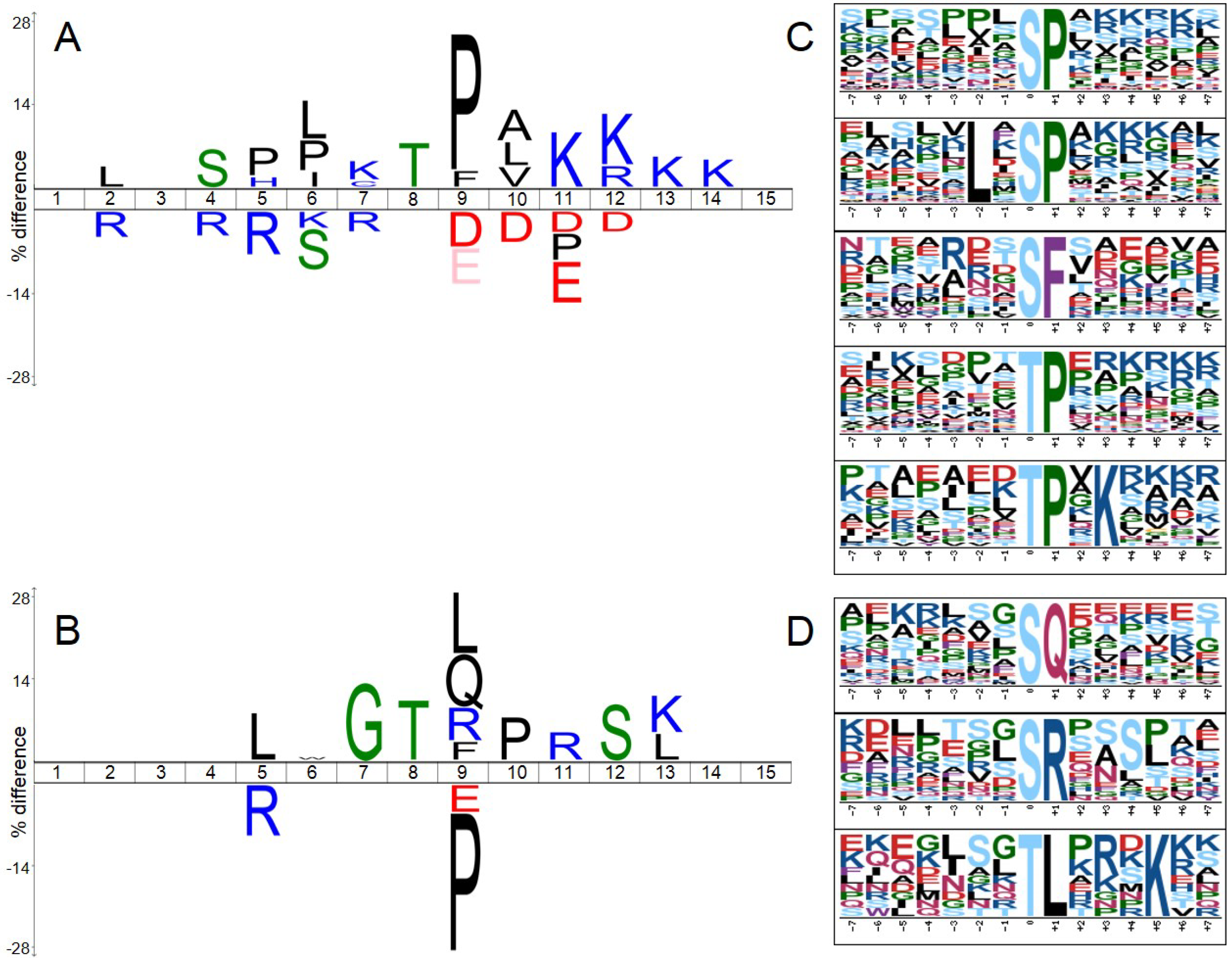
2.5. Significantly Down-Regulated Phosphorylation Sites Comprised SP/TP Motifs, while SQ/TQ Were amongst Up-Regulated Ones

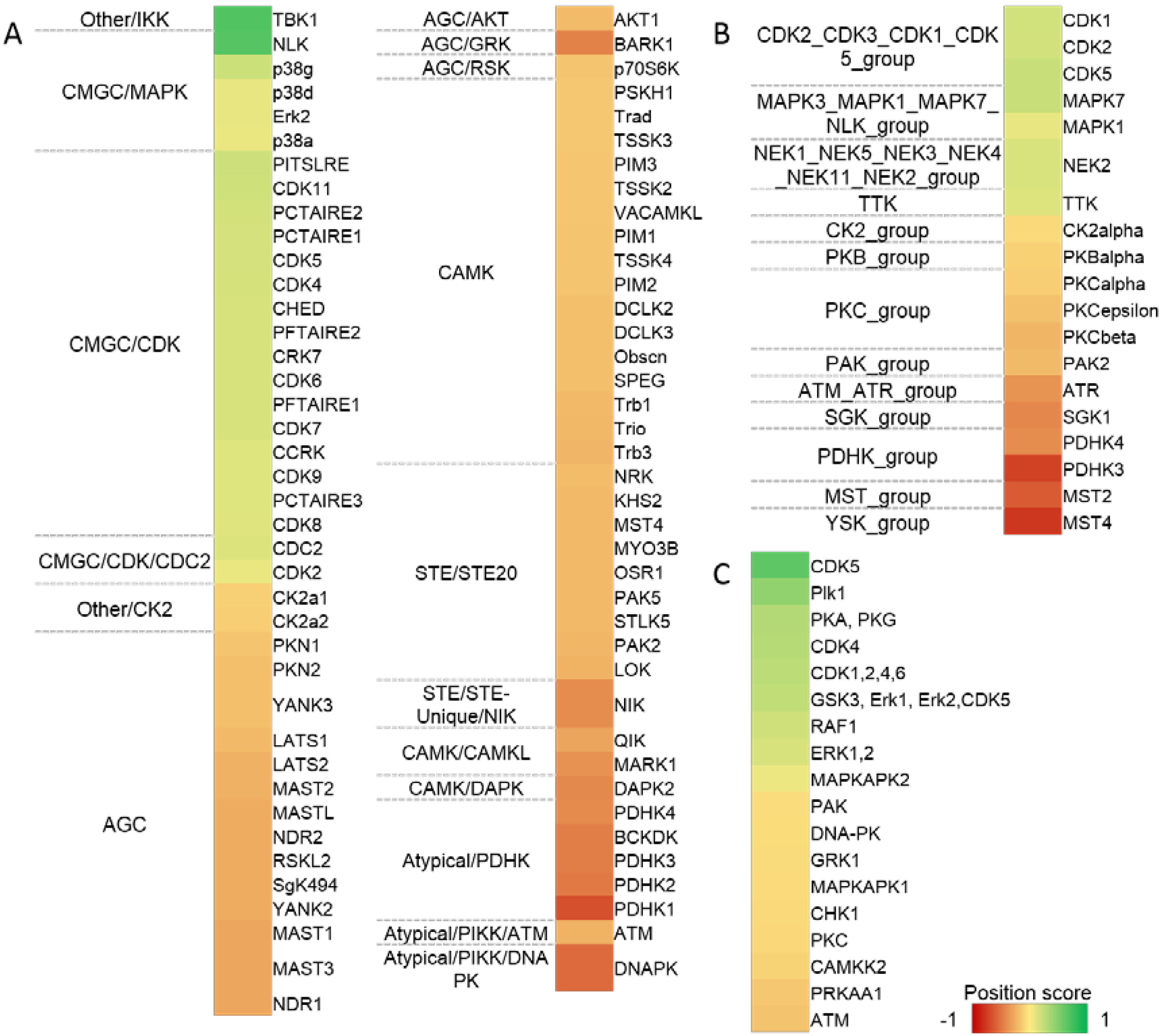
2.6. Kinase Activity Analyses Predicts Various Kinase Substrates to Be Regulated

3. Discussion
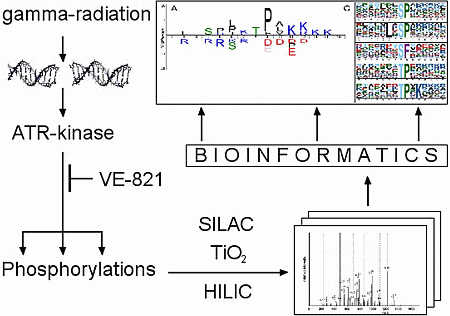
4. Experimental Section
4.1. Cell Culture and Culture Conditions
4.2. Gamma-Ray Irradiation
4.3. Cell Lysis and Protein Digest
4.4. Electrophoresis and Western Blotting
4.5. Hydrophilic Interaction Liquid Chromatography Fractionation and Enrichment of Phosphopeptides
4.6. Mass Spectrometric Analysis
4.7. Data Processing
4.8. Bioinformatic Analysis
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tichý, A.; Vávrová, J.; Pejchal, J.; Rezácová, M. Ataxia-telangiectasia mutated kinase (ATM) as a central regulator of radiation-induced DNA damage response. Acta Med. Hradec Králove 2010, 53, 13–17. [Google Scholar]
- Gamper, A.M.; Rofougaran, R.; Watkins, S.C.; Greenberger, J.S.; Beumer, J.H.; Bakkenist, C.J. ATR kinase activation in G1 phase facilitates the repair of ionizing radiation-induced DNA damage. Nucleic Acids Res. 2013, 41, 10334–10344. [Google Scholar]
- Charrier, J.-D.; Durrant, S.J.; Golec, J.M.C.; Kay, D.P.; Knegtel, R.M.A.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar]
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299. [Google Scholar]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 2012, 13, 1072–1081. [Google Scholar] [CrossRef]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 2014, 40, 109–117. [Google Scholar]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Johnson, L.N. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009, 37, 627–641. [Google Scholar] [CrossRef]
- Tichy, A.; Salovska, B.; Rehulka, P.; Klimentova, J.; Vavrova, J.; Stulik, J.; Hernychova, L. Phosphoproteomics: Searching for a needle in a haystack. J. Proteomics 2011, 74, 2786–2797. [Google Scholar]
- Salovska, B.; Tichy, A.; Fabrik, I.; Rezacova, M.; Vavrova, J. Comparison of resins for metal oxide affinity chromatography with mass spectrometry detection for the determination of phosphopeptides. Anal. Lett. 2013, 46, 1505–1524. [Google Scholar] [CrossRef]
- Vávrová, J.; Zárybnická, L.; Lukášová, E.; Řezáčová, M.; Novotná, E.; Sinkorová, Z.; Tichý, A.; Pejchal, J.; Durišová, K. Inhibition of ATR kinase with the selective inhibitor VE-821 results in radiosensitization of cells of promyelocytic leukaemia (HL-60). Radiat. Environ. Biophys. 2013, 52, 471–479. [Google Scholar]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 2011, 39, D712–D717. [Google Scholar]
- Kamburov, A.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB—A database for integrating human functional interaction networks. Nucleic Acids Res. 2009, 37, D623–D628. [Google Scholar]
- Croft, D. Building models using Reactome pathways as templates. Methods Mol. Biol. 2013, 1021, 273–283. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef]
- Chou, M.F.; Schwartz, D. Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinforma. 2011, 2011. [Google Scholar] [CrossRef]
- Schwartz, D.; Gygi, S.P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 2005, 23, 1391–1398. [Google Scholar]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jørgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar]
- Song, C.; Ye, M.; Liu, Z.; Cheng, H.; Jiang, X.; Han, G.; Songyang, Z.; Tan, Y.; Wang, H.; Ren, J.; et al. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol. Cell. Proteomics 2012, 11, 1070–1083. [Google Scholar] [CrossRef]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar]
- Mareková, M.; Vávrová, J.; Vokurková, D. Monitoring of premitotic and postmitotic apoptosis in gamma-irradiated HL-60 cells by the mitochondrial membrane protein-specific monoclonal antibody APO2.7. Gen. Physiol. Biophys. 2003, 22, 191–200. [Google Scholar]
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [Google Scholar] [CrossRef]
- Myers, J.S.; Cortez, D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar]
- Smits, V.A.J.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol. 2000, 2, 672–676. [Google Scholar]
- Tsvetkov, L.; Stern, D.F. Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle Georget. Tex. 2005, 4, 166–171. [Google Scholar] [CrossRef]
- Fletcher, L.; Cerniglia, G.J.; Nigg, E.A.; Yend, T.J.; Muschel, R.J. Inhibition of centrosome separation after DNA damage: A role for Nek2. Radiat. Res. 2004, 162, 128–135. [Google Scholar]
- Mi, J.; Guo, C.; Brautigan, D.L.; Larner, J.M. Protein phosphatase-1alpha regulates centrosome splitting through Nek2. Cancer Res. 2007, 67, 1082–1089. [Google Scholar]
- Qin, B.; Gao, B.; Yu, J.; Yuan, J.; Lou, Z. Ataxia telangiectasia-mutated- and Rad3-related protein regulates the DNA damage-induced G2/M checkpoint through the Aurora A cofactor Bora protein. J. Biol. Chem. 2013, 288, 16139–16144. [Google Scholar] [CrossRef]
- Bertoli, C.; Klier, S.; McGowan, C.; Wittenberg, C.; de Bruin, R.A.M. Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell-cycle transcription. Curr. Biol. 2013, 23, 1629–1637. [Google Scholar]
- Lin, W.C.; Lin, F.T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001, 15, 1833–1844. [Google Scholar]
- Lau, E.; Tsuji, T.; Guo, L.; Lu, S.H.; Jiang, W. The role of pre-replicative complex (pre-RC) components in oncogenesis. FASEB J. 2007, 21, 3786–3794. [Google Scholar] [CrossRef]
- Bendall, S.C.; Hughes, C.; Stewart, M.H.; Doble, B.; Bhatia, M.; Lajoie, G.A. Prevention of amino acid conversion in SILAC experiments with embryonic stem cells. Mol. Cell. Proteomics. 2008, 7, 1587–1597. [Google Scholar]
- Ong, S.E.; Mann, M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 2006, 1, 2650–2660. [Google Scholar]
- Rogers, L.D.; Fang, Y.; Foster, L.J. An integrated global strategy for cell lysis, fractionation, enrichment and mass spectrometric analysis of phosphorylated peptides. Mol. Biosyst. 2010, 6, 822–829. [Google Scholar] [CrossRef]
- Yeung, Y.G.; Stanley, E.R. Rapid detergent removal from peptide samples with ethyl acetate for Mass Spectrometry Analysis. Curr. Protoc. Protein Sci. 2010, 2010. [Google Scholar] [CrossRef]
- McNulty, D.E.; Annan, R.S. Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol. Cell. Proteomics 2008, 7, 971–980. [Google Scholar] [CrossRef]
- Larsen, M.R.; Thingholm, T.E.; Jensen, O.N.; Roepstorff, P.; Jorgensen, T.J.D. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 2005, 4, 873–886. [Google Scholar]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Zhou, Y.; Cras-Méneur, C.; Ohsugi, M.; Stormo, G.D.; Permutt, M.A. A global approach to identify differentially expressed genes in cDNA (two-color) microarray experiments. Bioinformatics 2007, 23, 2073–2079. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar]
- Cox, J.; Mann, M. 1D and 2D annotation enrichment: A statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinfom. 2012, 13. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes . Nature 2007, 446, 153–158. [Google Scholar]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Šalovská, B.; Fabrik, I.; Ďurišová, K.; Link, M.; Vávrová, J.; Řezáčová, M.; Tichý, A. Radiosensitization of Human Leukemic HL-60 Cells by ATR Kinase Inhibitor (VE-821): Phosphoproteomic Analysis. Int. J. Mol. Sci. 2014, 15, 12007-12026. https://doi.org/10.3390/ijms150712007
Šalovská B, Fabrik I, Ďurišová K, Link M, Vávrová J, Řezáčová M, Tichý A. Radiosensitization of Human Leukemic HL-60 Cells by ATR Kinase Inhibitor (VE-821): Phosphoproteomic Analysis. International Journal of Molecular Sciences. 2014; 15(7):12007-12026. https://doi.org/10.3390/ijms150712007
Chicago/Turabian StyleŠalovská, Barbora, Ivo Fabrik, Kamila Ďurišová, Marek Link, Jiřina Vávrová, Martina Řezáčová, and Aleš Tichý. 2014. "Radiosensitization of Human Leukemic HL-60 Cells by ATR Kinase Inhibitor (VE-821): Phosphoproteomic Analysis" International Journal of Molecular Sciences 15, no. 7: 12007-12026. https://doi.org/10.3390/ijms150712007