PSNO: Predicting Cysteine S-Nitrosylation Sites by Incorporating Various Sequence-Derived Features into the General Form of Chou’s PseAAC

Abstract
:1. Introduction
2. Results and Discussion
2.1. The Feature Selection Results
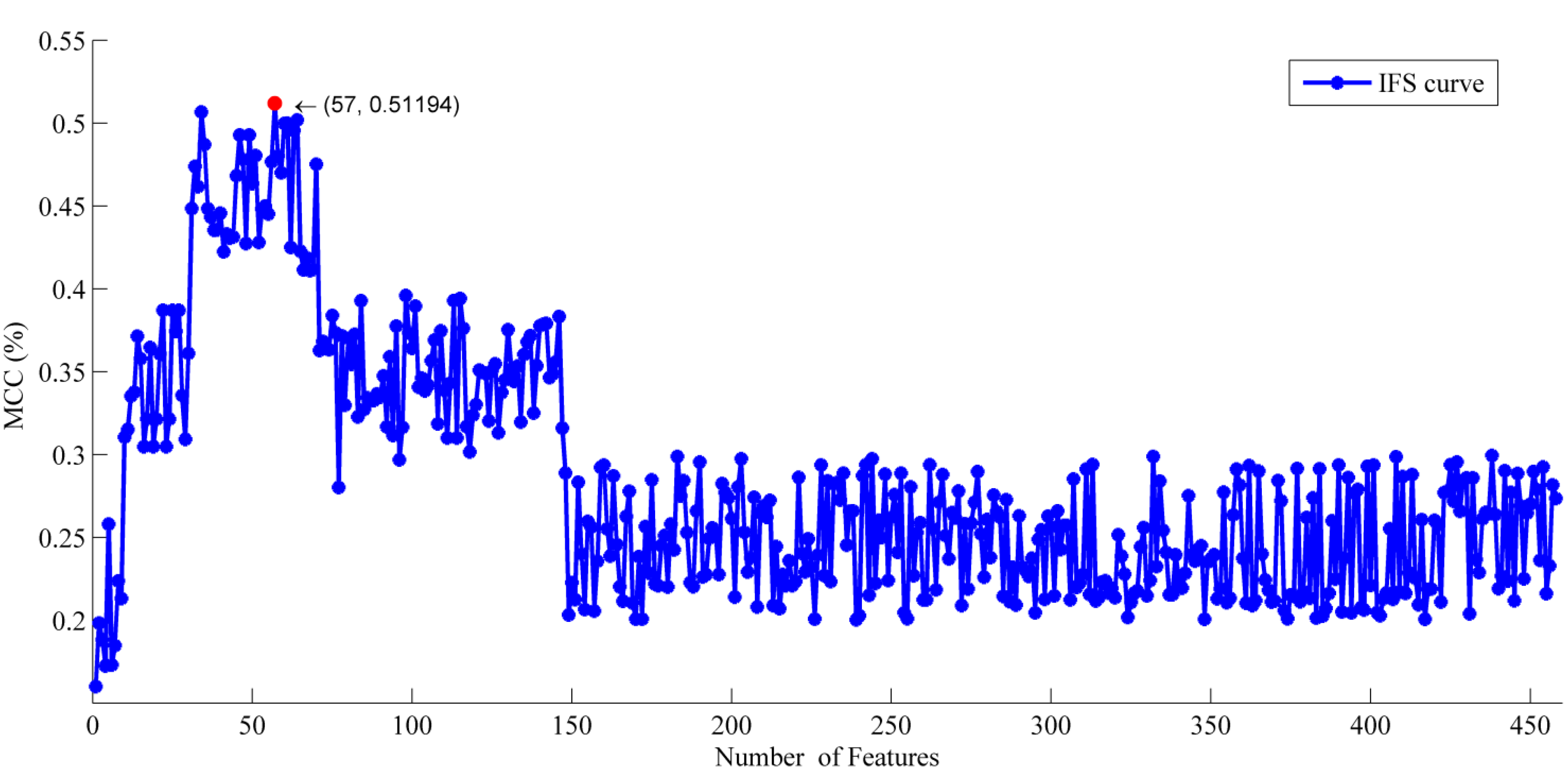
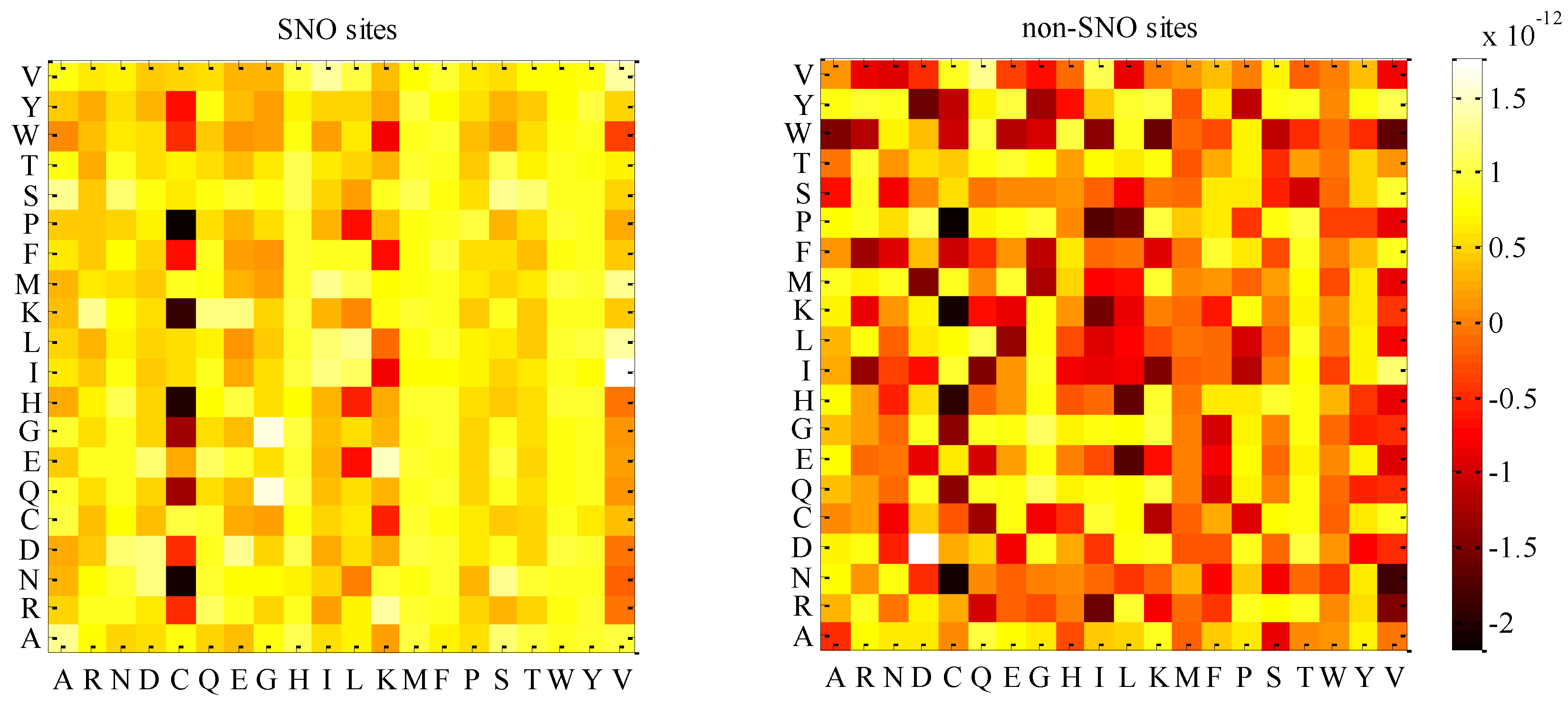
2.2. Analysis of the Optimal Feature Set

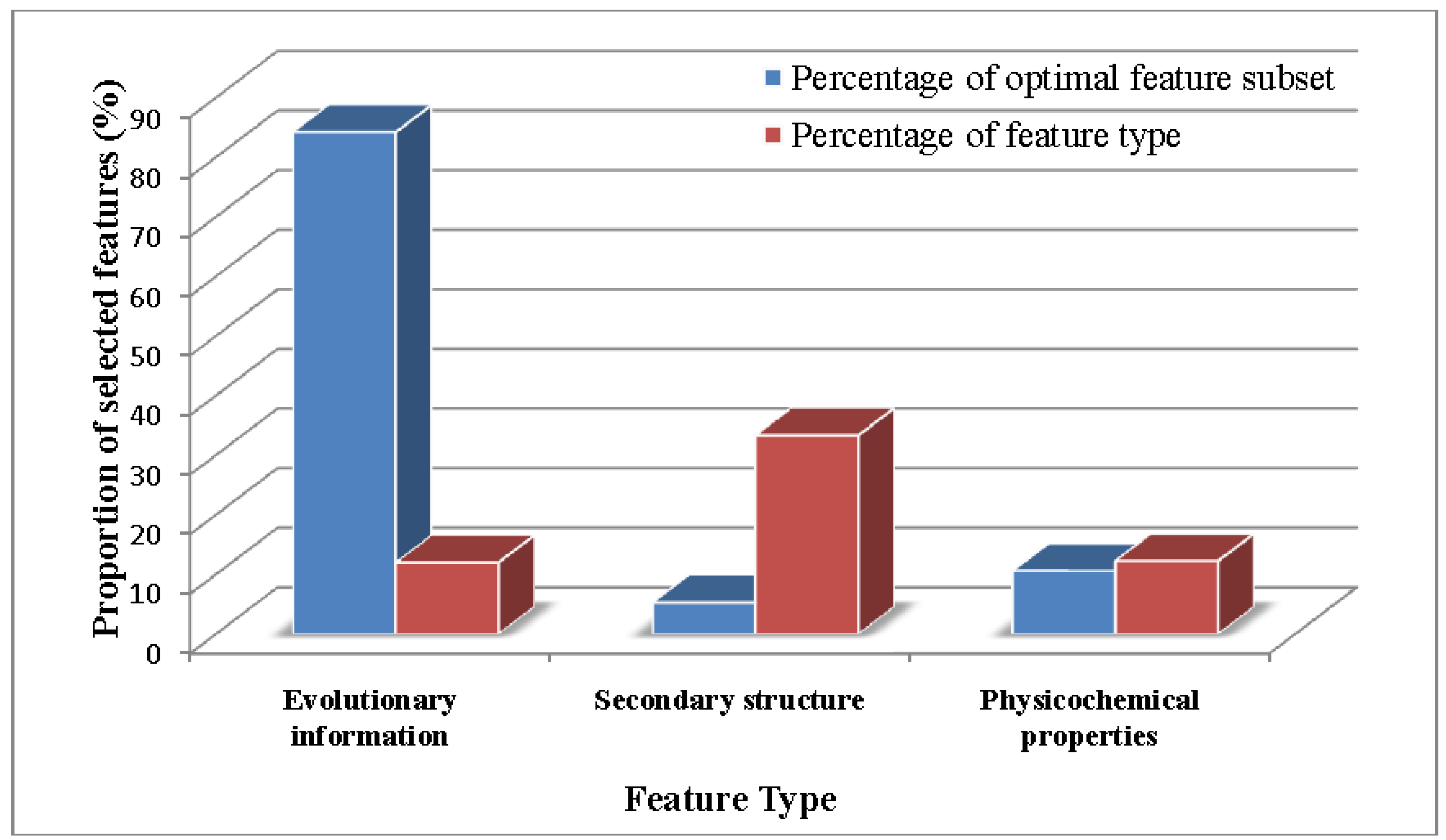
2.3. Comparison of PSNO with Other Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predictor | SN (%) | SP (%) | ACC (%) | MCC |
|---|---|---|---|---|
| GPS-SNO 1 | 18.88 | 89.63 | 56.07 | 0.1210 |
| GPS-SNO 2 | 28.04 | 81.98 | 56.39 | 0.1193 |
| GPS-SNO 3 | 45.01 | 73.33 | 59.90 | 0.1915 |
| iSNO-PseAAC | 67.01 | 68.15 | 67.62 | 0.3515 |
| PSNO | 74.15 | 77.04 | 75.67 | 0.5119 |
| Predictor | SN (%) | SP (%) | ACC (%) | MCC |
|---|---|---|---|---|
| GPS-SNO 1 | 44.5 | 81.0 | 64.7 | 0.28 |
| iSNO-PseAAC | 50.2 | 75.2 | 62.8 | 0.30 |
| iSNO-AAPair | 79.6 | 84.1 | 81.7 | 0.63 |
| PSNO | 87.7 | 85.0 | 86.2 | 0.72 |
| Predictor | SN (%) | SP (%) | ACC (%) | MCC |
|---|---|---|---|---|
| GPS-SNO 1 | 41.51 | 70.87 | 60.90 | 0.1244 |
| iSNO-PseAAC | 60.38 | 67.96 | 65.38 | 0.2722 |
| iSNO-AAPair | 66.04 | 66.02 | 66.03 | 0.3053 |
| PSNO | 79.25 | 67.96 | 71.79 | 0.4475 |
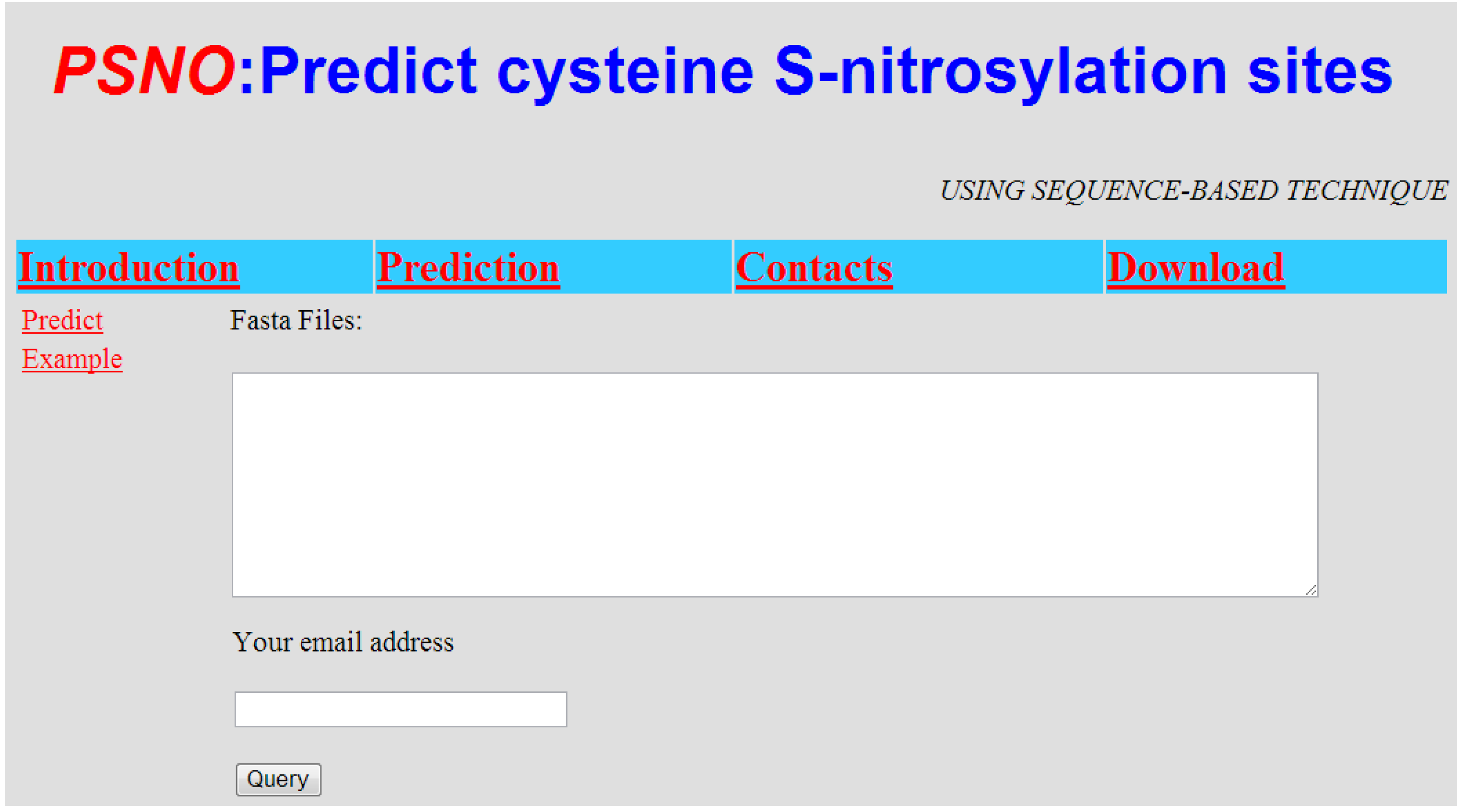
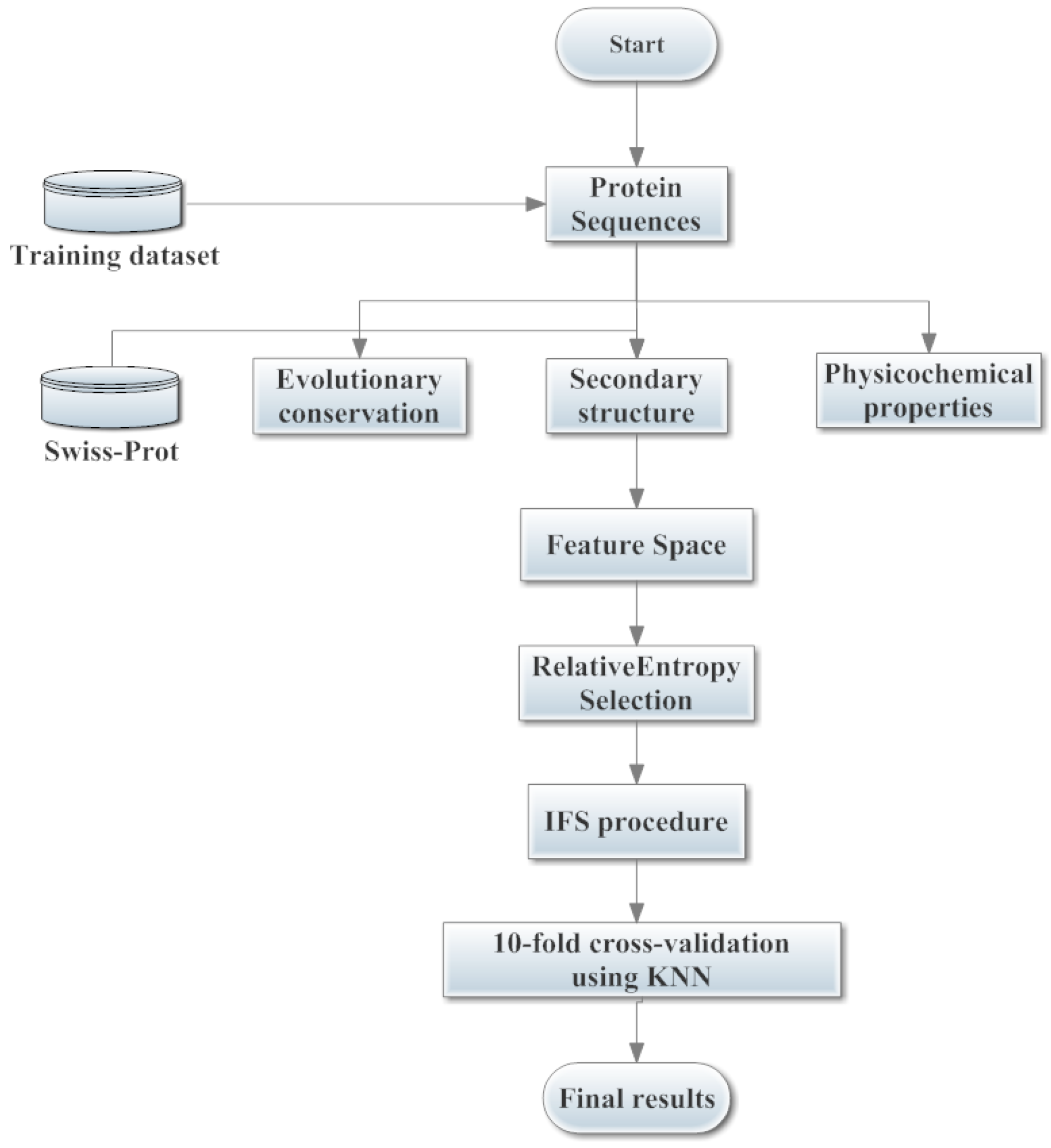
2.4. Implementation of PSNO Server

3. Materials and Methods
3.1. Benchmark Datasets
| Dataset | Proteins | Peptides | SNOs | Non-SNOs |
|---|---|---|---|---|
| Training dataset | 438 | 1541 | 731 | 810 |
| Xue’s independent dataset | 461 | 2302 | 2302 | 0 |
| Xu’s independent dataset | - | 181 | 81 | 100 |
| Our independent dataset | 20 | 156 | 53 | 103 |
3.2. Sample Formulation and Feature Construction
3.2.1. Features of Evolutionary Conservation Scores
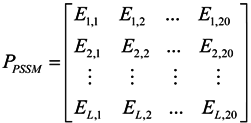

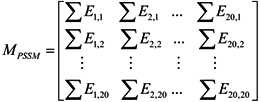
3.2.2. Features of Predicted Secondary Structure
3.2.3. Features of Physicochemical Properties
3.3. The Relative Entropy Selection
3.4. Incremental Feature Selection
3.5. K-Nearest Neighbor Algorithm
3.6. Assessment of Prediction Accuracy
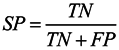
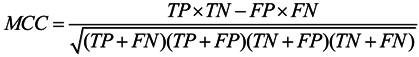
3.7. Cross-Validation Test

4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Foster, M.W.; Douglas, T.H.; Jonathan, S.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef]
- Foster, M.W.; Timothy, J.M.; Jonathan, S.S. S-nitrosylation in health and disease. Trends Mol. Med. 2003, 9, 160–168. [Google Scholar] [CrossRef]
- Aranda, E.; López-Pedrera, C.; de La Haba-Rodriguez, R.J.; Rodriguez-Ariza, A. Nitric oxide and cancer: The emerging role of S-nitrosylation. Curr. Mol. Med. 2012, 12, 50–67. [Google Scholar] [CrossRef]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Lipton, S.A. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef]
- Nakamura, T.; Cieplak, P.; Cho, D.H.; Godzik, A.; Lipton, S.A. S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 2006, 10, 573–578. [Google Scholar]
- Schonhoff, C.M.; Matsuoka, M.; Tummala, H.; Johnson, M.A.; Estevéz, A.G.; Wu, R.; Mannick, J.B. S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2404–2409. [Google Scholar]
- Lindermayr, C.; Saalbach, G.; Durner, J. Proteomic identification of S-nitrosylated proteins in Arabidopsis. Plant Physiol. 2005, 137, 921–930. [Google Scholar] [CrossRef]
- Cook, J.A.; Kim, S.Y.; Teague, D.; Krishna, M.C.; Pacelli, R.; Mitchell, J.B.; Wink, D.A. Convenient colorimetric and fluorometric assays for S-nitrosothiols. Anal. Biochem. 1996, 238, 150–158. [Google Scholar] [CrossRef]
- Gaston, B. Nitric oxide and thiol groups. Biochim. Biophys. Acta 1999, 1411, 323–333. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Snyder, S.H. The biotin switch method for the detection of S-nitrosylated proteins. Sci. Signal. 2001, 2001. [Google Scholar] [CrossRef]
- Hao, G.; Derakhshan, B.; Shi, L.; Campagne, F.; Gross, S.S. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc. Natl. Acad. Sci. USA 2006, 103, 1012–1017. [Google Scholar] [CrossRef]
- Forrester, M.T.; Thompson, J.W.; Foster, M.W.; Nogueira, L.; Moseley, M.A.; Stamler, J.S. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009, 27, 557–559. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, Z.; Gao, X.; Jin, C.; Wen, L.; Yao, X.; Ren, J. GPS-SNO: Computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PLoS One 2010, 5, e11290. [Google Scholar]
- Li, Y.X.; Yuan, H.S.; Ling, J.; Nai, Y.D. An efficient support vector machine approach for identifying protein S-nitrosylation sites. Protein Pept. Lett. 2011, 18, 573–587. [Google Scholar] [CrossRef]
- Xu, Y.; Ding, J.; Wu, L.Y.; Chou, K.C. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One 2013, 8, e55844. [Google Scholar]
- Xu, Y.; Shao, X.J.; Wu, L.Y.; Deng, N.Y.; Chou, K.C. iSNO-AAPair: Incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ 2013, 1, e171. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. Large-Scale plant protein subcellular location prediction. J. Cell. Biochem. 2007, 100, 665–678. [Google Scholar] [CrossRef]
- Chou, K.C.; Chun, T.Z. Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef]
- Li, B.Q.; Hu, L.L.; Niu, S.; Cai, Y.D.; Chou, K.C. Predict and analyze S-nitrosylation modification sites with the mRMR and IFS approaches. J. Proteomics 2012, 75, 1654–1665. [Google Scholar] [CrossRef]
- Chen, Y.J.; Ku, W.C.; Lin, P.Y.; Chou, H.C.; Khoo, K.H.; Chen, Y.J. S-alkylating labeling strategy for site-specific identification of the S-nitrosoproteome. J. Proteome Res. 2010, 9, 6417–6439. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Lin, H.; Ding, H.; Guo, F.B.; Zhang, A.Y.; Huang, J. Predicting subcellular localization of mycobacterial proteins by using Chou’s pseudo amino acid composition. Protein Pept. Lett. 2008, 15, 739–744. [Google Scholar] [CrossRef]
- Nanni, L.; Lumini, A.; Gupta, D.; Garg, A. Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of Chou’s pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 2012, 9, 467–475. [Google Scholar] [CrossRef]
- Zou, D.; He, Z.; He, J.; Xia, Y. Supersecondary structure prediction using Chou’s pseudo amino acid composition. J. Comput. Chem. 2011, 32, 271–278. [Google Scholar] [CrossRef]
- Sahu, S.S.; Panda, G. A novel feature representation method based on Chou’s pseudo amino acid composition for protein structural class prediction. Comput. Biol. Chem. 2010, 34, 320–327. [Google Scholar] [CrossRef]
- Qiu, J.D.; Huang, J.H.; Shi, S.P.; Liang, R.P. Using the concept of chous pseudo amino acid composition to predict enzyme family classes: An approach with support vector machine based on discrete wavelet transform. Protein Pept. Lett. 2010, 17, 715–722. [Google Scholar] [CrossRef]
- Zhou, X.B.; Chen, C.; Li, Z.C.; Zou, X.Y. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol. 2007, 248, 546–551. [Google Scholar] [CrossRef]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Xie, D.; Li, A.; Wang, M.; Fan, Z.; Feng, H. LOCSVMPSI: A web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST. Nucleic Acids Res. 2005, 33, W105–W110. [Google Scholar] [CrossRef]
- Mundra, P.; Kumar, M.; Kumar, K.K.; Jayaraman, V.K.; Kulkarni, B.D. Using pseudo amino acid composition to predict protein subnuclear localization: Approached with PSSM. Pattern Recognit. Lett. 2007, 28, 1610–1615. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. ProtIdent: A web server for identifying proteases and their types by fusing functional domain and sequential evolution information. Biochem. Biophys. Res. Commun. 2008, 376, 321–325. [Google Scholar] [CrossRef]
- Qiu, W.R.; Xiao, X.; Chou, K.C. iRSpot-TNCPseAAC: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int. J. Mol. Sci. 2014, 15, 1746–1766. [Google Scholar] [CrossRef]
- Altschul, S.F. Evaluating the statistical significance of multiple distinct local alignments. In Theoretical and Computational Methods in Genome Research; Springer US: New York, NY, USA, 1997; pp. 1–14. [Google Scholar]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Selvaraj, S. Importance of long-range interactions in protein folding. Biophys. Chem. 1999, 77, 49–68. [Google Scholar] [CrossRef]
- Gromiha, M.M. A statistical model for predicting protein folding rates from amino acid sequence with structural class information. J. Chem. Inf. Model. 2005, 45, 494–501. [Google Scholar] [CrossRef]
- Qian, J.; Miao, D.Q.; Zhang, Z.H.; Li, W. Hybrid approaches to attribute reduction based on indiscernibility and discernibility relation. Int. J. Approx. Reason. 2011, 52, 212–230. [Google Scholar] [CrossRef]
- Johnson, D.H.; Sinanovic, S. Symmetrizing the Kullback-Leibler Distance; Technical Report for Computer and Information Technology; Rice University: Houston, TX, USA, 2001. [Google Scholar]
- Keller, J.M.; Michael, R.G.; James, A.G. A fuzzy k-nearest neighbor algorithm. Syst. Man Cybern. IEEE Trans. 1985, 4, 580–585. [Google Scholar] [CrossRef]
- Mardia, K.V.; John, T.K.; John, M.B. Multivariate Analysis; Academic Press: London, UK, 1980. [Google Scholar]
- Read, C.B.; Samuel, K.; Norman, L.J. Encyclopedia of Statistical Sciences; Wiley: Hoboken, NJ, USA, 1982. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, J.; Zhao, X.; Sun, P.; Ma, Z. PSNO: Predicting Cysteine S-Nitrosylation Sites by Incorporating Various Sequence-Derived Features into the General Form of Chou’s PseAAC. Int. J. Mol. Sci. 2014, 15, 11204-11219. https://doi.org/10.3390/ijms150711204
Zhang J, Zhao X, Sun P, Ma Z. PSNO: Predicting Cysteine S-Nitrosylation Sites by Incorporating Various Sequence-Derived Features into the General Form of Chou’s PseAAC. International Journal of Molecular Sciences. 2014; 15(7):11204-11219. https://doi.org/10.3390/ijms150711204
Chicago/Turabian StyleZhang, Jian, Xiaowei Zhao, Pingping Sun, and Zhiqiang Ma. 2014. "PSNO: Predicting Cysteine S-Nitrosylation Sites by Incorporating Various Sequence-Derived Features into the General Form of Chou’s PseAAC" International Journal of Molecular Sciences 15, no. 7: 11204-11219. https://doi.org/10.3390/ijms150711204
APA StyleZhang, J., Zhao, X., Sun, P., & Ma, Z. (2014). PSNO: Predicting Cysteine S-Nitrosylation Sites by Incorporating Various Sequence-Derived Features into the General Form of Chou’s PseAAC. International Journal of Molecular Sciences, 15(7), 11204-11219. https://doi.org/10.3390/ijms150711204