Steatosis and Steatohepatitis: Complex Disorders

Abstract
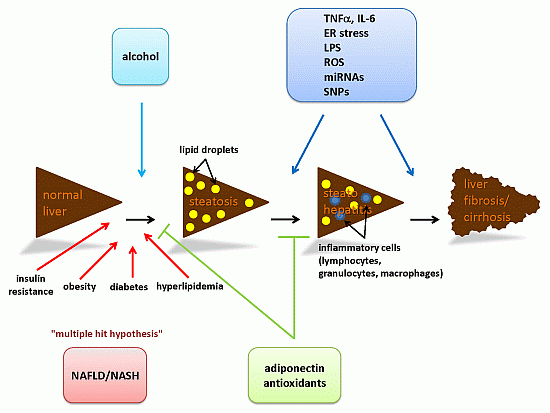
:
{kind=link}
{kind=link}
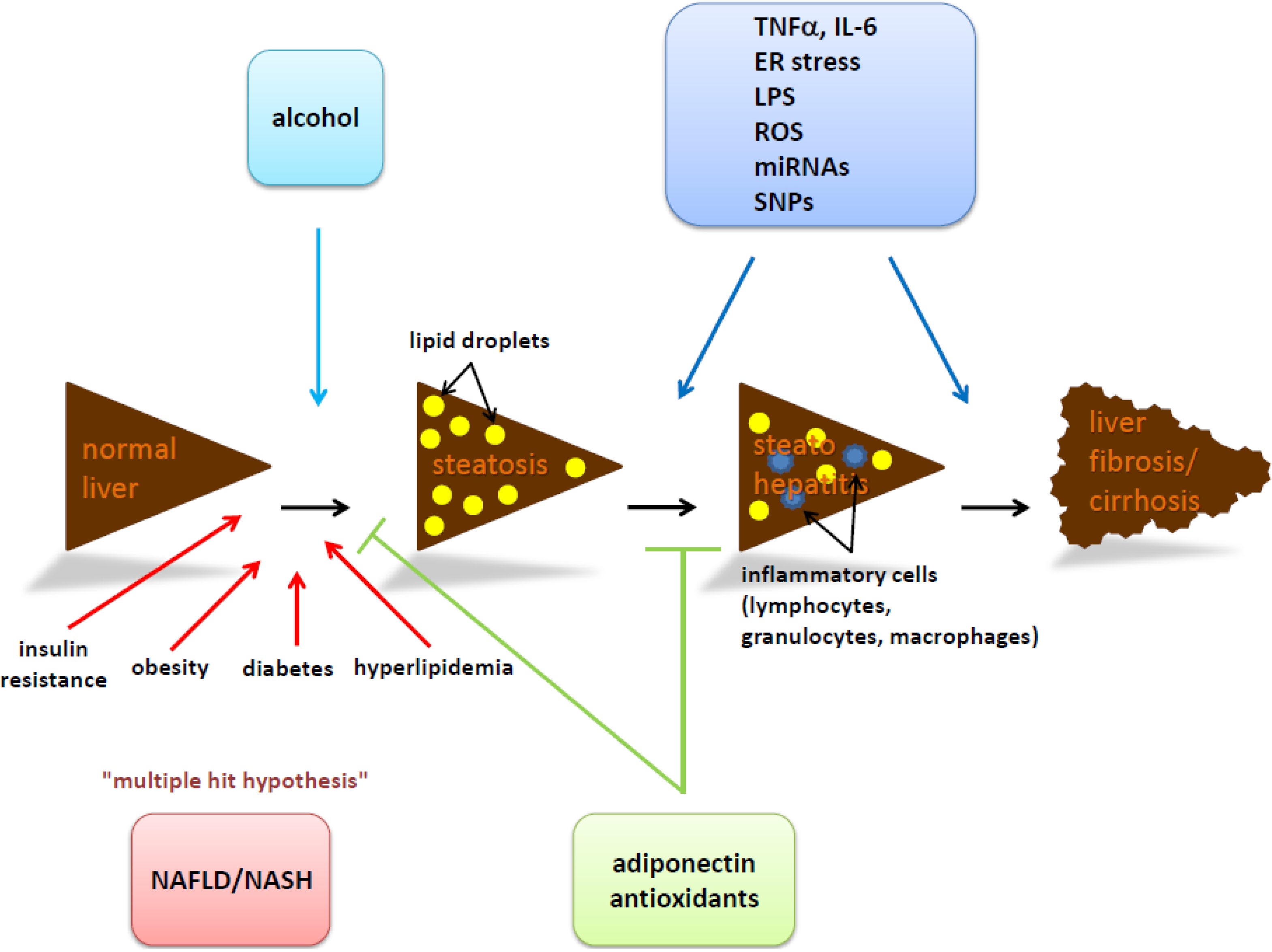{kind=link}
1. Introduction
2. Epidemiology and Risk Factors
3. Genomics and Epigenomics
4. Diagnosis
5. Molecular Pathology
6. Treatment and Prevention
7. Murine Non-Alcoholic Fatty Liver Disease (NAFLD) Models and Their Potential Human Relevance
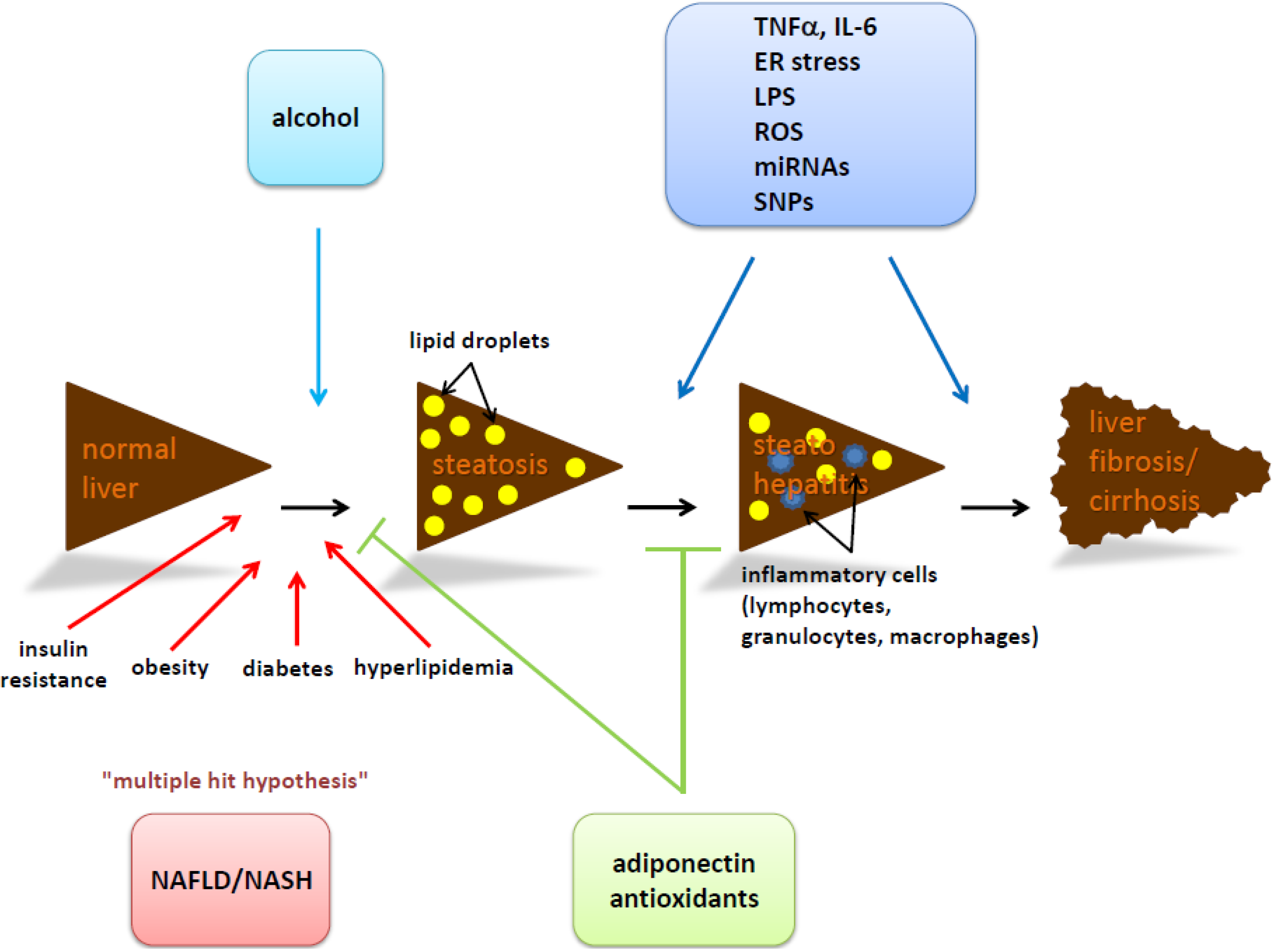
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Angulo, P. GI epidemiology: Non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef]
- Law, K.; Brunt, E.M. Nonalcoholic fatty liver disease. Clin. Liver Dis. 2010, 14, 591–604. [Google Scholar] [CrossRef]
- Oh, M.K.; Winn, J.; Poordad, F. Review article: Diagnosis and treatment of non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 28, 503–522. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2011, 9, 524–530. [Google Scholar]
- Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 195–203. [Google Scholar]
- Aly, F.Z.; Kleiner, D.E. Update on fatty liver disease and steatohepatitis. Adv. Anat. Pathol. 2011, 18, 294–300. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef]
- Ratziu, V.; Poynard, T. Assessing the outcome of nonalcoholic steatohepatitis? It’s time to get serious. Hepatology 2006, 44, 802–805. [Google Scholar] [CrossRef]
- Falck-Ytter, Y.; Younossi, Z.M.; Marchesini, G.; McCullough, A.J. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin. Liver Dis. 2001, 21, 17–26. [Google Scholar] [CrossRef]
- Powell, E.E.; Cooksley, W.G.; Hanson, R.; Searle, J.; Halliday, J.W.; Powell, L.W. The natural history of nonalcoholic steatohepatitis: A follow-up study of forty-two patients for up to 21 years. Hepatology 1990, 11, 74–80. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Deutsch, R.; Rauch, J.B.; Behling, C.; Newbury, R.; Lavine, J.E. Obesity, insulin resistance, and other clinicopathological correlates of pediatric nonalcoholic fatty liver disease. J. Pediatr. 2003, 143, 500–505. [Google Scholar] [CrossRef]
- Kelishadi, R.; Cook, S.R.; Adibi, A.; Faghihimani, Z.; Ghatrehsamani, S.; Beihaghi, A.; Salehi, H.; Khavarian, N.; Poursafa, P. Association of the components of the metabolic syndrome with non-alcoholic fatty liver disease among normal-weight, overweight and obese children and adolescents. Diabetol. Metab. Syndr. 2009, 1, 29. [Google Scholar]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar]
- Pais, R.; Pascale, A.; Fedchuck, L.; Charlotte, F.; Poynard, T.; Ratziu, V. Progression from isolated steatosis to steatohepatitis and fibrosis in nonalcoholic fatty liver disease. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 23–28. [Google Scholar] [CrossRef]
- Zois, C.-D.; Baltayiannis, G.-H.; Bekiari, A.; Goussia, A.; Karayiannis, P.; Doukas, M.; Demopoulos, D.; Mitsellou, A.; Vougiouklakis, T.; Mitsi, V.; et al. Steatosis and steatohepatitis in postmortem material from Northwestern Greece. World J. Gastroenterol. 2010, 16, 3944–3949. [Google Scholar] [CrossRef]
- Caballería, L.; Pera, G.; Auladell, M.A.; Torán, P.; Muñoz, L.; Miranda, D.; Alumà, A.; Casas, J.D.; Sánchez, C.; Gil, D.; et al. Prevalence and factors associated with the presence of nonalcoholic fatty liver disease in an adult population in Spain. Eur. J. Gastroenterol. Hepatol. 2010, 22, 24–32. [Google Scholar] [CrossRef]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Kojima, S.-I.; Watanabe, N.; Numata, M.; Ogawa, T.; Matsuzaki, S. Increase in the prevalence of fatty liver in Japan over the past 12 years: Analysis of clinical background. J. Gastroenterol. 2003, 38, 954–961. [Google Scholar]
- Amarapurkar, D.; Kamani, P.; Patel, N.; Gupte, P.; Kumar, P.; Agal, S.; Baijal, R.; Lala, S.; Chaudhary, D.; Deshpande, A. Prevalence of non-alcoholic fatty liver disease: Population based study. Ann. Hepatol. 2007, 6, 161–163. [Google Scholar]
- Fan, J.-G.; Farrell, G.C. Epidemiology of non-alcoholic fatty liver disease in China. J. Hepatol. 2009, 50, 204–210. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, K.M.; Lee, S.G.; Yu, E.; Lim, Y.-S.; Lee, H.C.; Chung, Y.-H.; Lee, Y.S.; Suh, D.-J. Prevalence and risk factors of non-alcoholic fatty liver disease in potential living liver donors in Korea: A review of 589 consecutive liver biopsies in a single center. J. Hepatol. 2007, 47, 239–244. [Google Scholar] [CrossRef]
- Kallwitz, E.R.; Kumar, M.; Aggarwal, R.; Berger, R.; Layden-Almer, J.; Gupta, N.; Cotler, S.J. Ethnicity and nonalcoholic fatty liver disease in an obesity clinic: The impact of triglycerides. Dig. Dis. Sci. 2008, 53, 1358–1363. [Google Scholar] [CrossRef]
- Wagenknecht, L.E.; Scherzinger, A.L.; Stamm, E.R.; Hanley, A.J.G.; Norris, J.M.; Chen, Y.-D.I.; Bryer-Ash, M.; Haffner, S.M.; Rotter, J.I. Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity 2009, 17, 1240–1246. [Google Scholar]
- Fischer, G.E.; Bialek, S.P.; Homan, C.E.; Livingston, S.E.; McMahon, B.J. Chronic liver disease among Alaska-Native people, 2003–2004. Am. J. Gastroenterol. 2009, 104, 363–370. [Google Scholar] [CrossRef]
- Bialek, S.R.; Redd, J.T.; Lynch, A.; Vogt, T.; Lewis, S.; Wilson, C.; Bell, B.P. Chronic liver disease among two American Indian patient populations in the southwestern United States, 2000–2003. J. Clin. Gastroenterol. 2008, 42, 949–954. [Google Scholar]
- Devadason, C.A.; Scheimann, A.O. Overview of screening methods for fatty liver disease in children. World J. Hepatol. 2012, 4, 1–4. [Google Scholar] [CrossRef]
- Hudson, O.D.; Nunez, M.; Shaibi, G.Q. Ethnicity and elevated liver transaminases among newly diagnosed children with type 2 diabetes. BMC Pediatr. 2012, 12, 174. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; McGreal, N.; Deutsch, R.; Finegold, M.J.; Lavine, J.E. Influence of gender, race, and ethnicity on suspected fatty liver in obese adolescents. Pediatrics 2005, 115, e561–e565. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, L.; Dai, H.; Chen, J.; Fang, L. Relationship between alanine aminotransferase levels and metabolic syndrome in nonalcoholic fatty liver disease. J. Zhejiang Univ. Sci. B 2008, 9, 616–622. [Google Scholar] [CrossRef]
- Sorrentino, P.; Tarantino, G.; Conca, P.; Perrella, A.; Terracciano, M.L.; Vecchione, R.; Gargiulo, G.; Gennarelli, N.; Lobello, R. Silent non-alcoholic fatty liver disease-a clinical-histological study. J. Hepatol. 2004, 41, 751–757. [Google Scholar] [CrossRef]
- Papatheodoridis, G.V.; Goulis, J.; Christodoulou, D.; Manolakopoulos, S.; Raptopoulou, M.; Andrioti, E.; Alexandropoulos, N.; Savvidou, S.; Papachristou, A.; Zervou, E.; et al. High prevalence of elevated liver enzymes in blood donors: Associations with male gender and central adiposity. Eur. J. Gastroenterol. Hepatol. 2007, 19, 281–287. [Google Scholar] [CrossRef]
- Carr, M.C.; Ayyobi, A.F.; Murdoch, S.J.; Deeb, S.S.; Brunzell, J.D. Contribution of hepatic lipase, lipoprotein lipase, and cholesteryl ester transfer protein to LDL and HDL heterogeneity in healthy women. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 667–673. [Google Scholar] [CrossRef]
- Rivera, C.A. Risk factors and mechanisms of non-alcoholic steatohepatitis. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2008, 15, 109–114. [Google Scholar]
- Elbers, J.M.; Asscheman, H.; Seidell, J.C.; Gooren, L.J. Effects of sex steroid hormones on regional fat depots as assessed by magnetic resonance imaging in transsexuals. Am. J. Physiol. 1999, 276, E317–E325. [Google Scholar]
- Haarbo, J.; Marslew, U.; Gotfredsen, A.; Christiansen, C. Postmenopausal hormone replacement therapy prevents central distribution of body fat after menopause. Metabolism. 1991, 40, 1323–1326. [Google Scholar]
- Toth, M.J.; Tchernof, A.; Sites, C.K.; Poehlman, E.T. Effect of menopausal status on body composition and abdominal fat distribution. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2000, 24, 226–231. [Google Scholar]
- Machado, M.; Marques-Vidal, P.; Cortez-Pinto, H. Hepatic histology in obese patients undergoing bariatric surgery. J. Hepatol. 2006, 45, 600–606. [Google Scholar] [CrossRef]
- Colicchio, P.; Tarantino, G.; del Genio, F.; Sorrentino, P.; Saldalamacchia, G.; Finelli, C.; Conca, P.; Contaldo, F.; Pasanisi, F. Non-alcoholic fatty liver disease in young adult severely obese non-diabetic patients in South Italy. Ann. Nutr. Metab. 2005, 49, 289–295. [Google Scholar] [CrossRef]
- Beymer, C.; Kowdley, K.V.; Larson, A.; Edmonson, P.; Dellinger, E.P.; Flum, D.R. Prevalence and predictors of asymptomatic liver disease in patients undergoing gastric bypass surgery. Arch. Surg. Chic. Ill 1960 2003, 138, 1240–1244. [Google Scholar]
- Boza, C.; Riquelme, A.; Ibañez, L.; Duarte, I.; Norero, E.; Viviani, P.; Soza, A.; Fernandez, J.I.; Raddatz, A.; Guzman, S.; et al. Predictors of nonalcoholic steatohepatitis (NASH) in obese patients undergoing gastric bypass. Obes. Surg. 2005, 15, 1148–1153. [Google Scholar] [CrossRef]
- Abrams, G.A.; Kunde, S.S.; Lazenby, A.J.; Clements, R.H. Portal fibrosis and hepatic steatosis in morbidly obese subjects: A spectrum of nonalcoholic fatty liver disease. Hepatology 2004, 40, 475–483. [Google Scholar]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Stranges, S.; Dorn, J.M.; Muti, P.; Freudenheim, J.L.; Farinaro, E.; Russell, M.; Nochajski, T.H.; Trevisan, M. Body fat distribution, relative weight, and liver enzyme levels: A population-based study. Hepatology 2004, 39, 754–763. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; de Michieli, F.; Rosina, F.; Orlandi, F.; Gambino, R. Nonalcoholic steatohepatitis versus steatosis: Adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology 2012, 56, 933–942. [Google Scholar] [CrossRef]
- Zhan, Y.-T.; An, W. Roles of liver innate immune cells in nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 4652–4660. [Google Scholar] [CrossRef]
- Copaci, I.; Micu, L.; Voiculescu, M. The role of cytokines in non-alcoholic steatohepatitis. A review. J. Gastrointest. Liver Dis. 2006, 15, 363–373. [Google Scholar]
- Syn, W.-K.; Jung, Y.; Omenetti, A.; Abdelmalek, M.; Guy, C.D.; Yang, L.; Wang, J.; Witek, R.P.; Fearing, C.M.; Pereira, T.A.; et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 2009, 137, 1478–1488. [Google Scholar] [CrossRef]
- Bohinc, B.N.; Diehl, A.M. Mechanisms of disease progression in NASH: New paradigms. Clin. Liver Dis. 2012, 16, 549–565. [Google Scholar] [CrossRef]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M. NASH CRN Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef]
- Cheung, O.; Sanyal, A.J. Role of microRNAs in non-alcoholic steatohepatitis. Curr. Pharm. Des. 2010, 16, 1952–1957. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs.”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Nakanishi, N.; Nakagawa, Y.; Tokushige, N.; Aoki, N.; Matsuzaka, T.; Ishii, K.; Yahagi, N.; Kobayashi, K.; Yatoh, S.; Takahashi, A.; et al. The up-regulation of microRNA-335 is associated with lipid metabolism in liver and white adipose tissue of genetically obese mice. Biochem. Biophys. Res. Commun. 2009, 385, 492–496. [Google Scholar] [CrossRef]
- Li, W.-Q.; Chen, C.; Xu, M.-D.; Guo, J.; Li, Y.-M.; Xia, Q.-M.; Liu, H.-M.; He, J.; Yu, H.-Y.; Zhu, L. The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats. FEBS J. 2011, 278, 1522–1532. [Google Scholar]
- Castro, R.E.; Ferreira, D.M.S.; Afonso, M.B.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.P. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 119–125. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Sumida, Y.; Umemura, A.; Matsuo, K.; Takahashi, M.; Takamura, T.; Yasui, K.; Saibara, T.; Hashimoto, E.; Kawanaka, M.; et al. Japan Study Group of Nonalcoholic Fatty Liver Disease. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS One 2012, 7, e38322. [Google Scholar]
- Rotman, Y.; Koh, C.; Zmuda, J.M.; Kleiner, D.E.; Liang, T.J. NASH CRN The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010, 52, 894–903. [Google Scholar] [CrossRef]
- Wang, C.-W.; Lin, H.-Y.; Shin, S.-J.; Yu, M.-L.; Lin, Z.-Y.; Dai, C.-Y.; Huang, J.-F.; Chen, S.-C.; Li, S.S.-L.; Chuang, W.-L. The PNPLA3 I148M polymorphism is associated with insulin resistance and nonalcoholic fatty liver disease in a normoglycaemic population. Liver Int. Off. J. Int. Assoc. Study Liver 2011, 31, 1326–1331. [Google Scholar]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.S.; Dongiovanni, P.; Patch, J.; Fracanzani, A.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef]
- Kotronen, A.; Johansson, L.E.; Johansson, L.M.; Roos, C.; Westerbacka, J.; Hamsten, A.; Bergholm, R.; Arkkila, P.; Arola, J.; Kiviluoto, T.; et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia 2009, 52, 1056–1060. [Google Scholar] [CrossRef]
- Johansson, L.E.; Hoffstedt, J.; Parikh, H.; Carlsson, E.; Wabitsch, M.; Bondeson, A.-G.; Hedenbro, J.; Tornqvist, H.; Groop, L.; Ridderstråle, M. Variation in the adiponutrin gene influences its expression and associates with obesity. Diabetes 2006, 55, 826–833. [Google Scholar] [CrossRef]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Mofrad, P.; Contos, M.J.; Haque, M.; Sargeant, C.; Fisher, R.A.; Luketic, V.A.; Sterling, R.K.; Shiffman, M.L.; Stravitz, R.T.; Sanyal, A.J. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003, 37, 1286–1292. [Google Scholar] [CrossRef]
- Joseph, A.E.; Dewbury, K.C.; McGuire, P.G. Ultrasound in the detection of chronic liver disease (the “bright liver”). Br. J. Radiol. 1979, 52, 184–188. [Google Scholar]
- Bydder, M.; Yokoo, T.; Hamilton, G.; Middleton, M.S.; Chavez, A.D.; Schwimmer, J.B.; Lavine, J.E.; Sirlin, C.B. Relaxation effects in the quantification of fat using gradient echo imaging. Magn. Reson. Imaging 2008, 26, 347–359. [Google Scholar]
- Hussain, H.K.; Chenevert, T.L.; Londy, F.J.; Gulani, V.; Swanson, S.D.; McKenna, B.J.; Appelman, H.D.; Adusumilli, S.; Greenson, J.K.; Conjeevaram, H.S. Hepatic fat fraction: MR imaging for quantitative measurement and display–Early experience. Radiology 2005, 237, 1048–1055. [Google Scholar] [CrossRef]
- Reeder, S.B.; Ranallo, F.; Taylor, A.J. CT and MRI for determining hepatic fat content. Am. J. Roentgenol. 2008, 190, W167, author reply W168. [Google Scholar] [CrossRef]
- Fitzpatrick, E.; Quaglia, A.; Vimalesvaran, S.; Basso, M.S.; Dhawan, A. Transient elastography is a useful noninvasive tool for the evaluation of fibrosis in paediatric chronic liver disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 72–76. [Google Scholar]
- Yin, M.; Talwalkar, J.A.; Glaser, K.J.; Manduca, A.; Grimm, R.C.; Rossman, P.J.; Fidler, J.L.; Ehman, R.L. Assessment of hepatic fibrosis with magnetic resonance elastography. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2007, 5, 1207–1213. [Google Scholar]
- Ratziu, V.; Massard, J.; Charlotte, F.; Messous, D.; Imbert-Bismut, F.; Bonyhay, L.; Tahiri, M.; Munteanu, M.; Thabut, D.; Cadranel, J.F.; et al. LIDO Study Group and CYTOL Study Group. Diagnostic value of biochemical markers (FibroTest-FibroSURE) for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2006, 6, 6. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Sozio, M.; Liangpunsakul, S.; Crabb, D. The role of lipid metabolism in the pathogenesis of alcoholic and nonalcoholic hepatic steatosis. Semin. Liver Dis. 2010, 30, 378–390. [Google Scholar] [CrossRef]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Lafontan, M. Fat cells: Afferent and efferent messages define new approaches to treat obesity. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 119–146. [Google Scholar] [CrossRef]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef]
- Diehl, A.M.; Li, Z.P.; Lin, H.Z.; Yang, S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 2005, 54, 303–306. [Google Scholar] [CrossRef]
- Van der Poorten, D.; Milner, K.-L.; Hui, J.; Hodge, A.; Trenell, M.I.; Kench, J.G.; London, R.; Peduto, T.; Chisholm, D.J.; George, J. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457. [Google Scholar] [CrossRef]
- Beasley, L.E.; Koster, A.; Newman, A.B.; Javaid, M.K.; Ferrucci, L.; Kritchevsky, S.B.; Kuller, L.H.; Pahor, M.; Schaap, L.A.; Visser, M.; et al. Health ABC study inflammation and race and gender differences in computerized tomography-measured adipose depots. Obesity 2009, 17, 1062–1069. [Google Scholar]
- Gabriely, I.; Ma, X.H.; Yang, X.M.; Atzmon, G.; Rajala, M.W.; Berg, A.H.; Scherer, P.; Rossetti, L.; Barzilai, N. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging an adipokine-mediated process? Diabetes 2002, 51, 2951–2958. [Google Scholar] [CrossRef]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Scalia, R. Adiponectin: A novel adipokine linking adipocytes and vascular function. J. Clin. Endocrinol. Metab. 2004, 89, 2563–2568. [Google Scholar] [CrossRef]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar]
- Louthan, M.V.; Barve, S.; McClain, C.J.; Joshi-Barve, S. Decreased serum adiponectin: An early event in pediatric nonalcoholic fatty liver disease. J. Pediatr. 2005, 147, 835–838. [Google Scholar]
- Crespo, J.; Fernández-Gil, P.; Hernández-Guerra, M.; Cayón, A.; Mayorga, M.; Domínguez-Diez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Are there predictive factors of severe liver fibrosis in morbidly obese patients with non-alcoholic steatohepatitis? Obes. Surg. 2001, 11, 254–257. [Google Scholar] [CrossRef]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar]
- Diehl, A.M. Tumor necrosis factor and its potential role in insulin resistance and nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 619–638. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar]
- Palmer, M.; Schaffner, F. Effect of weight reduction on hepatic abnormalities in overweight patients. Gastroenterology 1990, 99, 1408–1413. [Google Scholar]
- Park, H.S.; Kim, M.W.; Shin, E.S. Effect of weight control on hepatic abnormalities in obese patients with fatty liver. J. Korean Med. Sci. 1995, 10, 414–421. [Google Scholar]
- Ueno, T.; Sugawara, H.; Sujaku, K.; Hashimoto, O.; Tsuji, R.; Tamaki, S.; Torimura, T.; Inuzuka, S.; Sata, M.; Tanikawa, K. Therapeutic effects of restricted diet and exercise in obese patients with fatty liver. J. Hepatol. 1997, 27, 103–107. [Google Scholar]
- Harrison, S.A.; Fecht, W.; Brunt, E.M.; Neuschwander-Tetri, B.A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009, 49, 80–86. [Google Scholar] [CrossRef]
- Andersen, T.; Gluud, C.; Franzmann, M.B.; Christoffersen, P. Hepatic effects of dietary weight loss in morbidly obese subjects. J. Hepatol. 1991, 12, 224–229. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar]
- Narasimhan, S.; Gokulakrishnan, K.; Sampathkumar, R.; Farooq, S.; Ravikumar, R.; Mohan, V.; Balasubramanyam, M. Oxidative stress is independently associated with non-alcoholic fatty liver disease (NAFLD) in subjects with and without type 2 diabetes. Clin. Biochem. 2010, 43, 815–821. [Google Scholar] [CrossRef]
- Rezazadeh, A.; Yazdanparast, R.; Molaei, M. Amelioration of diet-induced nonalcoholic steatohepatitis in rats by Mn-salen complexes via reduction of oxidative stress. J. Biomed. Sci. 2012, 19, 26. [Google Scholar] [CrossRef]
- Lavine, J.E. Vitamin E treatment of nonalcoholic steatohepatitis in children: A pilot study. J. Pediatr. 2000, 136, 734–738. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. NASH CRN Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar]
- Tiikkainen, M.; Häkkinen, A.-M.; Korsheninnikova, E.; Nyman, T.; Mäkimattila, S.; Yki-Järvinen, H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes 2004, 53, 2169–2176. [Google Scholar] [CrossRef]
- Lehtovirta, M.; Forsén, B.; Gullström, M.; Häggblom, M.; Eriksson, J.G.; Taskinen, M.R.; Groop, L. Metabolic effects of metformin in patients with impaired glucose tolerance. Diabet. Med. J. Br. Diabet. Assoc. 2001, 18, 578–583. [Google Scholar] [CrossRef]
- Shields, W.W.; Thompson, K.E.; Grice, G.A.; Harrison, S.A.; Coyle, W.J. The effect of Metformin and standard therapy versus standard therapy alone in nondiabetic patients with insulin resistance and nonalcoholic steatohepatitis (NASH): A pilot trial. Ther. Adv. Gastroenterol. 2009, 2, 157–163. [Google Scholar] [CrossRef]
- Omer, Z.; Cetinkalp, S.; Akyildiz, M.; Yilmaz, F.; Batur, Y.; Yilmaz, C.; Akarca, U. Efficacy of insulin-sensitizing agents in nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2010, 22, 18–23. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Brunt, E.M.; Wehmeier, K.R.; Oliver, D.; Bacon, B.R. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-γ ligand rosiglitazone. Hepatology 2003, 38, 1008–1017. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef]
- Masterton, G.S.; Plevris, J.N.; Hayes, P.C. Review article: Omega-3 fatty acids—A promising novel therapy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 31, 679–692. [Google Scholar] [CrossRef]
- Li, W.; Zheng, L.; Sheng, C.; Cheng, X.; Qing, L.; Qu, S. Systematic review on the treatment of pentoxifylline in patients with non-alcoholic fatty liver disease. Lipids Health Dis. 2011, 10, 49. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Yuan, M. Inflammation and the IKKβ/IκB/NF-κB axis in obesity- and diet-induced insulin resistance. Int. J. Obes. 2003, 27, S49–S52. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.-W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar]
- Wueest, S.; Rapold, R.A.; Schumann, D.M.; Rytka, J.M.; Schildknecht, A.; Nov, O.; Chervonsky, A.V.; Rudich, A.; Schoenle, E.J.; Donath, M.Y.; et al. Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J. Clin. Investig. 2010, 120, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis—New insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Syn, W.-K.; Teaberry, V.; Choi, S.S.; Diehl, A.M. Similarities and differences in the pathogenesis of alcoholic and nonalcoholic steatohepatitis. Semin. Liver Dis. 2009, 29, 200–210. [Google Scholar] [CrossRef]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Nakamuta, M.; Kohjima, M.; Morizono, S.; Kotoh, K.; Yoshimoto, T.; Miyagi, I.; Enjoji, M. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2005, 16, 631–635. [Google Scholar]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar]
- Higuchi, N.; Kato, M.; Shundo, Y.; Tajiri, H.; Tanaka, M.; Yamashita, N.; Kohjima, M.; Kotoh, K.; Nakamuta, M.; Takayanagi, R.; et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2008, 38, 1122–1129. [Google Scholar]
- Caballero, F.; Fernández, A.; de Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef]
- Towle, H.C.; Kaytor, E.N.; Shih, H.-M. Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu. Rev. Nutr. 1997, 17, 405–433. [Google Scholar] [CrossRef]
- Iizuka, K.; Horikawa, Y. ChREBP: A glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr. J. 2008, 55, 617–624. [Google Scholar] [CrossRef]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar]
- Iizuka, K.; Miller, B.; Uyeda, K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E358–E364. [Google Scholar] [CrossRef]
- Gyamfi, M.A.; Wan, Y.-J.Y. Pathogenesis of alcoholic liver disease: The role of nuclear receptors. Exp. Biol. Med. 2010, 235, 547–560. [Google Scholar] [CrossRef]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by Liver X receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, L.; Hillgartner, F.B. SREBP-1 integrates the actions of thyroid hormone, insulin, cAMP, and medium-chain fatty acids on ACCα transcription in hepatocytes. J. Lipid Res. 2003, 44, 356–368. [Google Scholar] [CrossRef]
- Cha, J.-Y.; Repa, J.J. The Liver X Receptor (LXR) and hepatic lipogenesis. The Carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef]
- Mitro, N.; Mak, P.A.; Vargas, L.; Godio, C.; Hampton, E.; Molteni, V.; Kreusch, A.; Saez, E. The nuclear receptor LXR is a glucose sensor. Nature 2007, 445, 219–223. [Google Scholar] [CrossRef]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Sozio, M.; Crabb, D.W. Alcohol and lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E10–E16. [Google Scholar] [CrossRef]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar]
- García-Villafranca, J.; Guillén, A.; Castro, J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 2008, 90, 460–466. [Google Scholar] [CrossRef]
- Song, Z.; Deaciuc, I.; Zhou, Z.; Song, M.; Chen, T.; Hill, D.; McClain, C.J. Involvement of AMP-activated protein kinase in beneficial effects of betaine on high-sucrose diet-induced hepatic steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G894–G902. [Google Scholar] [CrossRef]
- Yang, J.; Maika, S.; Craddock, L.; King, J.A.; Liu, Z.-M. Chronic activation of AMP-activated protein kinase-α1 in liver leads to decreased adiposity in mice. Biochem. Biophys. Res. Commun. 2008, 370, 248–253. [Google Scholar] [CrossRef]
- Shang, J.; Chen, L.; Xiao, F.; Sun, H.; Ding, H.; Xiao, H. Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase. Acta Pharmacol. Sin. 2008, 29, 698–706. [Google Scholar] [CrossRef]
- Wada, S.; Yamazaki, T.; Kawano, Y.; Miura, S.; Ezaki, O. Fish oil fed prior to ethanol administration prevents acute ethanol-induced fatty liver in mice. J. Hepatol. 2008, 49, 441–450. [Google Scholar] [CrossRef]
- Kallwitz, E.R.; McLachlan, A.; Cotler, S.J. Role of peroxisome proliferators-activated receptors in the pathogenesis and treatment of nonalcoholic fatty liver disease. World J. Gastroenterol. 2008, 14, 22–28. [Google Scholar] [CrossRef]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef]
- Savage, D.B.; Tan, G.D.; Acerini, C.L.; Jebb, S.A.; Agostini, M.; Gurnell, M.; Williams, R.L.; Umpleby, A.M.; Thomas, E.L.; Bell, J.D.; et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-γ. Diabetes 2003, 52, 910–917. [Google Scholar]
- Molnar, A.; Haybaeck, J.; Lackner, C.; Strnad, P. The cytoskeleton in nonalcoholic steatohepatitis: 100 years old but still youthful. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 167–177. [Google Scholar]
- Haybaeck, J.; Stumptner, C.; Thueringer, A.; Kolbe, T.; Magin, T.M.; Hesse, M.; Fickert, P.; Tsybrovskyy, O.; Müller, H.; Trauner, M.; et al. Genetic background effects of keratin 8 and 18 in a DDC-induced hepatotoxicity and Mallory-Denk body formation mouse model. Lab. Investig. J. Tech. Methods Pathol. 2012, 92, 857–867. [Google Scholar] [CrossRef]
- Denk, H.; Stumptner, C.; Fuchsbichler, A.; Zatloukal, K. Alkoholische und nichtalkoholische Steatohepatitis. Pathology 2001, 22, 388–398. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Marí, M.; Colell, A.; Morales, A.; Fernandez-Checa, J.C. Metabolic therapy: Lessons from liver diseases. Curr. Pharm. Des. 2011, 17, 3933–3944. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bettermann, K.; Hohensee, T.; Haybaeck, J. Steatosis and Steatohepatitis: Complex Disorders. Int. J. Mol. Sci. 2014, 15, 9924-9944. https://doi.org/10.3390/ijms15069924
Bettermann K, Hohensee T, Haybaeck J. Steatosis and Steatohepatitis: Complex Disorders. International Journal of Molecular Sciences. 2014; 15(6):9924-9944. https://doi.org/10.3390/ijms15069924
Chicago/Turabian StyleBettermann, Kira, Tabea Hohensee, and Johannes Haybaeck. 2014. "Steatosis and Steatohepatitis: Complex Disorders" International Journal of Molecular Sciences 15, no. 6: 9924-9944. https://doi.org/10.3390/ijms15069924