Molecular Mechanisms of Liver Fibrosis in HIV/HCV Coinfection

 ,
, 
Abstract
:
1. Introduction
2. Pathophysiology of Liver Fibrosis in Chronic Hepatitis C Virus (HCV) Infection
2.1. Background Issues
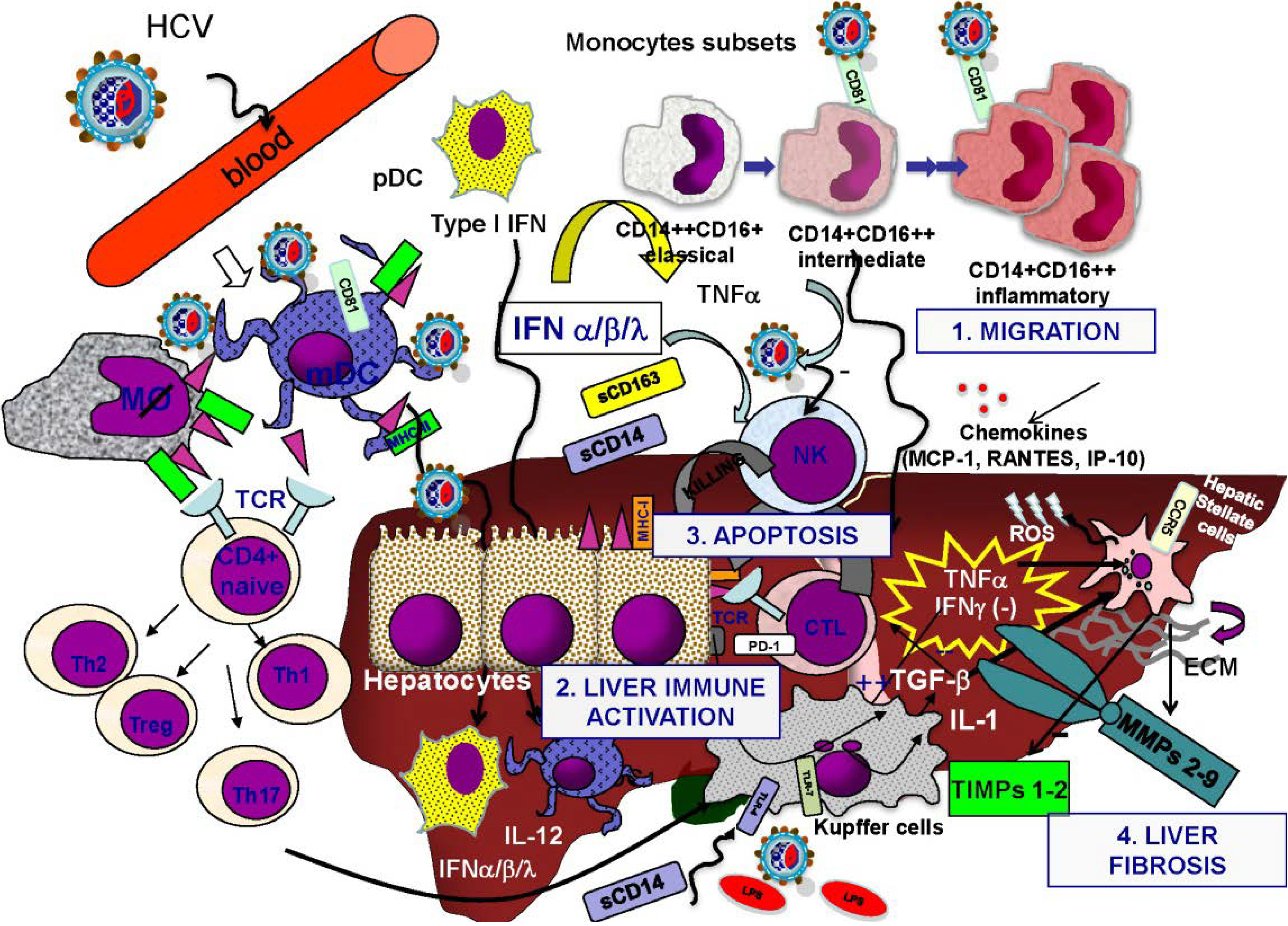
2.2. Interplay between HCV and Liver Inflammation and Fibrosis

3. Accelerated Liver Fibrosis in Human Immunodeficiency Virus (HIV)/HCV Coinfection
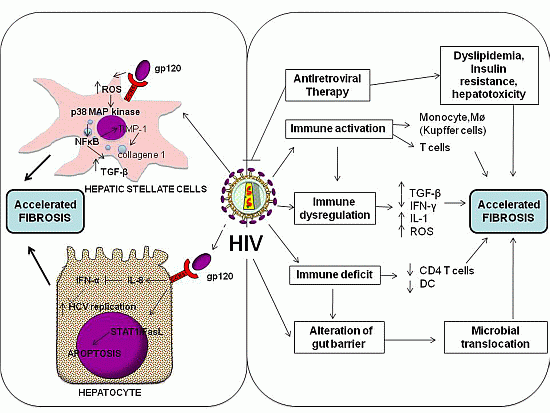
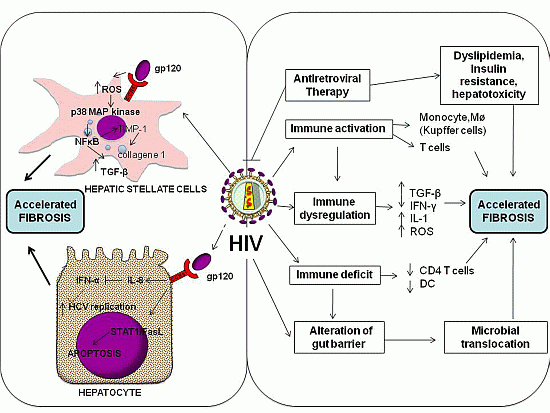
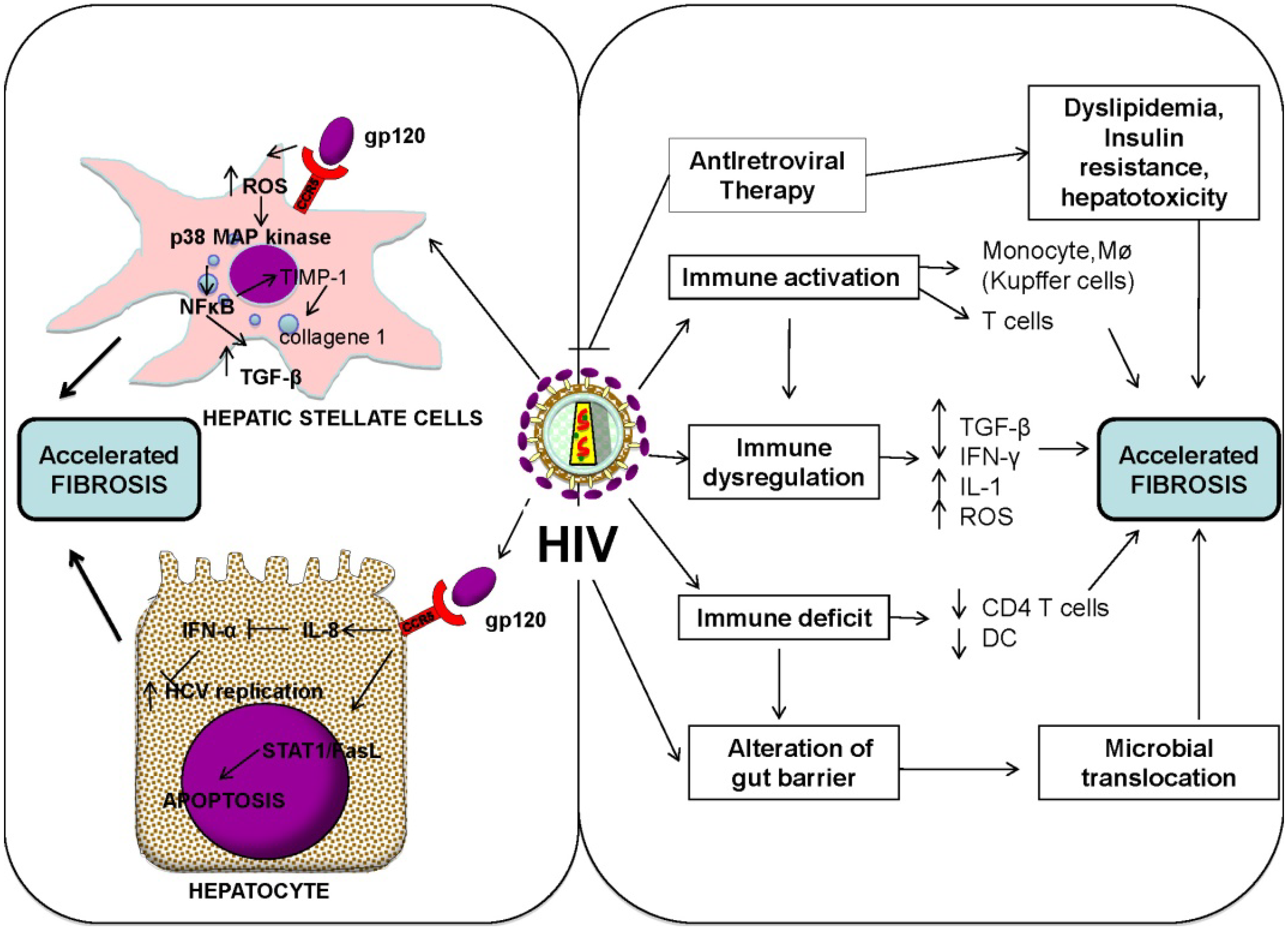
3.1. Direct Effects of HIV

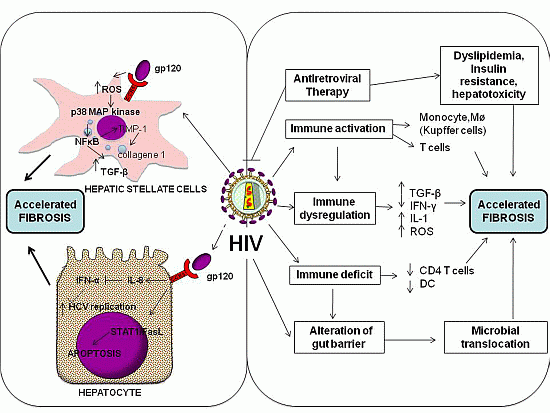{kind=link}
{kind=link}
{kind=link}
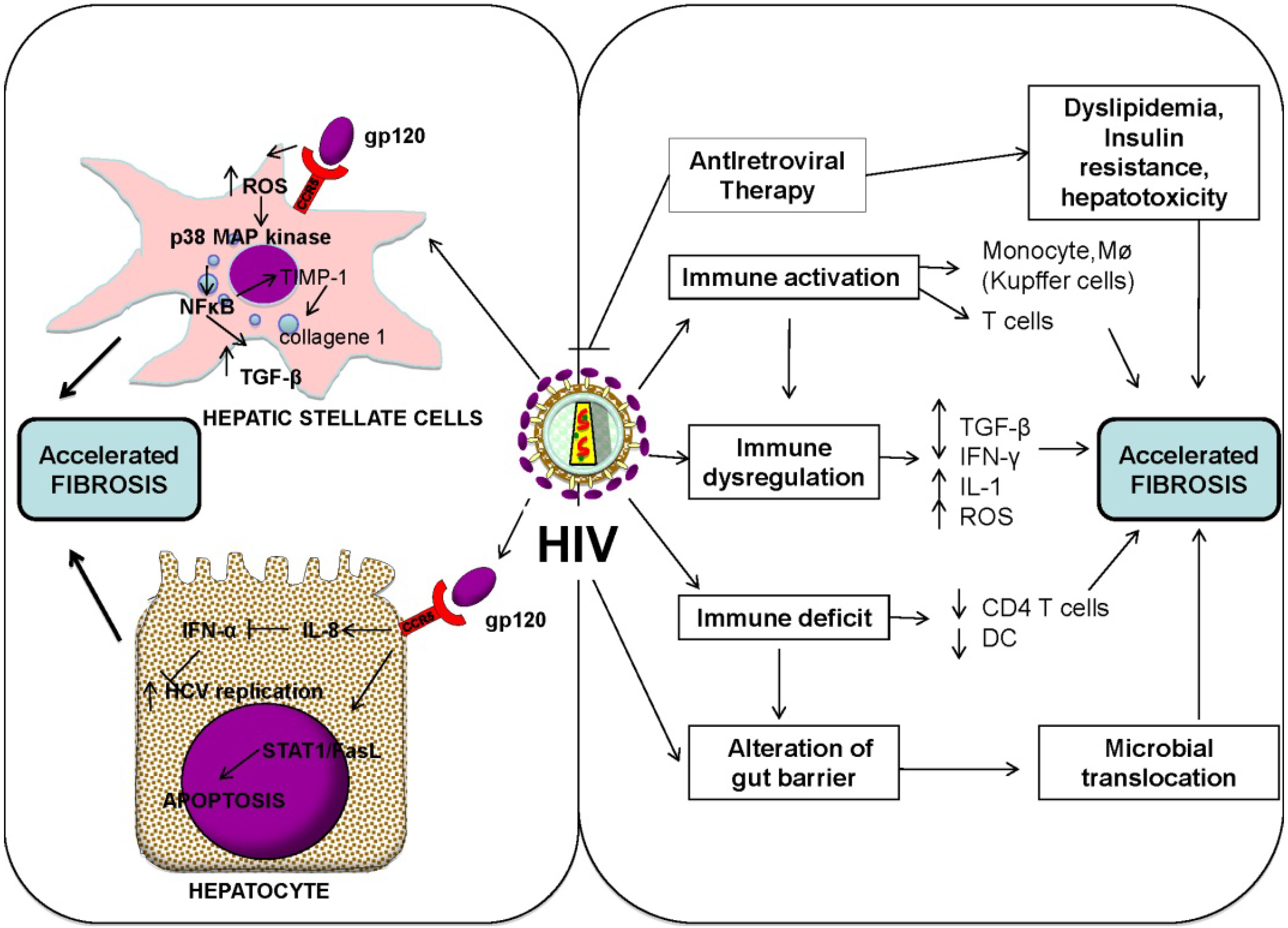{kind=link}
{kind=link}
| Markers | HIV | HCV | HCV/HIV |
|---|---|---|---|
| Immune cells | |||
| NK | ↓ | ↓ | ↓↓ |
| DC | ↓↓ | ↓ | ↓↓↓ |
| CD4 T cell | ↓↓ | ↓ | ↓↓↓ |
| Immune activation | |||
| CD4 T cell DR/38+ | ++ | + | +++ |
| CD8 T cell DR/38+ | +++ | +++ | +++ |
| Macrophage (Kupffer cells) | + | ++ | +++ |
| Cytokines and chemokines | |||
| IP-10 | ++ | ++ | ++++ |
| IL-1β | + | + | ++ |
| IFN-γ | ↓ | ↓ | ↓ |
| TGF-β | + | ++ | +++ |
| TNF-α | ++ | + | +++ |
| MIP-1α | ↓↓↓ | ↓↓ | ↓↓ |
| MIP-1β | ↓↓↓ | ↓ | ↓↓ |
| RANTES | +++ | + | ++ |
| Microbial translocation | |||
| sCD14 | ++ | + | +++ |
| LPS | ++ | + | +++ |
| Fibrosis mediators | |||
| MMP | ++ | ++ | +++ |
| TIMPs | ++ | ++ | +++ |
| HA | + | ++ | +++ |
| Apoptosis and ROS | |||
| TRAIL/FAS | ++ | ++ | ++++ |
| ROS | ++ | ++ | +++ |
| Metabolic parameters | |||
| Insulin resistance | ++ | + | +++ |
| Adiponectin | ↓ | ↓↓ | ↓↓↓ |
| Resistin | + | ++ | +++ |
| Leptin | + | ++ | +++ |
3.2. HIV-Associated Dysregulation of the Immune Response and Cytokine Network
3.3. Role of Immune Activation and Microbial Translocation
4. Dysregulation of Matrix Metalloproteinases and Role of Fibrosis Biomarkers
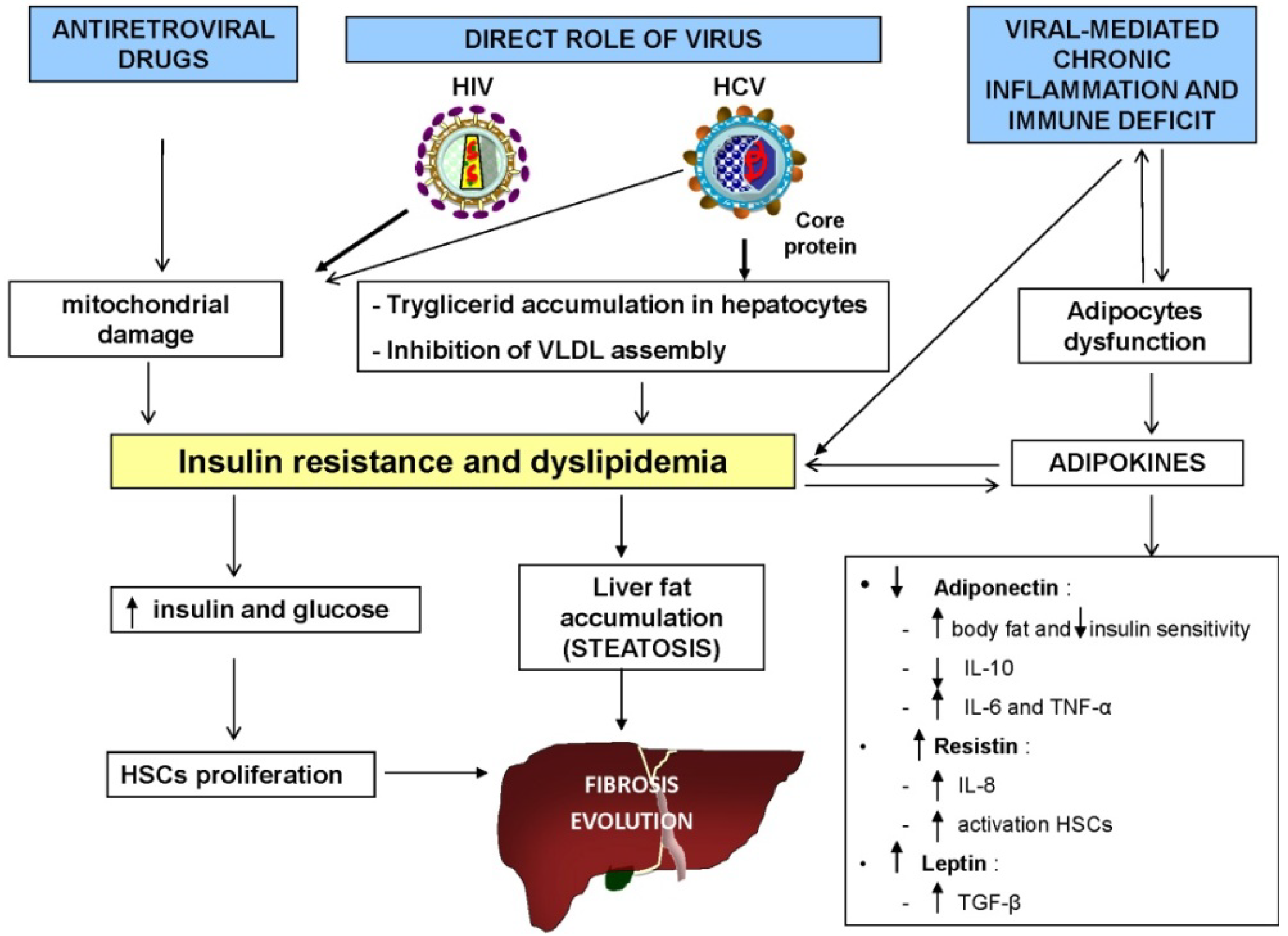
4.1. Role of Metabolic Alterations
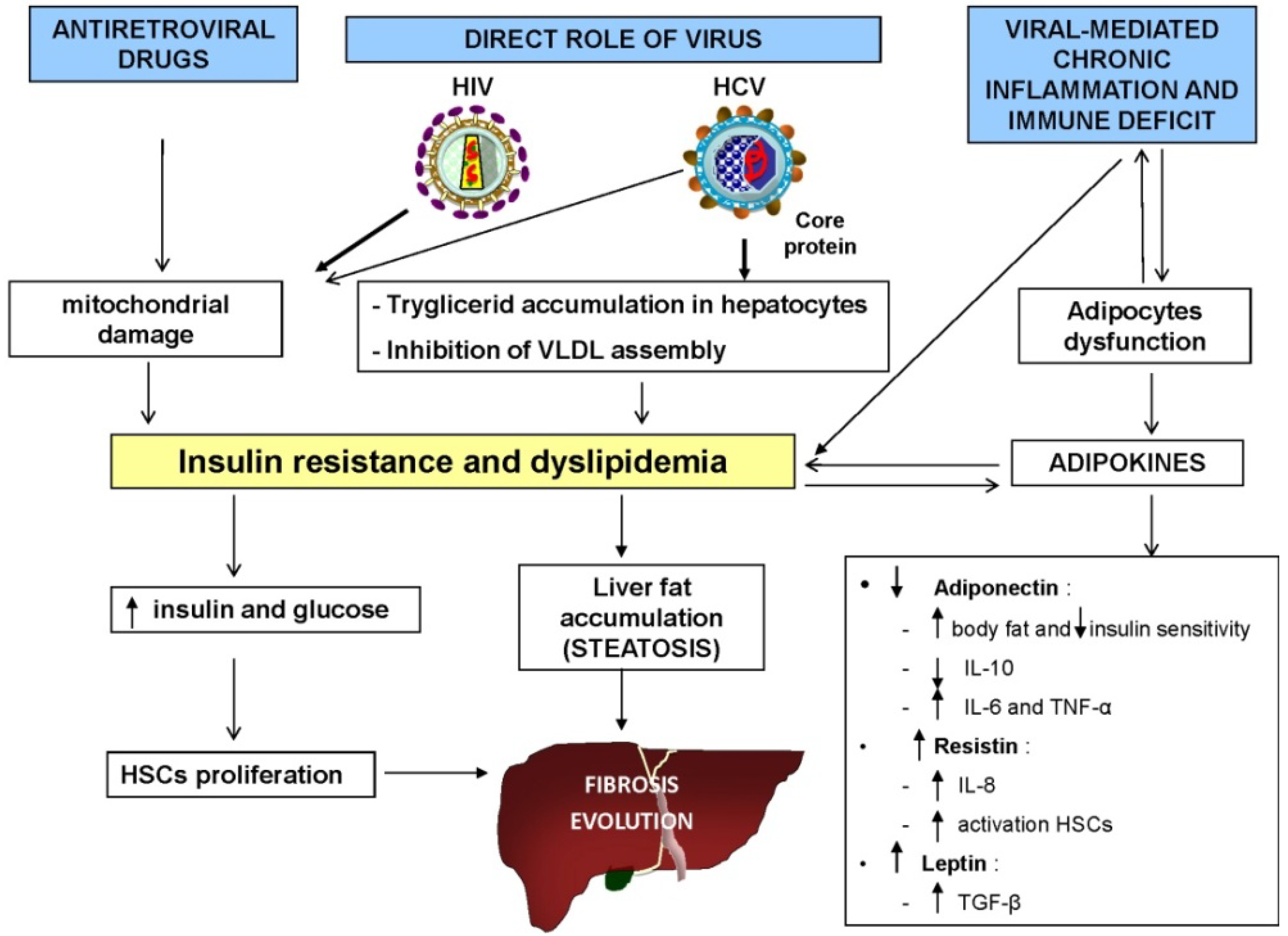
4.2. Role of Alcohol and Drug Use
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, A.Y.; Chung, R.T. Coinfection with HIV-1 and HCV—A one-two punch. Gastroenterology 2009, 137, 795–781. [Google Scholar]
- Chen, J.Y.; Feeney, E.R.; Chung, R.T. HCV and HIV co-infection: Mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2014. [Google Scholar] [CrossRef]
- Hernandez, M.D.; Sherman, K.E. HIV/HCV coinfection natural history and disease progression. Curr. Opin. HIV AIDS 2011, 6, 478–482. [Google Scholar] [CrossRef]
- Benhamou, Y.; Bochet, M.; di Martino, V.; Charlotte, F.; Azria, F.; Coutellier, A.; Vidaud, M.; Bricaire, F.; Opolon, P.; Katlama, C.; et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. Hepatology 1999, 30, 1054–1058. [Google Scholar] [CrossRef]
- Kovacs, A.; Al-Harthi, L.; Christensen, S.; Mack, W.; Cohen, M.; Landay, A. CD8+ T cell activation in women coinfected with human immunodeficiency virus type 1 and hepatitis C virus. J. Infect. Dis. 2008, 197, 1402–1407. [Google Scholar]
- Justice, A.C.; Lasky, E.; McGinnis, K.A.; Skanderson, M.; Conigliano, J.; Fultz, S.L.; Crothers, K.; Rabeneck, L.; Rodriguez-Barradas, M.; Weissman, S.B. Medical disease and alcohol use among veterans with human immunodeficiency infection: A comparison of disease measurement strategies. Med. Care 2006, 44, 52–60. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of accelerated fibrosis in HIV/HCV co-infection. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Schuppan, D.; Ruehl, M.; Somasundaran, R.; Hahn, E.G. Matrix as modulator of stellate cell and hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef]
- Bataller, R.; Gines, P. New therapeutic strategies in liver fibrosis: Pathogenic basis. Med. Clin. 2002, 118, 339–346. [Google Scholar] [CrossRef]
- Hui, A.; Friedman, S.L. Molecular basis of hepatic fibrosis. Expert Rev. Mol. Med. 2003, 5, 1–23. [Google Scholar]
- Zhao, Q.; Qin, C.Y.; Zhao, Z.H.; Fan, Y.C.; Wang, K. Epigenetic modifications in hepatic stellate cells contribute to liver fibrosis. Tohoku J. Exp. Med. 2013, 229, 35–43. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Tsukamoto, H. Epigenetic mechanism of stellate cell trans-differentiation. J. Hepatol. 2007, 46, 352–353. [Google Scholar] [CrossRef]
- Rossmanith, W.; Schulte-Hermann, R. Biology of transforming growth factor beta in hepatocarcinogenesis. Microsc. Res. Tech. 2001, 52, 430–436. [Google Scholar] [CrossRef]
- Matsuzaki, K. Modulation of TGF-β signaling during progression of chronic liver diseases. Front. Biosci. 2009, 14, 2923–2934. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef]
- Zavadil, J.; Bottinger, E.P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef]
- Proell, V.; Carmona-Cuenca, I.; Murillo, M.M.; Huber, H.; Fabregat, I.; Mikulits, W. TGF-β dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp. Hepatol. 2007, 6, 1. [Google Scholar] [CrossRef]
- Seki, E.; de Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Moreno, M.; Bataller, R. Cytokines and renin-angiotensin system signaling in hepatic fibrosis. Clin. Liver Dis. 2008, 12, 825–852. [Google Scholar] [CrossRef]
- Pinzani, M.; Gesualdo, L.; Sabbah, G.M.; Abboud, H.E. Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J. Clin. Investig. 1989, 84, 1786–1793. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; van Roeyen, C.R.; Ostendorf, T.; Floege, J.; Gressner, A.M.; Weiskirchen, R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J. Hepatol. 2007, 46, 1064–1074. [Google Scholar] [CrossRef]
- Pinzani, M.; Milani, S.; Herbst, H.; de Franco, R.; Grappone, C.; Gentilini, A.; Caligiuri, A.; Pellegrini, G.; Ngo, D.V.; Romanelli, R.G.; et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 1996, 148, 785–800. [Google Scholar]
- Wong, L.; Yamasaki, G.; Johnson, R.J.; Friedman, S.L. Induction of β-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Investig. 1994, 94, 1563–1569. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Zhu, N.L.; Asahina, K.; Mann, D.A.; Mann, J. Epigenetic cell fate regulation of hepatic stellate cells. Hepatol. Res. 2011, 41, 675–682. [Google Scholar] [CrossRef]
- Ramachandran, P.; Iredale, J.P. Macrophages: Central regulators of hepatic fibrogenesis and fibrosis resolution. J. Hepatol. 2012, 56, 1417–1419. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef]
- Durante-Mangoni, E.; Wang, R.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H. Hepatic CD1d expression in hepatitis C virus infection and recognition by resident proinflammatory CD1d-reactive T cells. J. Immunol. 2004, 173, 2159–2166. [Google Scholar] [CrossRef]
- Hammam, O.; Mahmoud, O.; Zahran, M.; Aly, S.; Hosny, K.; Helmy, A.; Anas, A. The role of Fas/Fas ligand system in the pathogenesis of liver cirrhosis and hepatocellular carcinoma. Hepat. Mon. 2012, 12, e6132. [Google Scholar]
- Losikoff, P.T.; Self, A.A.; Gregory, S.H. Dendritic cells, regulatory T cells and the pathogenesis of chronic hepatitis C. Virulence 2012, 3, 610–620. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Norkina, O.; Kodys, K.; Catalano, D.; Bakis, G.; Marshall, C.; Mandrekar, P.; Szabo, G. Viral and host factors induce macrophage activation and loss of Toll Like Receptor tolerance in chronic HCV infection. Gastroenterology 2007, 133, 1627–1636. [Google Scholar] [CrossRef]
- Coquillard, G.; Patterson, B.K. Determination of hepatitis C virus-infected, monocyte lineage reservoirs in individuals with or without HIV coinfection. J. Infect. Dis. 2009, 200, 947–954. [Google Scholar] [CrossRef]
- Gerlach, J.T.; Diepolder, H.M.; Jung, M.C.; Gruener, N.H.; Schraut, W.W.; Zachoval, R.; Hoffmann, R.; Schirren, C.A.; Santantonio, T.; Pape, G.R. Recurrence of hepatitis C virus after loss of virus specific CD4+ T-cell response in acute hepatitis C. Gastroenterology 1999, 117, 933–941. [Google Scholar] [CrossRef]
- Lechner, F.; Wong, D.K.; Dunbar, P.R.; Chapman, R.; Chung, R.T.; Dohrenwend, P.; Robbins, G.; Phillips, R.; Klenerman, P.; Walker, B.D. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000, 191, 1499–1512. [Google Scholar] [CrossRef]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef]
- Harvey, C.E.; Post, J.J.; Palladinetti, P.; Freeman, A.J.; Ffrench, R.A.; Kumar, R.K.; Marinos, G.; Lloyd, A.R. Expression of the chemokine IP-10 (CXCL10) by hepatocytes in chronic hepatitis C virus infection correlates with histological severity and lobular inflammation. J. Leukoc. Biol. 2003, 74, 360–369. [Google Scholar]
- Thio, C.L.; Gao, X.; Goedert, J.J.; Vlahov, D.; Nelson, K.E.; Hilgartner, M.W.; O’Brien, S.J.; Karacki, P.; Astemborski, J.; Carrington, M.; et al. HLA-Cw*04 and hepatitis C virus persistence. J. Virol. 2002, 76, 4792–4797. [Google Scholar] [CrossRef]
- McKiernan, S.M.; Hagan, R.; Curry, M.; McDonald, G.S.; Kelly, A.; Nolan, N.; Walsh, A.; Hegarty, J.; Lawlor, E.; Kelleher, D. Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology 2004, 40, 108–114. [Google Scholar]
- Thursz, M.; Yallop, R.; Goldin, R.; Trepo, C.; Thomas, H.C. Influence of MHC class II genotype on outcome of infection with hepatitis C virus. Lancet 1999, 354, 2119–2124. [Google Scholar] [CrossRef]
- Harris, R.A.; Sugimoto, K.; Kaplan, D.E.; Ikeda, F.; Kamoun, M.; Chang, K.M. Human leukocyte antigen class II associations with hepatitis C virus clearance and virus-specific CD4 T cell response among Caucasians and African Americans. Hepatology 2008, 48, 70–79. [Google Scholar] [CrossRef]
- Apolinario, A.; Majano, P.L.; Alvarez-Pèrez, E.; Saez, A.; Lozano, C.; Vargas, J.; Garcìa-Monzòn, C. Increased expression of T cell chemokines and their receptors in chronic hepatitis C: Relationship with the histological activity of liver disease. Am. J. Gastroenterol. 2002, 97, 2861–2870. [Google Scholar] [CrossRef]
- Wang, J.; Holmes, T.H.; Cheung, R.; Greenberg, H.B.; He, X.S. Expression of chemokine receptors on intrahepatic and peripheral lymphocytes in chronic hepatitis C infection: Its relationship to liver inflammation. J. Infect. Dis. 2004, 190, 989–997. [Google Scholar] [CrossRef]
- Kusano, F.; Tanaka, Y.; Marumo, F.; Sato, C. Expression of C–C chemokines is associated with portal and periportal inflammation in the liver of patients with chronic hepatitis C. Lab. Investig. 2000, 80, 415–422. [Google Scholar] [CrossRef]
- Nishitsuji, H.; Funami, K.; Shimizu, Y.; Ujino, S.; Sugiyama, K.; Seya, T.; Takaku, H.; Shimotohno, K. Hepatitis C virus infection induces inflammatory cytokines and chemokines mediated by the cross talk between hepatocytes and stellate cells. J. Virol. 2013, 87, 8169–8178. [Google Scholar] [CrossRef]
- Dolganiuc, A.; Oak, S.; Kodys, K.; Golenbock, D.T.; Finberg, R.W.; Kurt-Jones, E.; Szabo, G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004, 127, 1513–1524. [Google Scholar] [CrossRef]
- Zeremski, M.; Petrovic, L.M.; Talal, A.H. The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J. Viral. Hepat. 2007, 14, 675–687. [Google Scholar]
- Wald, O.; Weiss, I.D.; Galun, E.; Peled, A. Chemokines in hepatitis C virus infection: Pathogenesis, prognosis and therapeutics. Cytokine 2007, 39, 50–62. [Google Scholar] [CrossRef]
- Larrea, E.; Garcia, N.; Qian, C.; Civeira, M.P.; Prieto, J. Tumor necrosis factor α gene expression and the response to interferon in chronic hepatitis C. Hepatology 1996, 23, 210–217. [Google Scholar]
- Nelson, D.R.; Lim, H.L.; Marousis, C.G.; Fang, J.W.; Davis, G.L.; Shen, L.; Urdea, M.S.; Kolberg, J.A.; Lau, J.Y. Activation of tumor necrosis factor-alpha system in chronic hepatitis C virus infection. Dig. Dis. Sci. 1997, 42, 2487–2494. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–887. [Google Scholar]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar]
- Knolle, P.A.; Gerken, G. Local control of the immune response in the liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef]
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar]
- Szabo, G.; Mandrekar, P.; Dolganiuc, A. Innate immune response and hepatic inflammation. Semin. Liver Dis. 2007, 27, 339–350. [Google Scholar] [CrossRef]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor κB-dependent manner. Gastroenterology 2010, 138, 2509–2518. [Google Scholar] [CrossRef]
- Lin, W.; Weinberg, E.M.; Tai, A.W.; Peng, L.F.; Brockman, M.A.; Kim, K.A.; Kim, S.S.; Borges, C.B.; Shao, R.X.; Chung, R.T. HIV increases HCV replication in a TGF-β1-dependent manner. Gastroenterology 2008, 134, 803–811. [Google Scholar] [CrossRef]
- Presser, L.D.; Haskett, A.; Waris, G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-β1: Role of TGF-β1 in HCV replication. Virology 2011, 412, 284–296. [Google Scholar] [CrossRef]
- Shin, J.Y.; Hur, W.; Wang, J.S.; Jang, J.W.; Kim, C.W.; Bae, S.H.; Jang, S.K.; Yang, S.H.; Sung, Y.C.; Kwon, O.J.; et al. HCV core protein promotes liver fibrogenesis via up-regulation of CTGF with TGF-β1. Exp. Mol. Med. 2005, 37, 138–145. [Google Scholar] [CrossRef]
- Taniguchi, H.; Kato, N.; Otsuka, M.; Goto, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Hepatitis C virus core protein up-regulates transforming growth factor-beta 1 transcription. J. Med. Virol. 2004, 72, 52–59. [Google Scholar] [CrossRef]
- Jaeschke, H. Inflammation in response to hepatocellular apoptosis. Hepatology 2002, 35, 964–966. [Google Scholar] [CrossRef]
- Galle, P.R.; Hofmann, W.J.; Walczak, H.; Schaller, H.; Otto, G.; Stremmel, W.; Krammer, P.H.; Runkel, L. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J. Exp. Med. 1995, 182, 1223–1230. [Google Scholar] [CrossRef]
- Yoon, J.; Gores, G. Death receptor-mediated apoptosis and the liver. J. Hepatol. 2002, 37, 400–410. [Google Scholar] [CrossRef]
- Fischer, R.; Cariers, A.; Reinehr, R.; Häussinger, D. Caspase 9-dependent killing of hepatic stellate cells by activated Kupffer cells. Gastroenterology 2002, 123, 845–861. [Google Scholar] [CrossRef]
- Kojima, Y.; Suzuki, S.; Tsuchiya, Y.; Konno, H.; Baba, S.; Nakamura, S. Regulation of pro-inflammatory and anti-inflammatory cytokine responses by Kupffer cells in endotoxin-enhanced reperfusion injury after total hepatic ischemia. Transpl. Int. 2003, 16, 231–240. [Google Scholar] [CrossRef]
- El Bassiouny, A.E.; El Bassiouni, N.E.; Nosseir, M.M.; Zoheiry, M.M.; El-Ahwany, E.G.; Salah, F.; Omran, Z.S.; Ibrahim, R.A. Circulating and hepatic Fas expression in HCV-induced chronic liver disease and hepatocellular carcinoma. Medscape J. Med. 2008, 10, 130. [Google Scholar]
- Heydtmann, M. Macrophages in hepatitis B and hepatitis C virus infections. J. Virol. 2009, 83, 2796–2802. [Google Scholar] [CrossRef]
- Chen, J.J.; Sun, Y.; Nabel, G.J. Regulation of the proinflammatory effects of Fas ligand (CD95L). Science 1998, 282, 1714–1717. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar]
- Blackard, J.T.; Hiasa, Y.; Smeaton, L.; Jamieson, D.J.; Rodriguez, I.; Mayer, K.H.; Chung, R.T. Compartmentalization of hepatitis C virus (HCV) during HCV/HIV coinfection. J. Infect. Dis. 2007, 195, 1765–1773. [Google Scholar] [CrossRef]
- Blackard, J.T.; Sherman, K.E. HCV/ HIV co-infection: Time to re-evaluate the role of HIV in the liver? J. Viral. Hepat. 2008, 15, 323–330. [Google Scholar]
- Blackard, J.T.; Welge, J.A.; Taylor, L.E.; Mayer, K.H.; Kleir, R.S.; Celentano, D.D.; Jamieson, D.J.; Gardner, L.; Sheman, K.E. HCV monoinfected, HIV monoinfected, HIV/HCV coinfected and HIV-seronegative/HCV-seronegative women. Clin. Infect. Dis. 2011, 52, 674–680. [Google Scholar] [CrossRef]
- Han, S.H.; Kim, S.U.; Kim, C.O.; Jeong, S.J.; Park, J.Y.; Choi, J.Y.; Kim, D.Y.; Ahn, S.H.; Song, Y.G.; et al. Abnormal liver stiffness assessed using transient elestography (Fibroscan) in HIV-infected patients without HBV/HCV coinfection receiving combined antiretroviral treatment. PLoS One 2013, 8, e52720. [Google Scholar] [CrossRef]
- Hasson, H.; Merli, M.; Galli, L.; Gallotta, G.; Carbone, A.; Messina, E.; Bagaglio, S.; Morsica, G.; Salpietro, S.; Castagna, A.; et al. Non-invasive fibrosis biomarkers—APRI and forms are associated with stiffness in HIV-monoinfected patients receiving antiretroviral drugs. Liver Int. 2013, 33, 1113–1120. [Google Scholar] [CrossRef]
- Merchante, N.; Pérez-Chamacho, I.; Mira, J.A.; Rivero, A.; Macìas, J.; Camacho, A.; Gòmez-Mateos, J.; Garcìa-Làzaro, M.; Torre-Cisneros, J.; Pineda, J.A. Prevalence and risk factors for abnormal liver stiffness in HIV-infected patients without viral hepatitis coinfection: Role of didanosine. Antivir. Ther. 2010, 15, 753–763. [Google Scholar] [CrossRef]
- Kovari, H.; Ledergerber, B.; Battegay, M.; Rauch, A.; Hirschel, B.; Foguena, A.K.; Vernazza, P.; Bernasconi, E.; Mueller, N.J.; Webr, R. Incidence and risk factors for chronic elevation of alanine aminotransferase levels in HIV-infected persons without hepatitis B or C virus co-infection. Clin. Infect. Dis. 2010, 50, 502–511. [Google Scholar] [CrossRef]
- Bräu, N.; Salvatore, M.; Ríos-Bedoya, C.F.; Fernández-Carbia, A.; Paronetto, F.; Rodríguez-Orengo, J.F.; Rodríguez-Torres, M. Slower fibrosis progression in HIV/HCV-coinfected patients with successful HIV suppression using antiretroviral therapy. J. Hepatol. 2006, 44, 47–55. [Google Scholar] [CrossRef]
- Vlahakis, S.; Villasis-Keever, A.; Gomez, T.S.; Bren, G.D.; Paya, C.V. Human immunodeficiency virus-induced apoptosis of human hepatocytes via CXCR4. J. Infect. Dis. 2003, 188, 1455–1460. [Google Scholar] [CrossRef]
- Munshi, N.; Balasubramanian, A.; Koziel, M.; Ganjub, R.K.; Groopmanb, J.E. Hepatitis C and human immunodeficiency virus envelope proteins cooperatively induce hepatocytic apoptosis via an innocent bystander mechanism. J. Infect. Dis. 2003, 188, 1192–1204. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Ganju, R.; Groopman, J.E. HCV and HIV envelope proteins collaboratively mediate IL-8 secretion through activation of p38 MAP kinase and SHP2 in hepatocytes. J. Biol. Chem. 2003, 278, 35755–35766. [Google Scholar] [CrossRef]
- Yoong, K.; Afford, S.C.; Jones, R.; Aujla, P.; Qin, S.; Price, K.; Hubscher, S.G.; Adams, D.H. Expression and function of CXC and CC chemokines in human malignant liver tumors: A role for human monokine induced by g-interferon in lymphocyte recruitment to hepatocellular carcinoma. Hepatology 1999, 30, 100–111. [Google Scholar] [CrossRef]
- Wout, A.B.; Ran, L.J.; Kuiken, C.L.; Kootstra, N.A.; Pals, S.T.; Schuitemaker, H. Analysis of the temporal relationchip between human immunodeficiency virus type 1 quasispecies in sequential blood samples and various organs obtained at autopsy. J. Virol. 1998, 72, 488–496. [Google Scholar]
- Tuyama, A.C.; Hong, F.; Saiman, Y.; Wang, C.; Ozkok, D.; Mosoian, A.; Chen, P.; Chen, B.K.; Klotman, M.E.; Bansal, M.B. Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: Implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatology 2010, 52, 612–622. [Google Scholar] [CrossRef]
- Rotman, Y.; Liang, T.J. Coinfection with hepatitis C virus and human immunodeficiency virus: Virological, immunological, and clinical out-comes. J. Virol. 2009, 83, 7366–7374. [Google Scholar] [CrossRef]
- Lin, W.; Wu, G.; Li, S.; Weinberg, E.M.; Kumthip, K.; Peng, L.F.; Màndez-Navarro, J.; Chen, W.C.; Jilg, N.; Zhao, H.; et al. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFκB. J. Biol. Chem. 2011, 286, 2665–2674. [Google Scholar] [CrossRef]
- Bruno, R.; Galastri, S.; Sacchi, P.; Cima, S.; Caligiuri, A.; DeFranco, R.; Milani, S.; Gessani, S.; Fantuzzi, L.; Liotta, F.; et al. gp 120 modulates the biology of human hepatic stellate cells: A link between HIV infection and liver fibrogenesis. Gut 2010, 59, 513–520. [Google Scholar] [CrossRef]
- Hong, F.; Tuyama, A.; Lee, T.F.; Loke, J.; Agarwal, R.; Cheng, X.; Garg, A.; Fiel, M.I.; Schwartz, M.; Walewski, J.; et al. Hepatic stellate cells express functional CXCR4: Role in stromal cell-derived factor-1α-mediated stellate cell activation. Hepatology 2009, 49, 2055–2067. [Google Scholar] [CrossRef]
- Babu, C.K.; Suwansrinon, K.; Bren, G.D.; Badley, A.D.; Rizza, S.A. HIV induces TRAIL sensitivity in hepatocytes. PLoS One 2009, 4, e4623. [Google Scholar] [CrossRef]
- Tuyama, A.C.; Hong, F.; Schecter, A.D.; Mosoian, A.; Chen, B.K.; Chen, P.; Klotman, M.E.; Bansal, M.B. HIV entry and replication in stellate cells promotes cellular activation and fibrogenesis: Implications for hepatic fibrosis in HIV/HCV coinfection. Hepatology 2007, 46, 291. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Bataller, R.; Brenner, D.A. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, 949–958. [Google Scholar]
- Marra, F.; Valente, A.J.; Pinzani, M.; Abboud, H.E. Cultured human liver fat-storing cells produce monocyte chemotactic protein-1. Regulation by proinflammatory cytokines. J. Clin. Investig. 1993, 92, 1674–1680. [Google Scholar] [CrossRef]
- Efsen, E.; Bonacchi, A.; Pastacaldi, S.; Valente, A.J.; Wenzel, U.O.; Tosti-Guerra, C.; Pinzani, M.; Laffi, G.; Abboud, H.E.; Gentilini, P.; et al. Agonist-specific regulation of monocyte chemoattractant protein-1 expression by cyclooxygenase metabolites in hepatic stellate cells. Hepatology 2001, 33, 713–721. [Google Scholar] [CrossRef]
- Marra, F.; de Franco, R.; Grappone, C.; Milani, S.; Pastacaldi, S.; Pinzani, M.; Romanelli, R.G.; Laffi, G.; Gentilini, P. Increased expression of monocyte chemotactic protein-1 during active hepatic fibrogenesis: Correlation with monocyte infiltration. Am. J. Pathol. 1998, 152, 423–430. [Google Scholar]
- Balasubramanian, A.; Ganju, R.K.; Groopman, J.E. Signal transducer and activator of transcription factor 1 mediates apoptosis induced by hepatitis C virus and HIV envelope proteins in hepatocytes. J. Infect. Dis. 2006, 194, 670–681. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef]
- Seki, E.; de Minicis, S.; Gwak, G.Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Investig. 2009, 119, 1858–1870. [Google Scholar]
- Ochoa-Callejero, L.; Perez-Martinez, L.; Rubio-Mediavilla, S.; Oteo, J.A.; Martìnez, A.; Blanco, J.R. Maraviroc, a CCR5 antagonist, prevents development of hepatocellular carcinoma in a mouse model. PLoS One 2013, 8, e53992. [Google Scholar]
- Affo, S.; Bataller, R. RANTES antagonism: A promising approach to treat chronic liver diseases. J. Hepatol. 2011, 55, 936–938. [Google Scholar] [CrossRef]
- Berres, M.L.; Koenen, R.R.; Rueland, A.; Zaldivar, M.M.; Heinrichs, D.; Sahin, H.; Schmitz, P.; Streetz, K.L.; Berg, T.; Gassler, N.; et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J. Clin. Investig. 2010, 120, 4129–4140. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef]
- Fatkenheuer, G.; Pozniak, A.L.; Johnson, M.A.; Plettenberg, A.; Staszewski, S.; Hoepelman, A.I.; Saag, M.S.; Goebel, F.D.; Rockstroh, J.K.; Dezube, B.J.; et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 2005, 11, 1170–1172. [Google Scholar] [CrossRef]
- Macias, J.; Viloria, M.M.; Rivero, A.; de los Santos, I.; Màrquez, M.; Postilla, J.; di Lello, F.; Camacho, A.; Sanz-Sanz, J.; Ojeda, G.; et al. Lack of short-term increase in serum mediators of fibrogenesis and in non-invasive markers of liver fibrosis in HIV/hepatitis C virus-coinfected patients starting maraviroc-based antiretroviral therapy. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2083–2088. [Google Scholar] [CrossRef]
- Glässner, A.; Eisenhardt, M.; Kokordelis, P.; Krämer, B.; Wolter, F.; Nischalke, H.D.; Boesecke, C.; Sauerbruch, T.; Rockstroh, J.K.; Spengler, U.; et al. Impaired CD4+ T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J. Hepatol. 2013, 59, 427–433. [Google Scholar] [CrossRef]
- Mehal, W.Z.; Friedman, S.L. The Role of Inflammation and Immunity in the Pathogenesis of Liver Fibrosis. In Liver Immunology; Humana Press: Clifton, NJ, USA, 2007; pp. 111–121. [Google Scholar]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of TGF-β in hepatic fibrosis. Front. Biosci. 2002, 7, 793–807. [Google Scholar] [CrossRef]
- Berenguer, J.; Fernandez-Rodriguez, A.; Jimenez-Sousa, M.A.; Cosìn, J.; Zarate, P.; Micheloud, D.; Lòpez, J.C.; Miralles, P.; Resino, S. High plasma CXCL10 levels are associated with HCV-genotype 1, and higher insulin resistance, fibrosis, and HIV viral load in HIV/HCV coinfected patients. Cytokine 2012, 57, 25–29. [Google Scholar] [CrossRef]
- Sultana, C.; Erscoiu, S.M.; Grancea, C.; Ceausu, E.; Ruta, S. Predictors of chronic hepatitis C evolution in HIV co-infected patients from Romania. Hepat. Mon. 2013, 13, e8611. [Google Scholar]
- Cardin, R.; Saccoccio, G.; Masutti, F.; Bellentani, S.; Farinati, F.; Tiribelli, C. DNA oxidative damage in leukocytes correlates with the severity of HCV-related liver disease: Validation in an open population study. J. Hepatol. 2001, 34, 587–592. [Google Scholar] [CrossRef]
- Qu, J.; Zhang, Q.; Li, Y.; Liu, W.; Chen, L.; Zhu, Y.; Wu, J. The Tat protein of human immunodeficiency virus-1 enhances HCV replication through interferon γ-inducible protein-10. BMC Immunol. 2012, 13, 15. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 and the pathogenesis of the acute-phase response. N. Engl. J. Med. 1984, 311, 1413–1418. [Google Scholar] [CrossRef]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef]
- Chakraborty, S.; Kaushik, D.K.; Gupta, M.; Basu, A. Inflammasome signaling at the heart of central nervous system pathology. J. Neurosci. Res. 2010, 88, 1615–1631. [Google Scholar]
- Artlett, C.M.; Sassi-Gaha, S.; Rieger, J.L.; Boesteanu, A.C.; Feghali-Bostwick, C.A.; Katsikis, P.D. The inflammasome activating caspase-1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011, 63, 3563–3574. [Google Scholar] [CrossRef]
- Daheshia, M.; Yao, J.Q. The interleukin 1β pathway in the pathogenesis of osteoarthritis. J. Rheumatol. 2008, 35, 2306–2312. [Google Scholar] [CrossRef]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göss, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschoppo, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef]
- Kanneganti, T.D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef]
- Montserret, R.; Saint, N.; Vanbelle, C.; Salvay, A.G.; Simorre, J.P.; Ebel, C.; Sapay, N.; Renisio, J.G.; Böckmann, A.; Steinmann, E.; et al. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J. Biol. Chem. 2010, 285, 31446–31461. [Google Scholar] [CrossRef]
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241. [Google Scholar] [CrossRef]
- Han, D.W. Intestinal endotoxemia as a pathogenetic mechamism in liver failure. World J. Gastroenterol. 2002, 8, 961–965. [Google Scholar]
- De-Oca, M.M.; Marquez, M.; Soto, M.J.; Rodriguez-Ramos, C.; Terron, A.; Vergara, A.; Arizcorreta, A.; Fernandez-Gutierrez, C.; Giron-González, J.A. Bacterial translocation in HIV-infected patients with HCV cirrhosis: Implications in hemodynamic alterations and mortality. J. Acquir. Immune Defic. Syndr. 2011, 56, 42–47. [Google Scholar]
- Guzman-Fulgencio, M.; Jimenez, J.L.; Berenguer, J.; Fernández-Rodríguez, A.; López, J.C.; Cosín, J.; Miralles, P.; Micheloud, D.; Muňoz-Fernández, M.A.; Resino, S. Plasma IL-6 and IL-9 predict failure of interferon-α plus ribavirin therapy in HIV/HCV coinfected patients. J. Antimicrob. Chemother. 2012, 67, 1238–1245. [Google Scholar] [CrossRef]
- Mattapallil, J.J.; Douek, D.C.; Hill, B.; Nishimura, Y.; Martin, M.; Roederer, M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 2005, 434, 1093–1097. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar]
- Landmann, R.; Knopf, H.P.; Link, S.; Sansano, S.; Schumann, R.; Zimmerli, W. Human monocyte CD14 is upregulated by lipopolysaccharide. Infect. Immun. 1996, 64, 1762–1769. [Google Scholar]
- Sandler, N.G.; Koh, C.; Roque, A.; Eccleston, J.L.; Siegel, R.B.; Demino, M.; Kleiner, D.E.; Deeks, S.G.; Liang, T.J.; Heller, T.; et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 2011, 141, 1220–1230. [Google Scholar] [CrossRef]
- Marchetti, G.; Cozzi-Lepri, A.; Merlini, E.; Bellistrì, G.M.; Castagna, A.; Galli, M.; Verucchi, G.; Antinori, A.; Costantini, A.; Giacometti, A.; et al. Microbial translocation predicts disease progression of HIV-infected antiretroviral-naive patients with high CD4+ cell count. AIDS 2011, 25, 1385–1394. [Google Scholar] [CrossRef]
- Page, E.E.; Nelson, M.; Kelleher, P. HIV and hepatitis C coinfection: Pathogenesis and microbial translocation. Curr. Opin. HIV AIDS 2011, 6, 472–477. [Google Scholar] [CrossRef]
- Balagopal, A.; Philp, F.H.; Astemborski, J.; Block, T.M.; Mehta, A.; Long, R.; Kirk, G.D.; Mehta, S.H.; Cox, A.L.; Thomas, D.L.; et al. Human immunodeficiency virus-related microbial translocation and progression of hepatitis C. Gastroenterology 2008, 135, 226–233. [Google Scholar] [CrossRef]
- Henderson, N.C.; Iredale, J.P. Liver fibrosis: Cellular mechanisms of progression and resolution. Clin. Sci. 2007, 112, 265–280. [Google Scholar] [CrossRef]
- Godichaud, S.; Krisa, S.; Couronnè, B.; Dubuisson, L.; Mérillon, J.M.; Desmoulière, A.; Rosenbaum, J. Deactivation of cultured human liver myofibroblasts by trans-resveratrol, a grapevine-derived polyphenol. Hepatology 2000, 31, 922–931. [Google Scholar] [CrossRef]
- Mastroianni, C.M.; Liuzzi, M.G.; D’Ettorre, G.; Lichtner, M.; Forcina, G.; di Campli, N.F.; Riccio, P.; Vullo, V. Matrix metalloproteinase-9 and tissue inhibitors of matrix metalloproteinase-1 in plasma of patients co-infected with HCV and HIV. HIV Clin. Trials. 2002, 3, 310–315. [Google Scholar] [CrossRef]
- Puoti, M.; Bonacini, M.; Spinetti, A.; Putzolu, V.; Govindarajan, S.; Zaltron, S.; Favret, M.; Callea, F.; Gargiulo, F.; Donato, F.; et al. Liver fibrosis progression is related to CD4 cell depletion in patients coinfected with hepatitis C virus and human immunodeficiency virus. J. Infect. Dis. 2001, 183, 134–137. [Google Scholar] [CrossRef]
- Mastroianni, C.M.; Liuzzi, G.M. Matrix metalloproteinase dysregulation in HIV infection: Implications for therapeutic strategies. Trends Mol. Med. 2007, 13, 449–459. [Google Scholar] [CrossRef]
- Eriksson, S.; Fraser, J.R.; Laurent, T.C.; Pertoft, H.; Smedsrød, B. Endothelial cells are a site of uptake and degradation of hyaluronic acid in the liver. Exp. Cell. Res. 1983, 144, 223–228. [Google Scholar] [CrossRef]
- Peters, L.; Mocroft, A.; Soriano, V.; Rockstroh, J.; Rauch, A.; Karlsson, A.; Knysz, B.; Pradier, C.; Zilmer, K.; Lundgren, J.D.; et al. Hyaluronic acid levels predict risk of hepatic encephalopathy and liver-related death in HIV/viral hepatitis coinfected patients. PLoS One 2013, 8, e64283. [Google Scholar] [CrossRef]
- Duong, M.; Petit, J.M.; Piroth, L.; Grappin, M.; Buisson, M.; Chavanet, P.; Hillon, P.; Portier, H. Association between insulin resistance and hepatitis C virus chronic infection in HIV-hepatitis C virus-coinfected patients undergoing antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2001, 27, 245–250. [Google Scholar] [CrossRef]
- Grigorescu, M.; Radu, C.; Crişan, D.; Grigorescu, M.D.; Şerban, A.; Neculoiu, D.; Rusu, M.; Acalovschi, M. Metabolic syndrome, insulin resistance and adiponectin level in patients with chronic hepatitis C. J. Gastrointest. Liver Dis. 2008, 17, 147–154. [Google Scholar]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef]
- Ryan, P.; Berenguer, J.; Michelaud, D.; Miralles, P.; Bellòn, J.M.; Alvarez, E.; Catàlan, P.; Sànchez-Conde, M.; Resino, S. Insulin resistance is associated with advanced liver fibrosis and high body mass index in HIV/HCV-coinfected patients. J. Acquir. Immune Defic. Syndr. 2009, 50, 109–110. [Google Scholar]
- Carper, M.J.; Cade, W.T.; Cam, M.; Zhang, S.; Shalev, A; Yarasheski, K.E.; Ramanadham, S. HIV-protease inhibitors induce expression of suppressor of cytokine signaling-1 in insulin-sensitive tissues and promote insulin resistance and type 2 diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 558–567. [Google Scholar] [CrossRef]
- Capel, E.; Auclair, M.; Caron-Debarle, M.; Capeau, J. Effect of ritonavir-boosted darunavir, atazanavir and lopinavir on adipose functions and insulin sensitivity in murine and human adipocyte. Antivir. Ther. 2012, 17, 549–556. [Google Scholar]
- Van Vonderen, M.G.; Blmer, R.M.; Hassink, E.A.; Sutinen, J.; Ackermans, M.T.; van Agtmael, M.A.; Yki-Jarvinen, H.; Danner, S.A.; Serlie, M.J.; Sauerwein, H.P.; et al. Insulin sensitivity in multiple pathways is differently affected during zidovudine/lamivudine-containing compared with NRTI-sparing combination antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2010, 53, 186–193. [Google Scholar]
- Hruz, PW. Molecular mechanisms for insulin resistance in treated HIV-infection. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 459–468. [Google Scholar] [CrossRef]
- Hull, M.W.; Rollet, K.; Moodie, E.E.; Walmsley, S.; Cox, J.; Potter, M.; Cooper, C.; Pick, N.; Saeed, S.; Klein, M.B.; et al. Insulin resistance is associated with progression to hepatic fibrosis in a cohort of HIV/hepatitis C virus-coinfected patients. AIDS 2012, 26, 1789–1794. [Google Scholar]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Hui, J.M.; Sud, A.; Farrell, G.C.; Bandara, P.; Byth, K.; Kench, J.G.; McCaughan, G.W.; George, J. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression. Gastroenterology 2003, 125, 1695–1704. [Google Scholar] [CrossRef]
- Ratziu, V.; Munteanu, M.; Charlotte, F.; Bonyhay, L.; Poynard, T. LIDO Study Group. Fibrogenic impact of high serum glucose in chronic hepatitis C. J. Hepatol. 2003, 39, 1049–1055. [Google Scholar] [CrossRef]
- Hickman, I.J.; Powell, E.E.; Prins, J.B.; Clouston, A.D.; Ash, S.; Purdie, D.M.; Jonsson, J.R. In overweight patients with chronic hepatitis C, circulating insulin is associated with hepatic fibrosis: Implications for therapy. J. Hepatol. 2003, 39, 1042–1048. [Google Scholar] [CrossRef]
- Paradis, V.; Perlemuter, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001, 34, 738–744. [Google Scholar] [CrossRef]
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J., Wang; Portincasa, P. Role of mitochondria in nonalcoholic fatty liver disease-from origin to propagation. Clin. Biochem. 2012, 45, 610–618. [Google Scholar] [CrossRef]
- Perez-Matute, P.; Perez-Martinez, L.; Blanco, J.R.; Oteo, J.A. Role of mitochondria in HIV infection and associated metabolic disorders: Focus on nonalcoholic fatty liver disease and lipodystrophy syndrome. Oxid. Med. Cell. Longev. 2013, 2013, 493413. [Google Scholar]
- Maagaard, A.; Holberg-Petersen, M.; Løvgården, G.; Holm, M.; Pettersen, F.O.; Kvale, D. Distinct mechanisms for mitochondrial DNA loss in T and B lymphocytes from HIV-infected patients exposed to nucleoside reverse-transcriptase inhibitors and those naive to antiretroviral treatment. J. Infect. Dis. 2008, 198, 1474–1481. [Google Scholar] [CrossRef]
- De Castro, I.F.; Berenguer, J.; Micheloud, D.; Guzmàn-Fulgencio, M.; Cosìn, J.; Alvarez, E.; Lòpez, J.C.; Miralles, P.; Garcìa-Alvarez, M.; Resino, S. Serum levels of adipokines in HIV/HCV co-infected patients and their association with insulin resistance and liver disease severity. J. Infect. 2010, 61, 499–501. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Bertolani, C.; Marra, F. The role of adipokines in liver fibrosis. Pathophysiology 2008, 15, 91–101. [Google Scholar] [CrossRef]
- Bertolani, C.; Sancho-Bru, P.; Failli, P.; Bataller, R.; Aleffi, S.; DeFranco, R.; Mazzinghi, B.; Romagnani, P.; Milani, S.; Ginès, P.; et al. Resistin as an intrahepatic cytokine: Over-expression during chronic injury and induction of proinflammatory actions in hepatic stellate cells. Am. J. Pathol. 2006, 169, 2042–2053. [Google Scholar] [CrossRef]
- Wang, J.; Leclercq, I.; Brymora, J.M.; Xu, N.; Ramezani-Moghadam, M.; London, R.M.; Brigstock, D.; George, J. Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 2009, 137, 713–723. [Google Scholar] [CrossRef]
- Pateria, P.; de Boer, B.; MacQuillan, G. Liver abnormalities in drug and substance abuser. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 577–596. [Google Scholar] [CrossRef]
- Hézode, C.; Roudot-Thoraval, F.; Nguyan, S.; Grenard, P.; Julien, B.; Zafrani, E.S.; Pawlotsky, J.M.; Dhumeaux, D.; Lotersztajn, S.; Mallat, A. Daily cannabis smoking as a risk factor for progression of fibrosis in chronic hepatitis C. Hepatology 2005, 42, 63–71. [Google Scholar]
- Maurer, H.H. Chemistry, pharmacology, and metabolism of emerging drugs of abuse. Ther. Drug Monit. 2010, 60, 438–446. [Google Scholar]
- Kothur, R.; Marsh, F.; Posner, G. Liver function tests in nonparenteral cocaine user. Arch. Intern. Med. 1991, 151, 1126–1128. [Google Scholar] [CrossRef]
- Silva, M.O.; Roth, D.; Reddy, K.R.; Fernandez, J.A.; Albores-Saavedra, J.; Schiff, E.R. Hepatic dysfunction accompanying acute cocaine intoxication. J. Hepatol. 1991, 12, 312–315. [Google Scholar] [CrossRef]
- Kielland, K.B.; Delaviris, G.J.; Rodge, S.; Eide, T.J.; Amundsen, E.J.; Dalgard, O. Liver fibrosis progression at autopsy in injecting drug users infected by hepatitis C: A longitudinal long-term cohort study. J. Hepatol. 2014, 60, 260–266. [Google Scholar] [CrossRef]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef]
- Farfán Labonne, B.G.; Gutiérrez, M.; Gómez-Quiroz, L.E.; Konigsberg Fainstein, M.; Bucio, L.; Souza, V.; Flores, O.; Ortíz, V.; Hernández, E.; Kershenobich, D. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol. Toxicol. 2009, 25, 599–609. [Google Scholar] [CrossRef]
- Albano, E.; Vidali, M. Immune mechanisms in alcoholic liver disease. Genes Nutr. 2010, 5, 141–147. [Google Scholar] [CrossRef]
- Mello, T.; Ceni, T.; Surrenti, C.; Galli, A. Alcohol induced hepatic fibrosis: Role of acetaldehyde. Mol. Asp. Med. 2008, 29, 17–21. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Gao, B. Abrogation of the antifibrotic effects of natural killer cells/interferon-γ contributes to alcohol acceleration of liver fibrosis. Gastroenterology 2008, 134, 248–258. [Google Scholar] [CrossRef]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mastroianni, C.M.; Lichtner, M.; Mascia, C.; Zuccalà, P.; Vullo, V. Molecular Mechanisms of Liver Fibrosis in HIV/HCV Coinfection. Int. J. Mol. Sci. 2014, 15, 9184-9208. https://doi.org/10.3390/ijms15069184
Mastroianni CM, Lichtner M, Mascia C, Zuccalà P, Vullo V. Molecular Mechanisms of Liver Fibrosis in HIV/HCV Coinfection. International Journal of Molecular Sciences. 2014; 15(6):9184-9208. https://doi.org/10.3390/ijms15069184
Chicago/Turabian StyleMastroianni, Claudio M., Miriam Lichtner, Claudia Mascia, Paola Zuccalà, and Vincenzo Vullo. 2014. "Molecular Mechanisms of Liver Fibrosis in HIV/HCV Coinfection" International Journal of Molecular Sciences 15, no. 6: 9184-9208. https://doi.org/10.3390/ijms15069184
APA StyleMastroianni, C. M., Lichtner, M., Mascia, C., Zuccalà, P., & Vullo, V. (2014). Molecular Mechanisms of Liver Fibrosis in HIV/HCV Coinfection. International Journal of Molecular Sciences, 15(6), 9184-9208. https://doi.org/10.3390/ijms15069184