Does Prop-2-ynylideneamine, HC≡CCH=NH, Exist in Space? A Theoretical and Computational Investigation


Abstract
:
1. Introduction
2. Results and Discussion
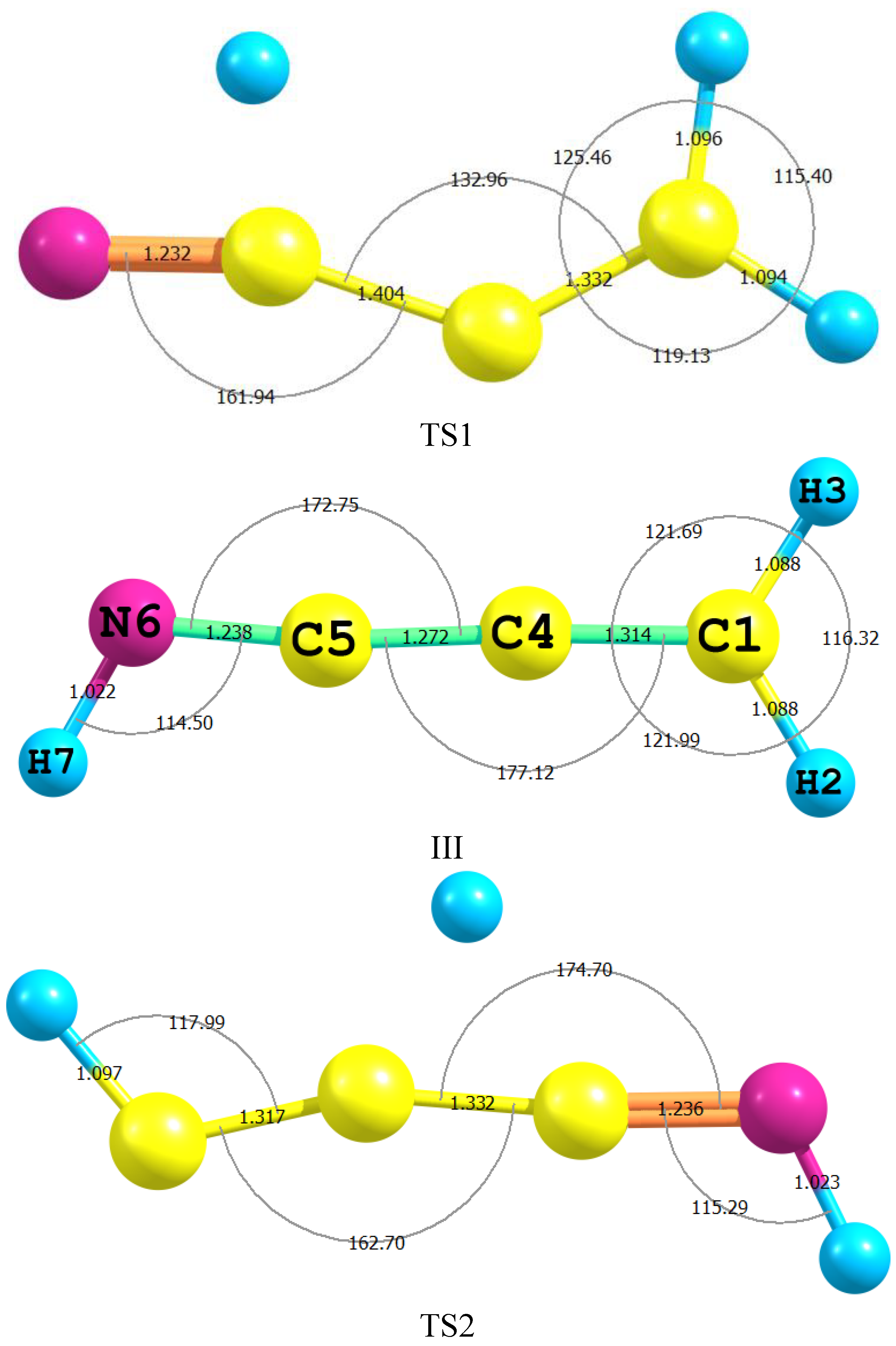
2.1. Molecular Structure
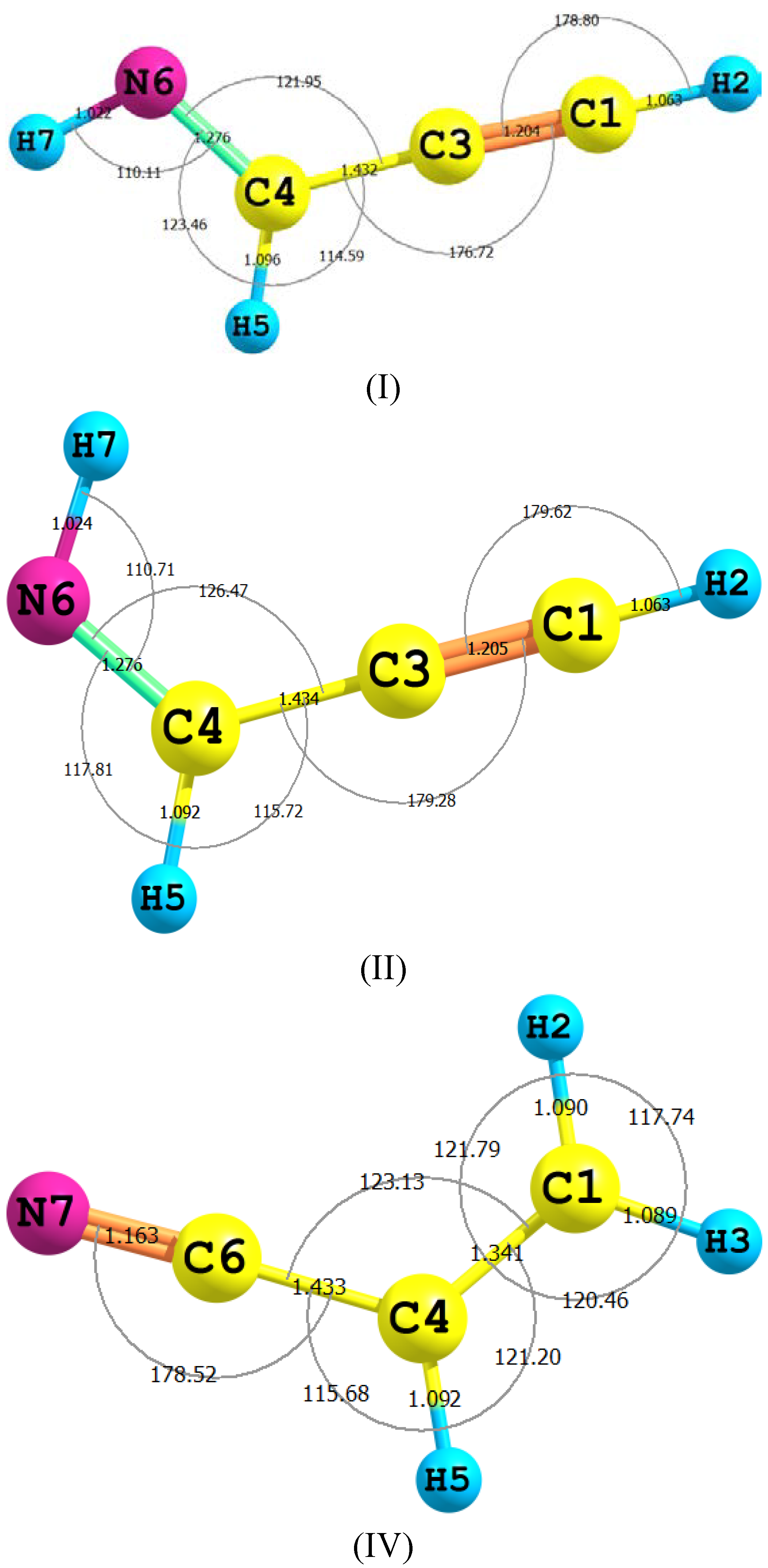

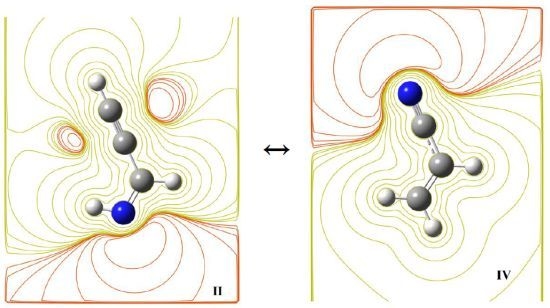
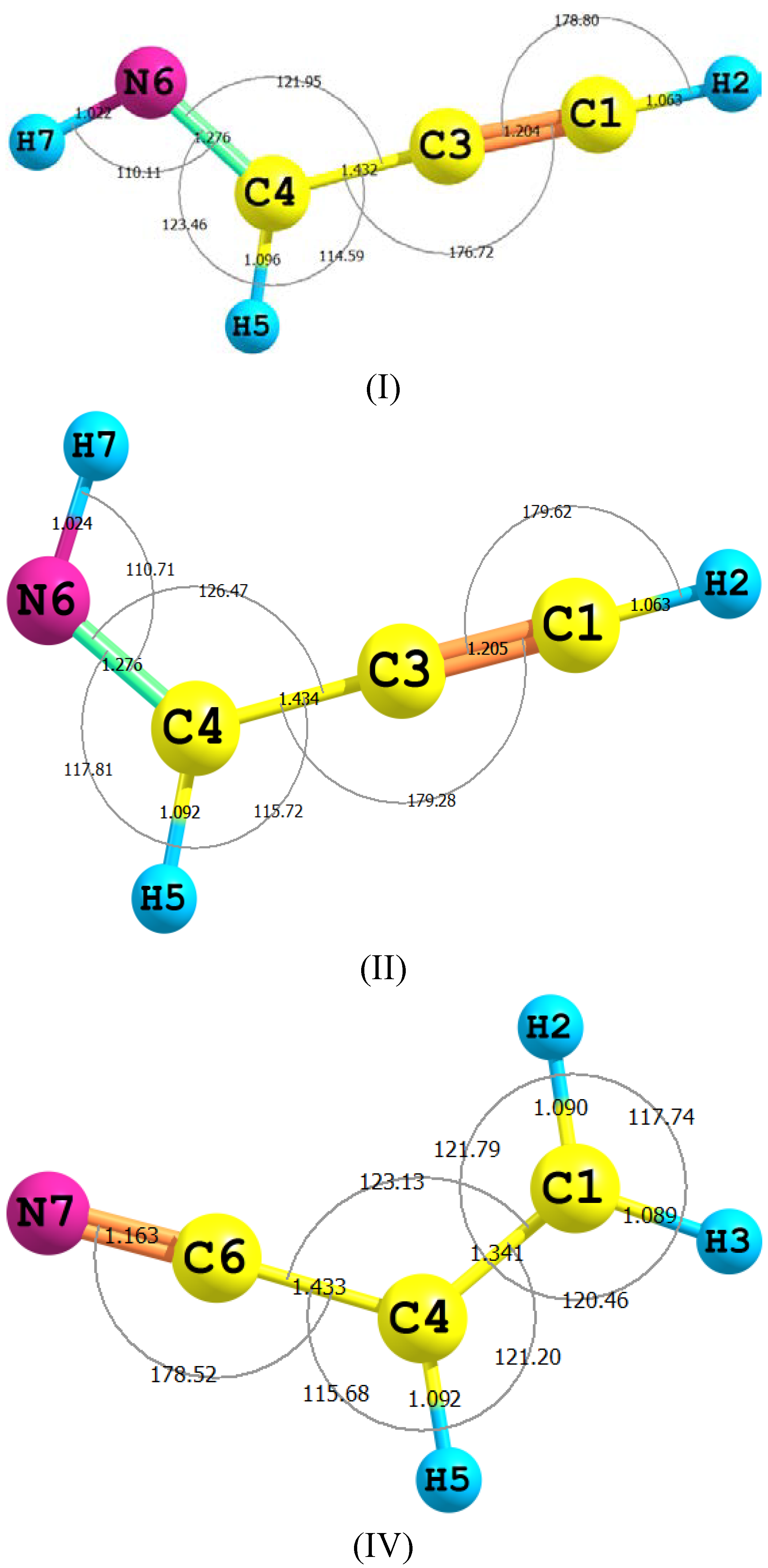
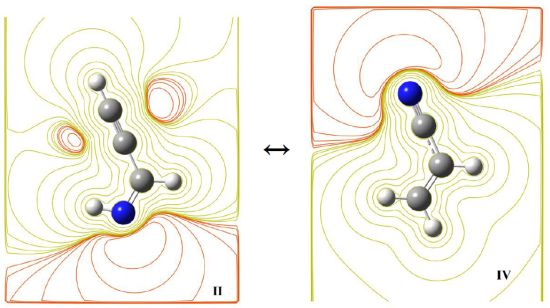{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Basis Set | H-C | C≡C | C-C | C=N | NH | CH | CCN | CNH | µ/Debye |
|---|---|---|---|---|---|---|---|---|---|---|
| MP2 | 6-311++G** | 1.065 | 1.221 | 1.438 | 1.288 | 1.024 | 1.095 | 120.95 | 108.61 | 2.27 |
| B3LYP | 6-311++G** | 1.063 | 1.204 | 1.432 | 1.276 | 1.022 | 1.096 | 121.95 | 110.11 | 2.07 |
| CCSD | 6-311++G** | 1.066 | 1.212 | 1.448 | 1.281 | 1.023 | 1.095 | 121.13 | 109.11 | 2.05 |
| Experimental a | - | - | - | - | - | - | - | - | 1.90 | |
| MP2 | 6-311++G** | 1.066 | 1.222 | 1.440 | 1.289 | 1.025 | 1.091 | 125.79 | 108.82 | 2.58 |
| B3LYP | 6-311++G** | 1.063 | 1.205 | 1.434 | 1.276 | 1.023 | 1.091 | 126.45 | 110.71 | 2.41 |
| CCSD | 6-311++G** | 1.067 | 1.212 | 1.452 | 1.281 | 1.024 | 1.091 | 125.68 | 109.52 | 2.32 |
| Experimental b | 1.057 | 1.207 | 1.431 | 1.286 | 1.039 | 1.101 | 125.38 | 108.89 | 2.15 | |
| Method Basis Set | MP2 | B3LYP | CCSD | Expt. a,b | |||
|---|---|---|---|---|---|---|---|
| 6-311++G** | aug-cc-pvdz | 6-311++G** | aug-cc-pvdz | 6-311++G** | aug-cc-pvdz | ||
| C-Ht | 1.085 | 1.092 | 1.083 | 1.089 | 1.086 | 1.093 | 1.097 |
| C-Hc | 1.084 | 1.092 | 1.083 | 1.090 | 1.086 | 1.094 | 1.093 |
| C=C | 1.344 | 1.353 | 1.335 | 1.341 | 1.342 | 1.351 | 1.343 |
| C-Hu | 1.086 | 1.093 | 1.085 | 1.092 | 1.086 | 1.094 | 1.085 |
| C-C | 1.435 | 1.443 | 1.428 | 1.433 | 1.445 | 1.454 | 1.429 |
| C≡N | 1.177 | 1.189 | 1.156 | 1.163 | 1.162 | 1.172 | 1.160 |
| CCHt | 120.28 | 120.17 | 120.56 | 120.46 | 120.4 | 120.4 | 118.5 |
| CCHc | 121.44 | 121.32 | 121.79 | 121.78 | 121.7 | 121.6 | 120.3 |
| C=CHu | 121.39 | 121.31 | 121.24 | 121.21 | 121.9 | 121.8 | 121.6 |
| CCC | 122.11 | 122.20 | 123.11 | 123.11 | 122.1 | 122.1 | 122.2 |
| CCN | 179.05 | 179.10 | 178.72 | 178.46 | 179.0 | 179.0 | 178.4 |
| µ/Debye | 4.47 | 4.51 | 4.05 | 4.04 | 3.90 | 3.95 | 3.92 |
| Parameter | II | TS2 | III | TS1 | IV |
|---|---|---|---|---|---|
| H2–C1 | 1.063 (1.057) | 1.097 | 1.088 | 1.095 | 1.090 (1.089) |
| C1–C3 | 1.205 (1.207) | 1.317 | 1.314 | 1.332 | 1.341 (1.343) |
| C3–C4 | 1.434 (1.431) | 1.332 | 1.314 | 1.332 | 1.433 (1.429) |
| C4–N6 | 1.276 (1.286) | 1.236 | 1.238 | 1.232 | 1.163 (1.163) |
| N6–H7 | 1.024 (1.039) | 1.023 | 1.022 | 1.585 | - |
| C4–H5 | 1.092 (1.101) | 1.433 | - | 1.096 | 1.092(1.086) |
| C4C5N6 | 126.5 (125.3) | 174.7 | 172.75 | 161.9 | 178.4(178.4) |
| C5N6H7 | 110.7 (108.9) | 45.8 | 114.2 | 45.8 | - |
| H3C1N7H9 | 180.00 | −179.9 | 0.00 | −179.9 | - |
| C4C1N7H9 | −0.004 | −179.9 | 180.0 | −179.9 | - |

2.2. Isomerism and Tautomerism
| Method | Basis Set | ΔE kJ/mol | ΔH kJ/mol | ΔS J/mol.K | ΔG kJ/mol | K |
|---|---|---|---|---|---|---|
| MP2 | 6-311++G** | −2.046 | −2.138 | −0.282 | −2.054 | 2.291 |
| aug-cc-pvdz | −3.707 | −3.452 | +2.484 | −4.192 | 5.425 | |
| B3LYP | 6-311++G** | −2.920 | −2.958 | +0.364 | −3.067 | 3.445 |
| aug-cc-pvdz | −3.063 | −2.954 | +1.375 | −3.364 | 3.885 | |
| CCSD | 6-311++G** | −1.707 | −1.757 | +0.126 | −1.795 | 2.063 |
| aug-cc-pvdz | −3.084 | −2.887 | +2.006 | −3.485 | 4.080 |
| Method | Basis Set | ΔE kJ/mol | ΔH kJ/mol | ΔS J/mol.K | ΔG kJ/mol | K |
|---|---|---|---|---|---|---|
| MP2 | 6-311++G** | −147.507 | −148.189 | −4.042 | −146.984 | 5.648 × 1025 |
| aug-cc-pvdz | −147.436 | −148.499 | −6.571 | −146.540 | 4.723 × 1025 | |
| B3LYP | 6-311++G** | −138.449 | −138.674 | −2.130 | −138.039 | 1.530 × 1024 |
| aug-cc-pvdz | −137.336 | −138.076 | −4.236 | −136.813 | 9.333 × 1023 | |
| CCSD | 6-311++G** | −147.535 | −148.073 | −3.029 | −147.170 | 6.088 × 1025 |
| aug-cc-pvdz | −148.375 | −149.304 | −5.618 | −147.629 | 7.326 × 1025 |
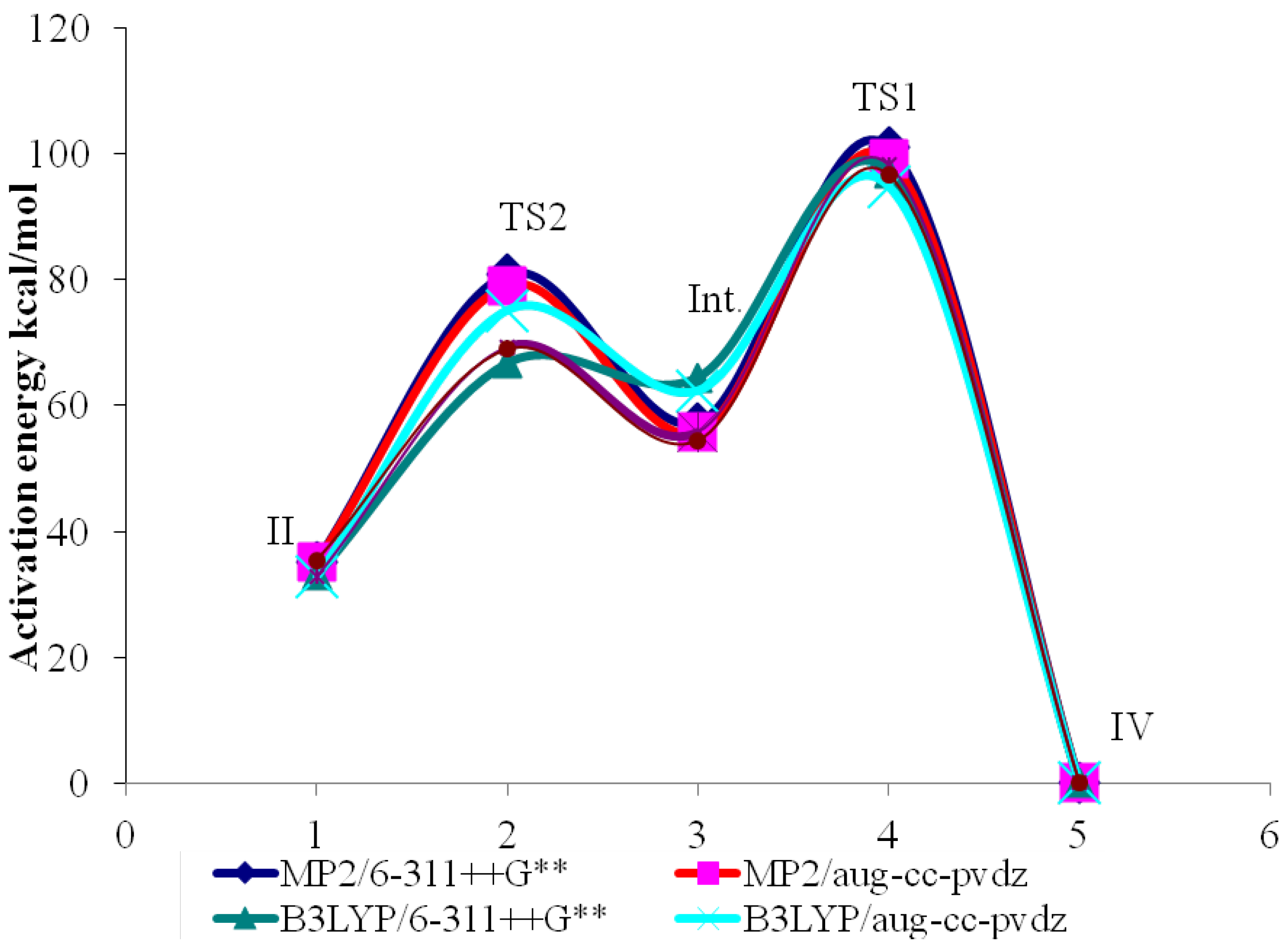
2.3. Activation Energies
| Method | Basis Set | II | TS1 | III | TS2 | IV |
|---|---|---|---|---|---|---|
| MP2 | 6-311++G** | −170.27730 | −170.148485 | −170.263702 | −170.172449 | −170.33348 |
| Activ. Energy | 80.83 | - | 57.26 | - | 101.05 | |
| aug-cc-pvdz | −170.24374 | −170.117638 | −170.230527 | −170.141694 | −170.29989 | |
| Activ. Energy | 79.13 | - | 55.74 | - | 99.27 | |
| B3LYP | 6-311++G** | −170.82975 | −170.723374 | −170.829238 | −170.728235 | −170.88290 |
| Activ. Energy | 66.75 | - | 63.38 | - | 97.05 | |
| aug-cc-pvdz | −170.80018 | −170.696274 | −170.799846 | −170.702204 | −170.85343 | |
| Activ. Energy | 65.20 | - | 61.27 | - | 94.90 | |
| CCSD | 6-311++G** | −170.29782 | −170.18736 | −170.286399 | −170.197429 | −170.35401 |
| Activ. Energy | 69.31 | - | 55.83 | - | 98.26 | |
| aug-cc-pvdz | −170.26630 | −170.15642 | −170.255619 | −170.168856 | −170.32282 | |
| Activ. Energy | 68.95 | - | 54.44 | - | 96.61 |
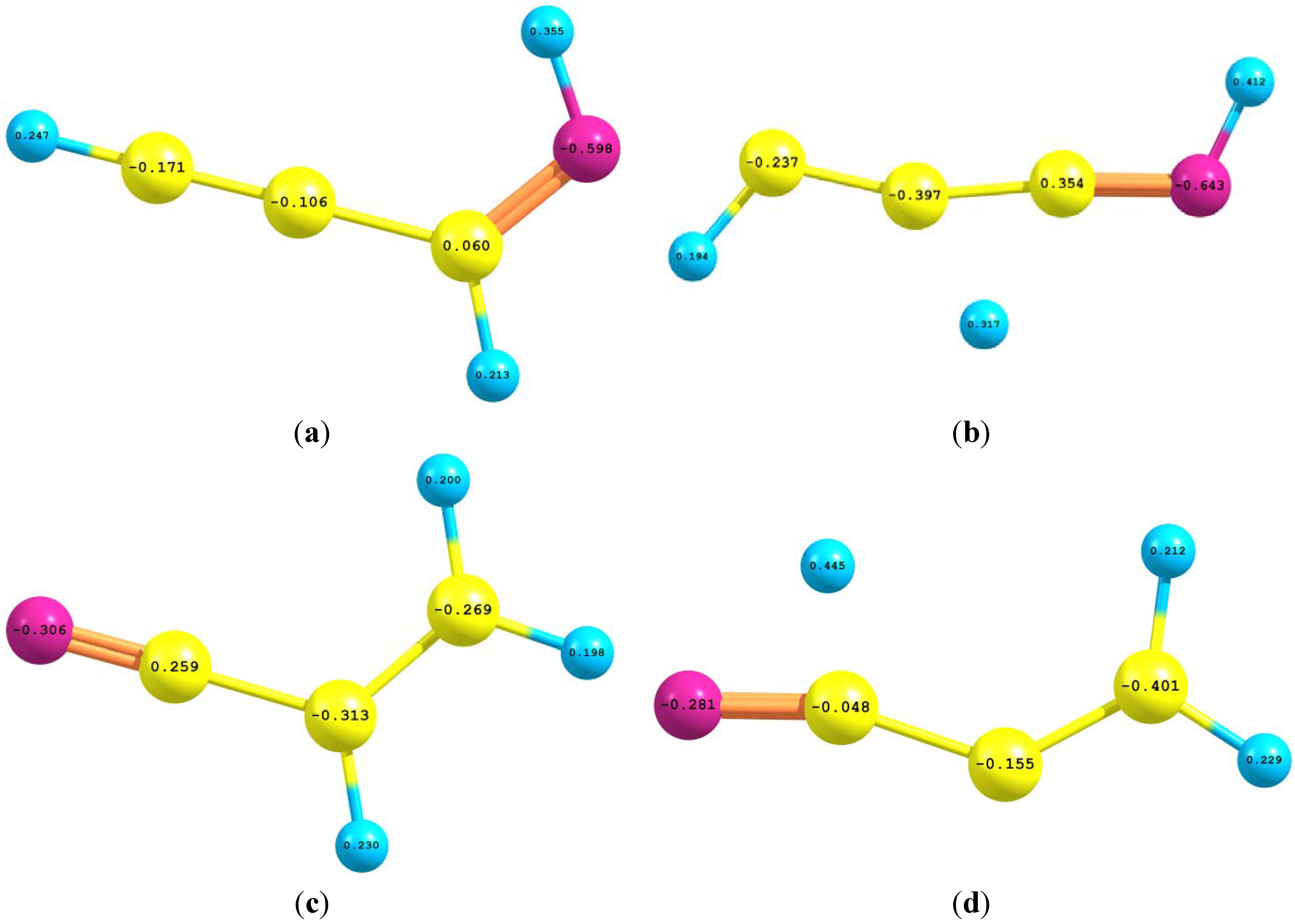
2.4. Natural Bond Orbital (NBO) Analyses

| Interaction | II | III | IV | Interaction | TS1 | TS2 |
|---|---|---|---|---|---|---|
| σC1-H2 → σ*C1-C3(σ*C4-H5) | 5.62 | 6.50 | (5.13) | σC1-C5 → σ*C3-N4 | 5.93 | 6.54 |
| σC1-H2 → σ*C3-C4 | 6.62 | 3.72 | 6.38 | πC1-C5 → π*C3-N4 | 25.16 | 53.53 |
| σC1-C3 → σ*C3-C4 | 6.31 | 11.74 | <0.50 | σC3-H2 → σ*C3-N4 | 119.30 | <0.50 |
| σC1-C3 → σ*C4-N6 | 0.67 | 5.68 | 5.28 | σC1-H7 → n*C3 | <0.50 | 269.74 |
| πC1-C3 → π*C4-N6 | 16.02 | 33.45 | 18.11 | σC5-H7 → σ*C1-C3 | 7.24 | 11.11 |
| σC3-c4 → σ*C1-C3 | 9.64 | 9.04 | <0.50 | nN → σ*C1-C3 | 11.76 | 10.71 |
| σC3-C4 → σ*C4-N6 | 1.22 | 7.33 | 4.84 | π*C3-N4 → π*C1-C5 | 40.66 | 28.89 |
| σC4-N6 → σ*C3-C4 | 1.49 | 15.31 | 4.52 | σ*C1-N4 → σ*C1-C3 | 30.39 | <0.50 |
| πC6-N7 → π*C1-C4 | <0.50 | <0.50 | 9.89 | σC1-C5 → σ*C1-C3 | <0.50 | 11.28 |
| nN → σ*C3-C4 | 13.03 | 5.95 | 12.26 | σ*C1-N4 → σC1-N4 | 10.04 | <0.50 |
| Total | 61.12 | 99.22 | 67.41 | Total | 251.48 | 393.30 |
| Level of Theory | Energy/a.u. | II | III | ΔE1 a | IV | ΔE2 b |
|---|---|---|---|---|---|---|
| RHF/6-311++G** | Total SCF energy (full) | −169.752021 | −169.737351 | +9.20 | −169.8113786 | +37.25 |
| Energy of Deletion (L) | −169.441285 | −169.302242 | +87.25 | −169.5367135 | +59.88 | |
| Delocalization energy | −0.310736 | −0.435109 | −78.05 | −0.274665 | −22.63 | |
| RHF/aug-cc-pvdz | Total SCF energy (full) | −169.729776 | −169.715694 | +8.84 | −169.7894057 | +37.42 |
| Energy of Deletion (L) | −169.439244 | −169.301429 | +86.48 | −169.5286064 | +56.08 | |
| Delocalization energy | −0.290532 | −0.414266 | −77.64 | −0.260799 | −18.66 | |
| B3LYP/6-311++G** | Total SCF energy (full) | −170.829746 | −170.829238 | +0.32 | −170.8829004 | +33.35 |
| Energy of Deletion (L) | −170.551258 | −170.431655 | +75.05 | −170.632217 | +50.80 | |
| Delocalization energy | −0.278488 | −0.397583 | −74.73 | −0.250683 | −17.45 | |
| B3LYP/aug-cc-pvdz | Total SCF energy (full) | −170.800179 | −170.799846 | +0.21 | −170.8534328 | +33.41 |
| Energy of Deletion (L) | −170.500599 | −170.420204 | +50.45 | −170.6127173 | +70.35 | |
| Delocalization energy | −0.299580 | −0.379641 | −50.24 | −0.240715 | −36.94 |
3. Computational Details
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Layer, R.W. The chemistry of imines. Chem. Rev. 1963, 63, 489–510. [Google Scholar] [CrossRef]
- Morin, M.S.T.; St-Cyr, D.J.; Arndtsen, B.A.; Krenske, E.H.; Houk, K.N. Modular mesoionics: understanding and controlling regioselectivity in 1,3-dipolar cycloadditions of münchnone derivatives. J. Am. Chem. Soc. 2013, 135, 17349–17358. [Google Scholar]
- Zhang, E.; Tian, H.; Xu, S.; Yu, X.; Xu, Q. Iron-catalyzed direct synthesis of imines from amines or alcohols and amines via aerobic oxidative reactions under air. Org. Lett. 2013, 15, 2704–2707. [Google Scholar] [CrossRef]
- Morin, M.S.T.; Lu, Y.; Black, D.A.; Arndtsen, B.A. Copper-catalyzed petasis-type reaction: A general route to α-substituted amides from imines, acid chlorides, and organoboron reagents. J. Org. Chem. 2012, 77, 2013–2017. [Google Scholar] [CrossRef]
- Warren, S.; Wyatt, P. Organic Synthesis: The Disconnection Approach, 2nd ed.; Wiley-Blackwell: Oxford, UK, 2008. [Google Scholar]
- Farkas, E.; Sunman, C.J. Chiral synthesis of doxpicomine. J. Org. Chem. 1985, 50, 1110–1112. [Google Scholar] [CrossRef]
- Green, S.; Herbst, E. Metastable Isomers: A new class of interstellar molecules. Astrophys. J. 1979, 229, 121–131. [Google Scholar] [CrossRef]
- Milligan, M.E. Infrared spectroscopic study of the photolysis of methyl azide and methyl-d 3 azide in solid argon and carbon dioxide. J. Chem. Phys. 1961, 35, 1491–1498. [Google Scholar] [CrossRef]
- Johnson, D.R.; Lovas, F.J. Microwave detection of the molecular transient methyleneimine (CH2=NH). Chem. Phys. Lett. 1972, 15, 65–68. [Google Scholar] [CrossRef]
- Godfrey, P.D.; Brown, R.D.; Robinson, B.J.; Sinclair, M.W. Discovery of interstellar methanimine (formaldimine). Astrophys. Lett. 1973, 13, 119–126. [Google Scholar]
- Jacox, M.E.; Milligan, D.E. Infrared study of the reactions of CH, and NH with C,H, and C2H, in solid argon. J. Am. Chem. Soc. 1963, 85, 278–282. [Google Scholar] [CrossRef]
- Rodler, M.; Brown, R.D.; Godfrey, P.D.; Tack, L.M. Generation, microwave spectrum and dipole moment of ketenimine. Chem. Phys. Lett. 1984, 110, 447–451. [Google Scholar] [CrossRef]
- Lovas, F.J.; Hollis, J.M.; Remijan, A.J.; Jewell, P.R. Detection of ketenimine (CH2=C=NH) in sagittarius B2(N) hot cores. Astrophys. J. Lett. 2006, 645, L137–L140. [Google Scholar] [CrossRef]
- Brown, R.D.; Rice, E.H.N.; Rodler, M. Ab initio studies of the structures and force fields of ketenimine and related molecules. Chem. Phys. 1985, 99, 347–356. [Google Scholar] [CrossRef]
- Kessler, M.; Ring, H.; Trambarulo, R.; Gordy, W. Microwave spectra and molecular structures of methyl cyanide and methyl isocyanide. Phys. Rev. 1950, 79, 54–56. [Google Scholar] [CrossRef]
- Matthews, H.E.; Sears, T.J. Detection of vinyl cyanide in TMC-1. Astrophys. J. 1984, 272, 149–153. [Google Scholar] [CrossRef]
- Pearson, J.C.; Müller, H.S.P. The submillimeter wave spectrum of isotopic methyl cyanide. Astrophys. J. 1996, 471, 1067–1072. [Google Scholar]
- Remijan, A.; Sutton, E.C.; Snyder, L.E.; Friedel, D.N.; Liu, S.-Y; Pei, C.-C. High-resolution observations of methyl cyanide (CH3CN) toward the hot core regions W51e1/e2. Astrophys. J. 2004, 606, 971–976. [Google Scholar]
- Cernicharo, J.; Kahane, C.; Guelin, M.; Gomez-Gonzalez, J. Tentative detection of CH3NC towards Sgr B2. Astron. Astrophys. 1988, 189, L1–L2. [Google Scholar]
- Remijan, A.; Hollis, J.M.; Lovas, F.J.; Plusquellic, D.F.; Jewell, P.R. Interstellar isomers: The importance of bonding energy differences. Astrophys. J. 2005, 632, 333–339. [Google Scholar] [CrossRef]
- Wilcox, W.S.; Goldstein, J.H.; Simmons, J.W. The microwave spectrum of vinyl cyanide. J. Chem. Phys. 1954, 22, 516–518. [Google Scholar]
- Costain, C.C.; Stoicheff, B.P. Microwave spectrum, molecular structure of vinyl cyanide and a summary of CC, CH bond lengths in simple molecules. J. Chem. Phys. 1959, 30, 777–782. [Google Scholar] [CrossRef]
- Demaison, J.; Cosleou, J.; Bocquet, R.; Lesarri, A.G. Submillimeter-wave spectrum and structure of acrylonitrile. J. Mol. Spectrosc. 1994, 167, 400–418. [Google Scholar] [CrossRef]
- Colmont, J.M.; Wlodarczak, G.; Priem, D.; Muller, H.S.P.; Tien, E.H.; Richards, R.J.; Gerry, M.C.L. Rotational spectra of selected isotopic species of vinyl cyanide: Molecular structure and quadrupole hyperfine structure. J. Mol. Spectrosc. 1997, 181, 330–344. [Google Scholar] [CrossRef]
- Stolze, M.; Sutter, D.H. Molecular g-values, magnetic susceptibility anisotropies, molecular electric quadrupole moments, improved molecular electric dipole moments and 14N-quadrupole coupling constants of acrylonitrile, H2C=CH-C≡N, and the magnetic susceptibility tensor of the nitrile group. Z. Naturforschung A 1985, 40, 998–1010. [Google Scholar]
- Nummelin, A.; Bergman, P. Vibrationally excited vinyl cyanide in Sgr B2(N). Astron. Astrophys. 1999, 341, L59–L60. [Google Scholar]
- Agundez, M.; Fonfria Exposito, J.P.; Cernicharo, J.; Pardo, J.R.; Guelin, M. Detection of circumstellar CH2CHCN, CH2CN, CH3CCH, and H2CS. Astron. Astrophys. 2008, 479, 493–501. [Google Scholar] [CrossRef]
- Kroto, H.W.; McNaughton, D.; Osman, O.I. The detection of the new molecule prop-2-ynylidineamine, HC≡CCH=NH by microwave spectroscopy. J. Chem. Soc. Chem. Commun. 1984, 993–994. [Google Scholar]
- Sugie, M.; Takeo, H.; Matsumura, C. Microwave spectra, nuclear quadrupole coupling constants, dipole moments, and rotational isomers of propargylimine. J. Mol. Spectrosc. 1985, 111, 83–92. [Google Scholar] [CrossRef]
- McNaughton, D.; Osman, O.I.; Kroto, H.W. The microwave spectrum and structure of Z-prop-2-ynylideneamine, HC≡CCH=NH. J. Mol. Struct. 1988, 190, 195–204. [Google Scholar] [CrossRef]
- Hamada, Y.; Takeo, M.H.; Matsumura, C. Pyrolysis of amines: Infrared spectrum of propargylimine. J. Mol. Spectrosc. 1984, 106, 175–185. [Google Scholar] [CrossRef]
- Osman, O.I. Microwave, Infrared and Photoelectron Studies of Unstable Molecules. Ph.D. Thesis, University of Sussex, Brighton, UK, 1986. [Google Scholar]
- Osman, O.I. The production of prop-2-ynylideneamine by thermolysis of N-chloropropargylamine, N-Fluoropropargylamine and N-Hydroxypropargylamine: A computational study. Mol. Phys. 2014, 112, 304–315. [Google Scholar] [CrossRef]
- Kroto, H.W.; Little, L.; McNaughton, D.; Osman, O.I. Microwave, photoelectron, infrared and radioastronomy study of prop-2-ynylidineamine H-C≡C-CH=NH. In Proceedings of the Ninth Colloquium on High Resolution Molecular Spectroscopy, Riccione, Italy, 16–20 September 1985; pp. 204–234.
- Basak, A.; Gupta, S.N.; Chakrabarty, K.; Das, G.K. New bimolecular mechanistic pathway for the 1,3-hydrogen Shift in allenamide and allene system: A theoretical prediction. Comput. Theor. Chem. 2013, 1007, 15–30. [Google Scholar]
- Watts, J.D.; Watts, D.J.; Huang, M-L. Theoretical study of the tautomerism, structures and vibrational frequencies of the phosphalkenes XP=C(CH3)2 (X = H, F, Cl, Br, OH, ArF (ArF = 2,6-(CF3)2C6H3)). J. Chem. Phys. A 2009, 113, 1886–1891. [Google Scholar] [CrossRef]
- Yasumoto, M.; Ueki, H.; Soloshonok, V.A. Thermal 1,3-proton shift reaction and its application for operationally convenient and improved synthesis of α-(trifluoromethyl)benzylamine). J. Fluor. Chem. 2007, 128, 736–739. [Google Scholar] [CrossRef]
- Osman, O.I.; Elroby, S.A.K.; Hilal, R.H.; Aziz, S.G. Theoretical characterization of gas-phase thermolysis products of ethane-1,2-diol, 2-chloroethanol and 2-fluoroethanol. Mol. Phys. 2013, 111, 643–659. [Google Scholar] [CrossRef]
- De Vicente, P.; Martin-Pintado, J.; Wilson, T.L. A hot ring in the Sgr B2 molecular cloud. In Proceedings Astronomical Society of the Pacific Conference Series; Astronomical Society of the Pacific: La Serena, Chile, 2007; pp. 64–67. [Google Scholar]
- Pratap, P.; Dickens, J.E.; Snell, R.L.; Miralles, M.P.; Bergin, E.A.; Irvine, W.N.; Schloerb, F.B. A study of the physics and chemistry of TMC1. Astrophys. J. 1997, 486, 862–885. [Google Scholar] [CrossRef]
- Zaleski, D.P.; Seifert, N.A.; Steber, A.L.; Muckle, M.T.; Loomis, R.A.; Corby, J.F.; Martinez, J.R.O.; Crabtree, K.N.; Jewell, P.R.; Hollis, J.M.; et al. Detection of E-cyanomethanimine toward sagittarius B2(N) in the green bank telescope primos survey. Astrophys. J. Lett. 2013, 765, L10:1–L10:6. [Google Scholar] [CrossRef]
- Loomis, R.A.; Zaleski, D.P.; Steber, A.L.; Neil, J.L.; Muckle, M.T.; Harris, B.J.; Martinez, J.R.O.; Jewell, P.R.; Lattanzi, V.; Hollis, J.M.; et al. The Detection of Interstellar Ethanimine (Ch3chnh) from Observations Taken during the Gbt Primos Survey. Astrophys. J. Lett. 2013, 765, L9:1–L9:7. [Google Scholar] [CrossRef]
- Reed, E.A.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Reed, E.A.; Weinhold, F. Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Song, L.; Lin, Y.; Wu, W.; Zhang, Q.; Mo, Y. Steric strain versus hyperconjugative stabilization in ethane congeners. J. Phys. Chem. A 2005, 109, 2310–2316. [Google Scholar] [CrossRef]
- Lovas, F.J.; Suenram, R.D.; Johnson, D.R.; Clark, F.O.; Tiemann, E. Pyrolysis of ethylamine. II. Synthesis and microwave spectrum of ethylidenimine (CH3CH = NH). J. Chem. Phys. 1980, 72, 4964–4972. [Google Scholar] [CrossRef]
- Guennec, M.L.; Wlodarczak, G.; Burie, J.; Demaison, J. Rotational spectrum of CH2DCN and structure of methyl cyanide. J. Mol. Spectrosc. 1992, 154, 305–323. [Google Scholar]
- Pophristic, V.; Goodman, L. Hyperconjugation not steric repulsion leads to the staggered structure of ethane. Nature 2001, 411, 565–568. [Google Scholar] [CrossRef]
- Glendenning, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1.; Gaussian Inc.: Pittsburg, PA, USA, 2001. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Peng, C.; Schlegel, H.B. Combining synchronous transit and quasi-newton methods to find transition states. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical-reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Geomodeling in GeoGraphix. Available online: http://www.chemcraftprog.com (accessed on 26 May 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Osman, O.I.; Elroby, S.A.; Aziz, S.G.; Hilal, R.H. Does Prop-2-ynylideneamine, HC≡CCH=NH, Exist in Space? A Theoretical and Computational Investigation. Int. J. Mol. Sci. 2014, 15, 11064-11081. https://doi.org/10.3390/ijms150611064
Osman OI, Elroby SA, Aziz SG, Hilal RH. Does Prop-2-ynylideneamine, HC≡CCH=NH, Exist in Space? A Theoretical and Computational Investigation. International Journal of Molecular Sciences. 2014; 15(6):11064-11081. https://doi.org/10.3390/ijms150611064
Chicago/Turabian StyleOsman, Osman I., Shaaban A. Elroby, Saadullah G. Aziz, and Rifaat H. Hilal. 2014. "Does Prop-2-ynylideneamine, HC≡CCH=NH, Exist in Space? A Theoretical and Computational Investigation" International Journal of Molecular Sciences 15, no. 6: 11064-11081. https://doi.org/10.3390/ijms150611064
APA StyleOsman, O. I., Elroby, S. A., Aziz, S. G., & Hilal, R. H. (2014). Does Prop-2-ynylideneamine, HC≡CCH=NH, Exist in Space? A Theoretical and Computational Investigation. International Journal of Molecular Sciences, 15(6), 11064-11081. https://doi.org/10.3390/ijms150611064