High-Salt Intake Suppressed MicroRNA-133a Expression in Dahl SS Rat Myocardium

 ,
,
Abstract
:
1. Introduction
2. Results
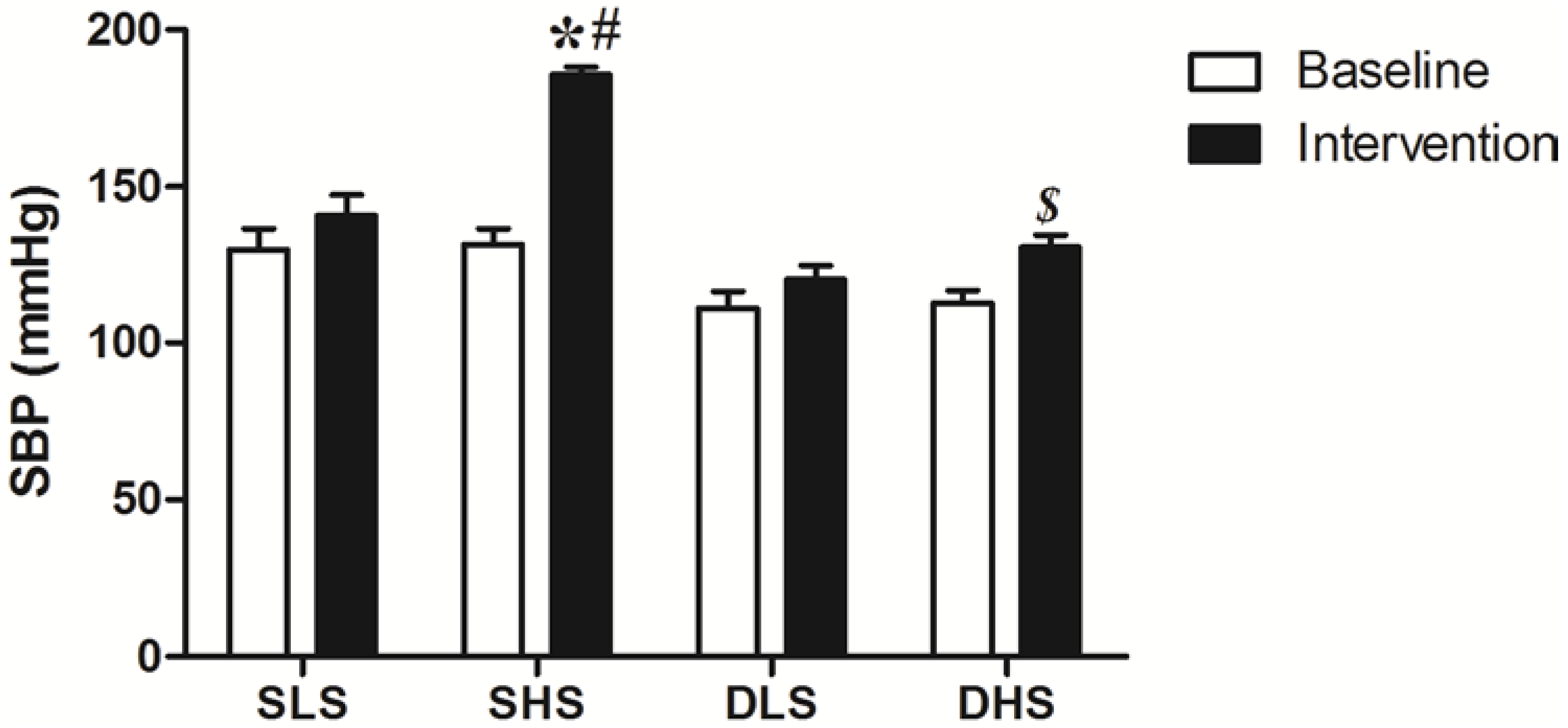
2.1. Effect of High-Salt Intake on Blood Pressure

2.2. Effects of High-Salt Intake on Left Ventricular Mass (LVW) and LVW Index (LVWI)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group (n = 8) | Body Weight (g) | LVM (mg) | LVMI (mg/g) |
|---|---|---|---|
| SLS | 263.53 ± 14.36 | 755.11 ± 29.84 | 2.87 ± 0.11 |
| SHS | 290.45 ± 10.25 | 932.72 ± 53.97 * | 3.21 ± 0.09 * |
| DLS | 250.37 ± 23.04 | 749.84 ± 49.36 | 2.84 ± 0.11 |
| DHS | 265.83 ± 10.93 | 802.87 ± 59.73 | 2.96 ± 0.12 |
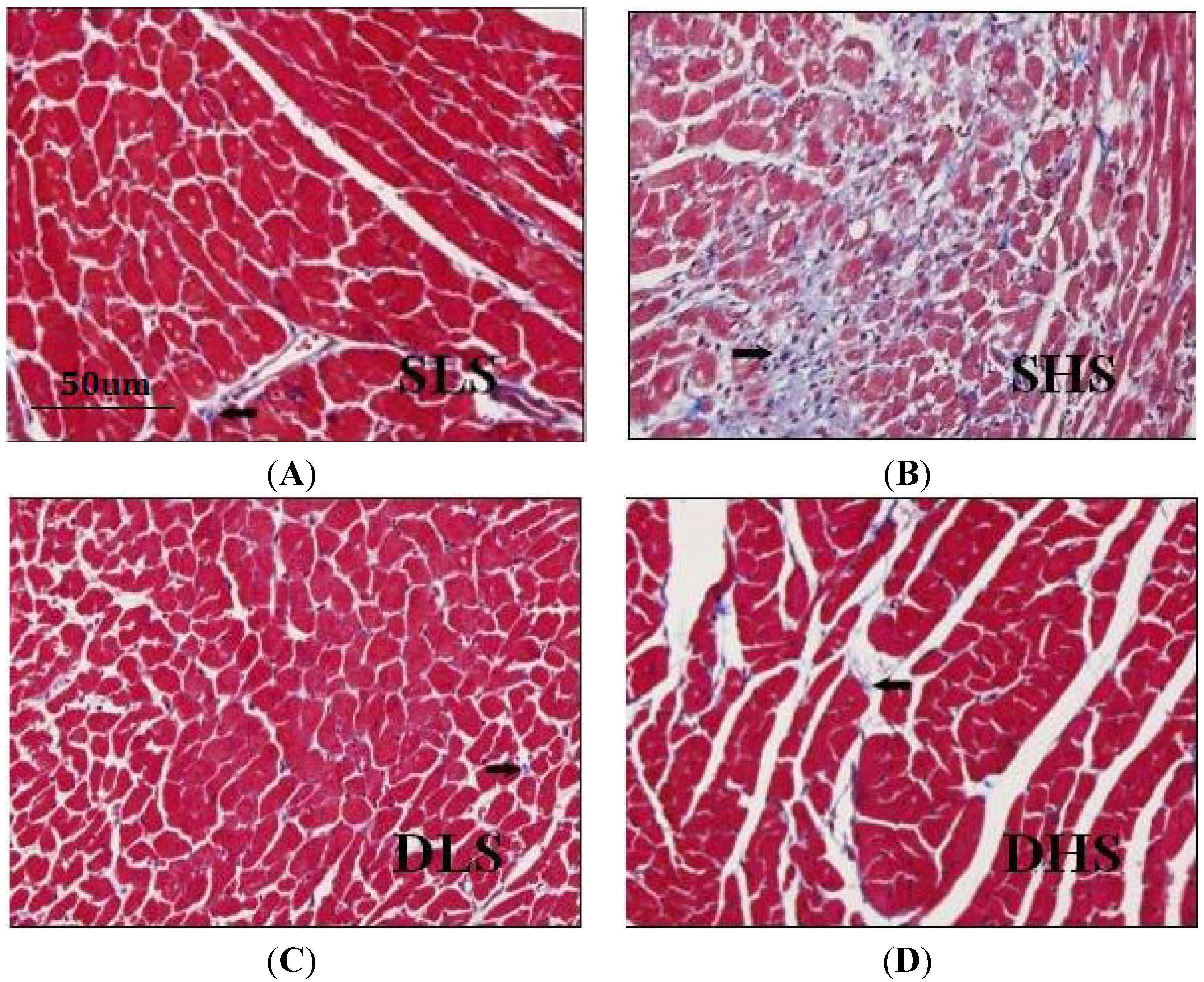
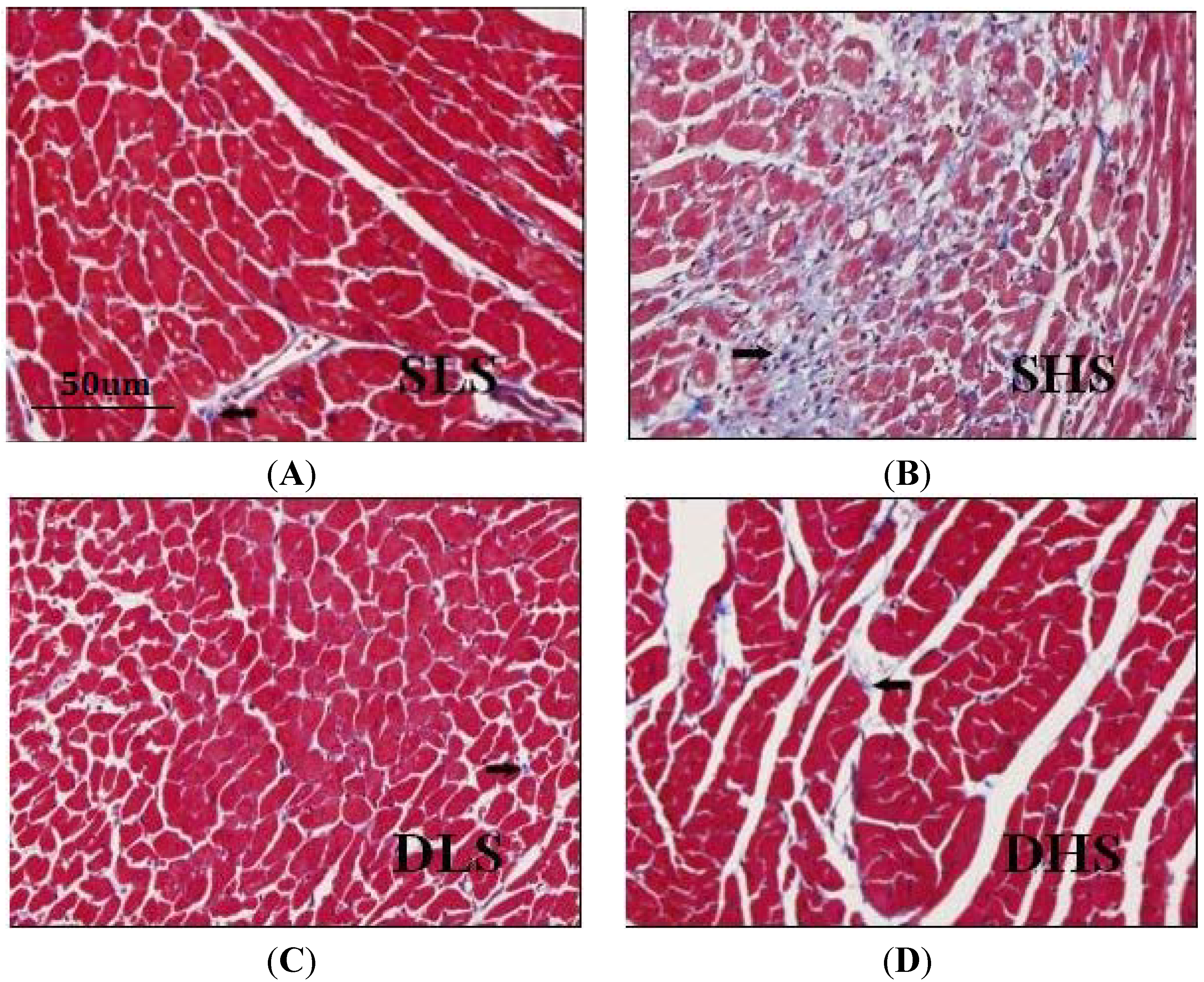
2.3. Changes in Collagen I after High-Salt Intervention

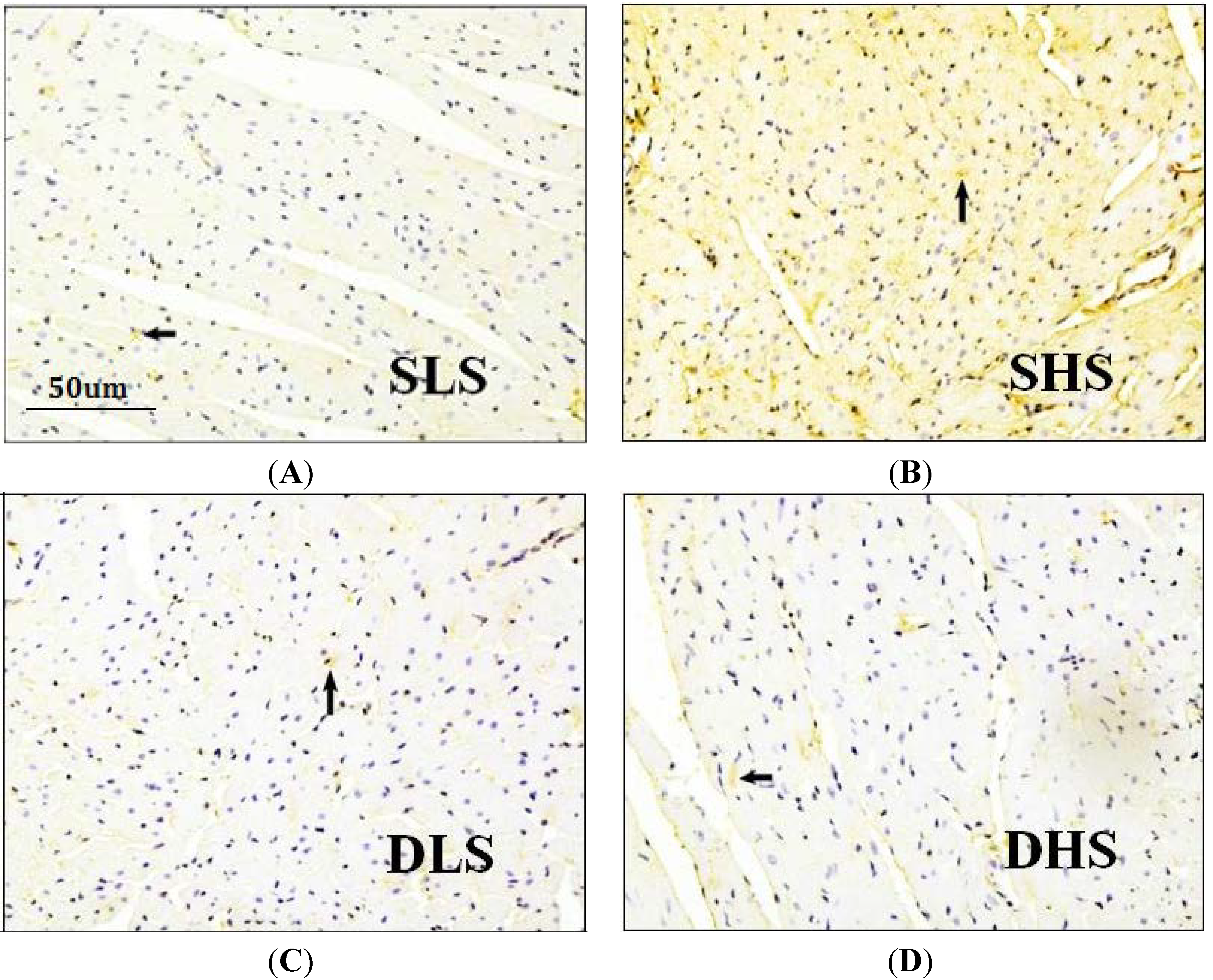
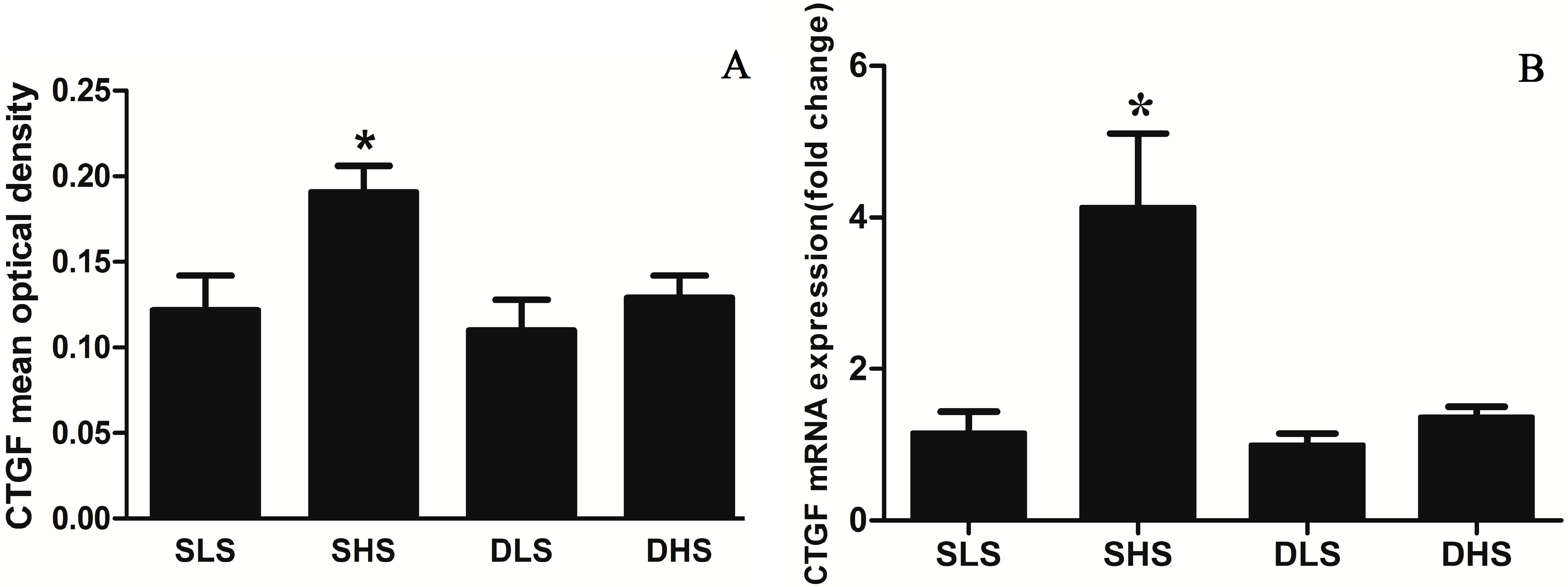
2.4. Effects of High-Salt Intake on Connective Tissue Growth Factor (CTGF) Expression
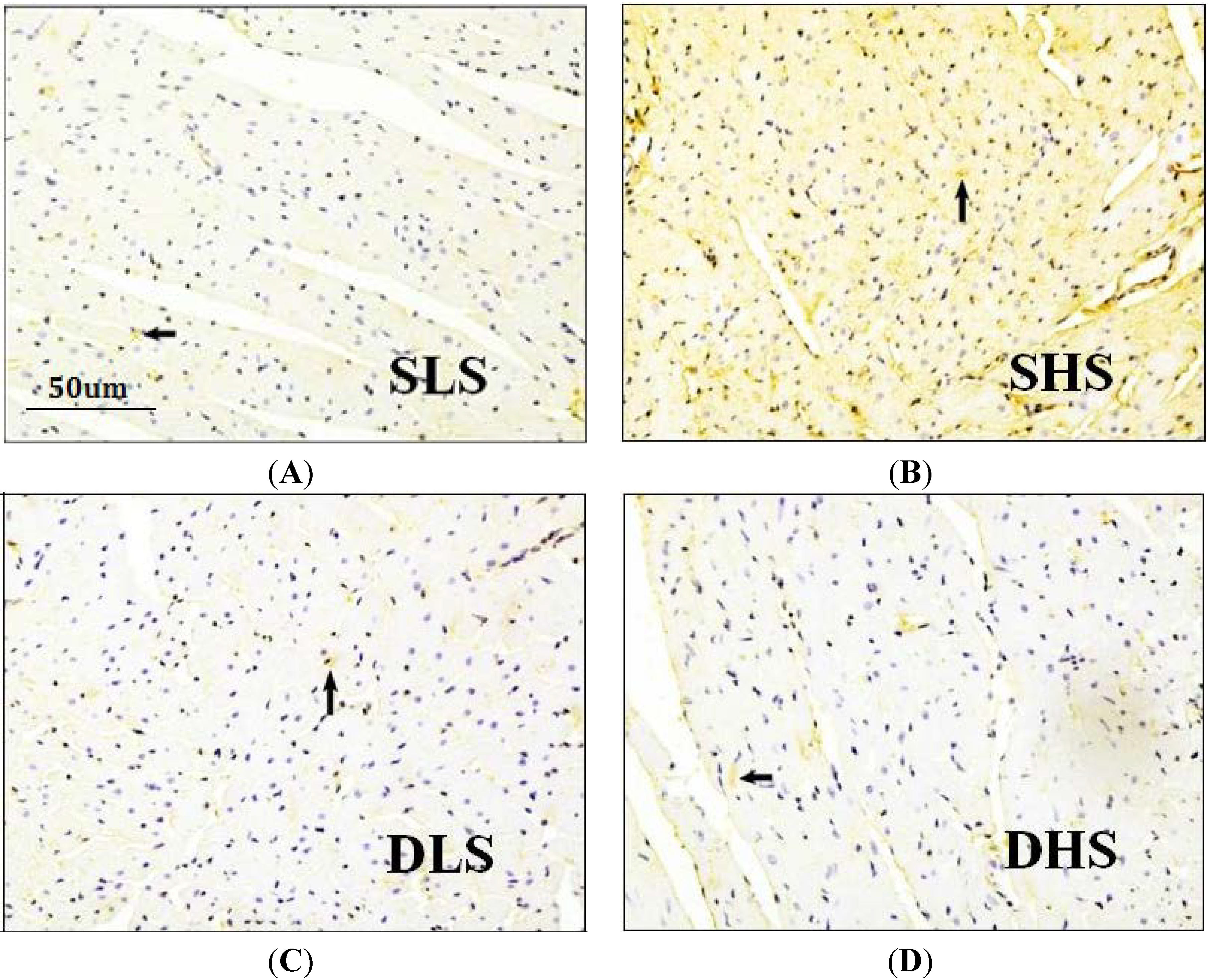
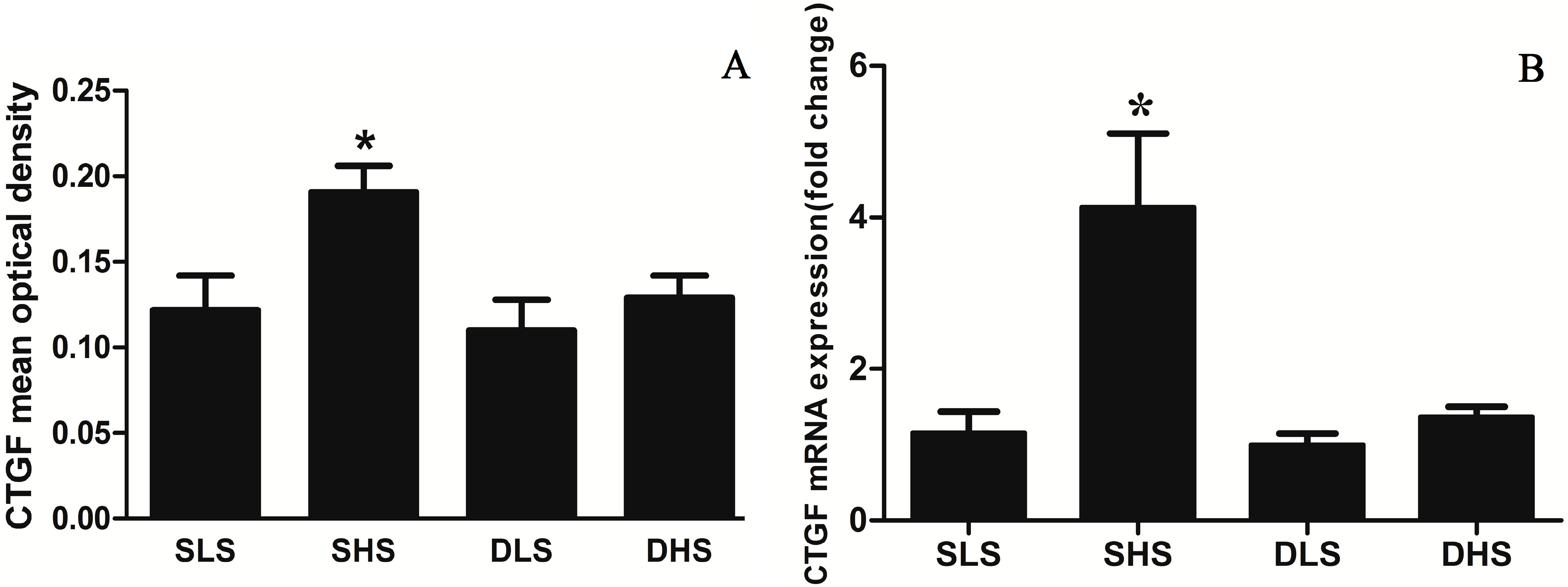
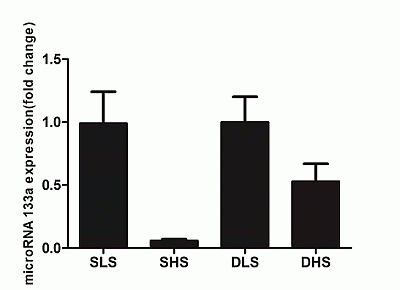
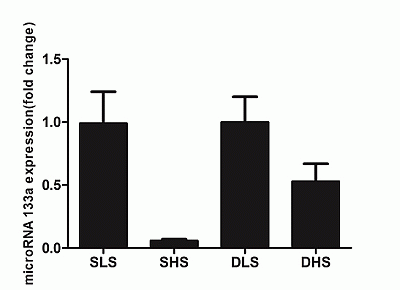
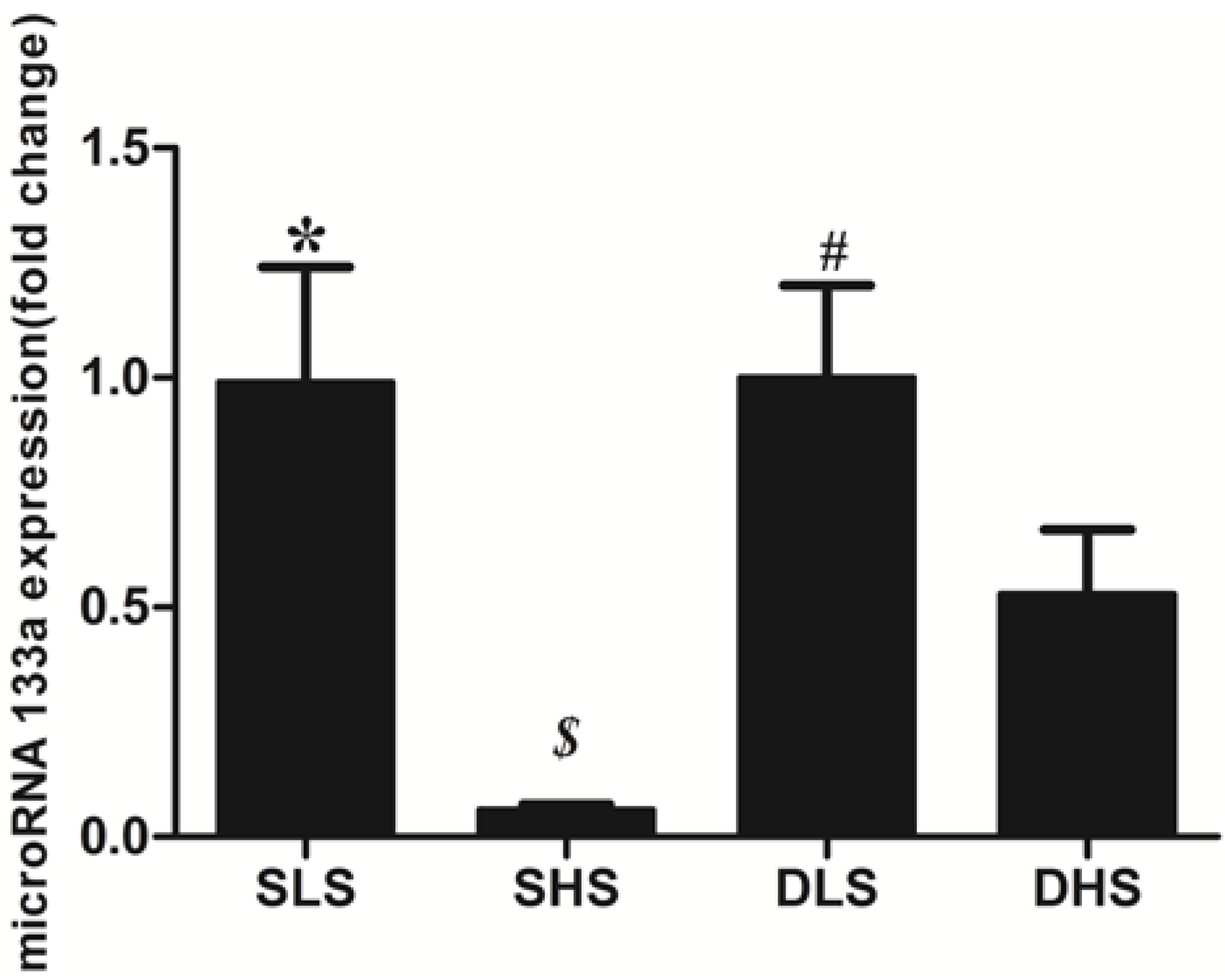
2.5. Changes in MicroRNA-133a after the Intervention
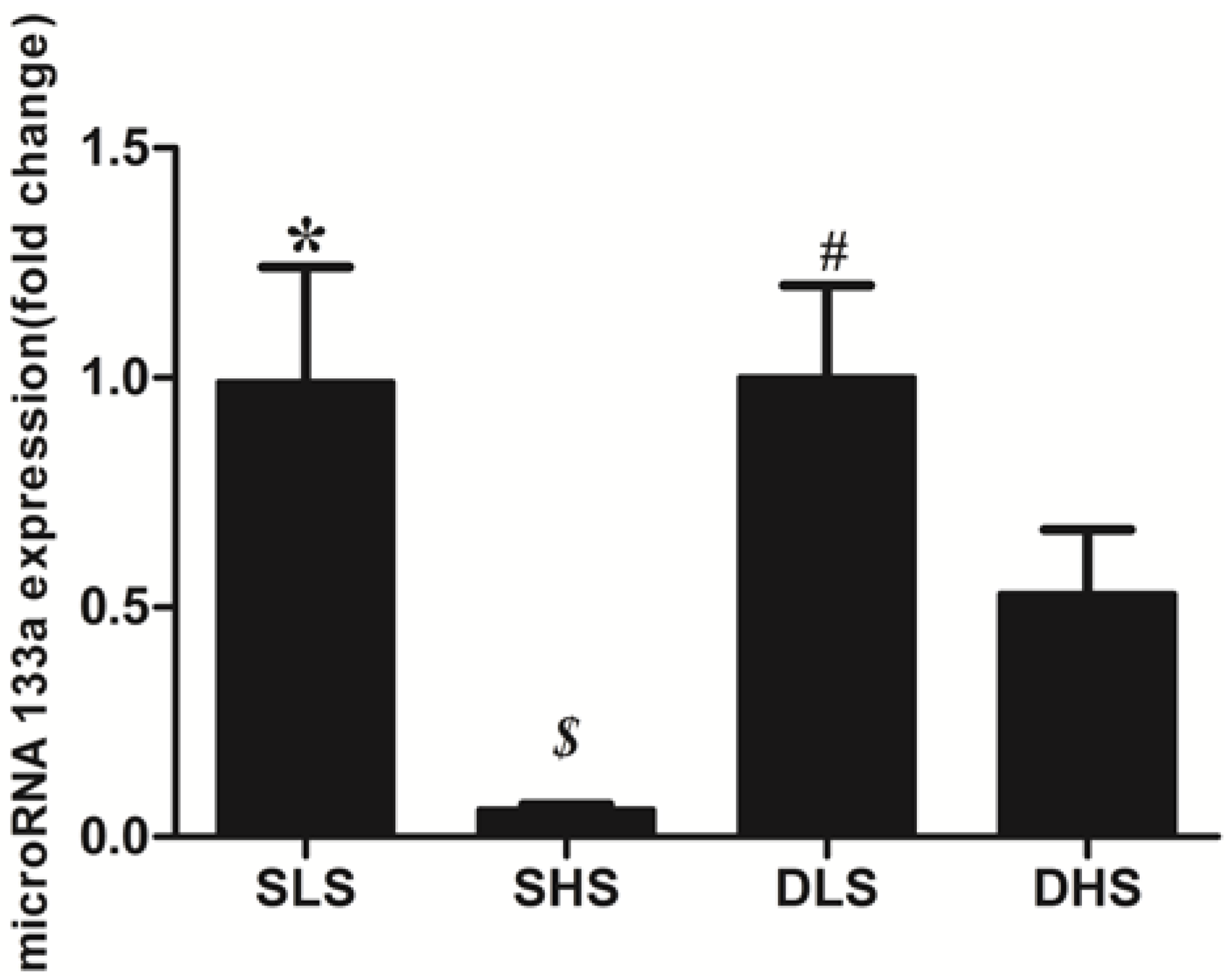
3. Discussion
4. Materials and Methods
4.1. Subjects and Protocol
4.2. Experimental Procedures
4.3. Histological Examination of the Heart
4.4. Immunohistochemistry
4.5. Real-Time Polymerase Chain Reaction (PCR)
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poteshkina, N.G. Salt consumption. Cardiovascular system as a target organ. Part III. Russ. J. Cardiol. 2012, 6, 84–90. [Google Scholar]
- Li, X.-Y.; Cai, X.-L.; Bian, P.-D.; Hu, L.-R. High salt intake and stroke: Meta-analysis of the epidemiologic evidence. CNS Neurosci. Ther. 2012, 18, 691–701. [Google Scholar] [CrossRef]
- Kimura, G.; Dohi, Y.; Fukuda, M. Salt sensitivity and circadian rhythm of blood pressure: The keys to connect ckd with cardiovasucular events. Hypertens. Res. 2010, 33, 515–520. [Google Scholar] [CrossRef]
- Young, M.J.; Rickard, A.J. Mechanisms of mineralocorticoid salt-induced hypertension and cardiac fibrosis. Mol. Cell. Endocrinol. 2012, 350, 248–255. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef]
- Xiao, J.; Luo, X.; Lin, H.; Zhang, Y.; Lu, Y.; Wang, N.; Zhang, Y.; Yang, B.; Wang, Z. MicroRNA miR-133 represses herg K+ channel expression contributing to QT prolongation in diabetic hearts. J. Biol. Chem. 2011, 286, 28656. [Google Scholar]
- Xu, C.; Lu, Y.; Pan, Z.; Chu, W.; Luo, X.; Lin, H.; Xiao, J.; Shan, H.; Wang, Z.; Yang, B. The muscle-specific micrornas miR-1 and miR-133 produce opposing effects on apoptosis by targeting Hsp60, Hsp70 and caspase-9 in cardiomyocytes. J. Cell Sci. 2007, 120, 3045–3052. [Google Scholar] [CrossRef]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar] [CrossRef]
- Care, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Shan, H.; Zhang, Y.; Lu, Y.; Pan, Z.; Cai, B.; Wang, N.; Li, X.; Feng, T.; Hong, Y.; Yang, B. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc. Res. 2009, 83, 465–472. [Google Scholar] [CrossRef]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of micrornas in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef]
- Castoldi, G.; di Gioia, C.R.T.; Bombardi, C.; Catalucci, D.; Corradi, B.; Gualazzi, M.G.; Leopizzi, M.; Mancini, M.; Zerbini, G.; Condorelli, G.; et al. mir-133a regulates collagen 1A1: Potential role of mir-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J. Cell. Physiol. 2012, 227, 850–856. [Google Scholar]
- Huang, B.S.; Leenen, F.H.H. Both brain angiotensin II and “ouabain” contribute to sympathoexcitation and hypertension in dahl s rats on high salt intake. Hypertension 1998, 32, 1028–1033. [Google Scholar] [CrossRef]
- Heggermont, W.A.; Heymans, S. MicroRNAs are involved in end-organ damage during hypertension. Hypertension 2012, 60, 1088–1093. [Google Scholar] [CrossRef]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; de Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar] [CrossRef]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.-H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and MEF2A genes. Mol. Cell. Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Lyon, A.R.; Volkmann, I.; Mills, A.M.; Bretthauer, J.; Pahuja, A.; Geers-Knoerr, C.; Kraft, T.; Hajjar, R.J.; Macleod, K.T.; et al. SERCA2A gene therapy restores microRNA-1 expression in heart failure via an Akt/FOXO3A-dependent pathway. Eur. Heart J. 2012, 33, 1067–1075. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Chen, M.M.; Lam, A.; Abraham, J.A.; Schreiner, G.F.; Joly, A.H. CTGF expression is induced by TGF-β in cardiac fibroblasts and cardiac myocytes: A potential role in heart fibrosis. J. Mol. Cell. Cardiol. 2000, 32, 1805–1819. [Google Scholar] [CrossRef]
- Varagic, J.; Ahmad, S.; Voncannon, J.L.; Moniwa, N.; Simington, S.W., Jr.; Brosnihan, B.K.; Gallagher, P.E.; Habibi, J.; Sowers, J.R.; Ferrario, C.M. Nebivolol reduces cardiac angiotensin II, associated oxidative stress and fibrosis but not arterial pressure in salt-loaded spontaneously hypertensive rats. J. Hypertens. 2012, 30, 1766–1774. [Google Scholar]
- Varagic, J.; Frohlich, E.D.; Susic, D.; Ahn, J.; Matavelli, L.; Lopez, B.; Diez, J. AT1 receptor antagonism attenuates target organ effects of salt excess in shrs without affecting pressure. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H853–H858. [Google Scholar] [CrossRef]
- Ferreira, D.N.; Katayama, I.A.; Oliveira, I.B.; Rosa, K.T.; Furukawa, L.N.S.; Coelho, M.S.; Casarini, D.E.; Heimann, J.C. Salt-induced cardiac hypertrophy and interstitial fibrosis are due to a blood pressure-independent mechanism in wistar rats. J. Nutr. 2010, 140, 1742–1751. [Google Scholar] [CrossRef]
- Bayorh, M.A.; Ganafa, A.A.; Emmett, N.; Socci, R.R.; Eatman, D.; Fridie, I.L. Alterations in aldosterone and angiotensin ii levels in salt-induced hypertension. Clin. Exp. Hypertens. 2005, 27, 355–367. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, T.-S.; Zhang, J.; Mu, J.-J.; Liu, F.-Q.; Yuan, Z.-Y.; Ren, K.-Y.; Wang, D. High-Salt Intake Suppressed MicroRNA-133a Expression in Dahl SS Rat Myocardium. Int. J. Mol. Sci. 2014, 15, 10794-10805. https://doi.org/10.3390/ijms150610794
Guo T-S, Zhang J, Mu J-J, Liu F-Q, Yuan Z-Y, Ren K-Y, Wang D. High-Salt Intake Suppressed MicroRNA-133a Expression in Dahl SS Rat Myocardium. International Journal of Molecular Sciences. 2014; 15(6):10794-10805. https://doi.org/10.3390/ijms150610794
Chicago/Turabian StyleGuo, Tong-Shuai, Jie Zhang, Jian-Jun Mu, Fu-Qiang Liu, Zu-Yi Yuan, Ke-Yu Ren, and Dan Wang. 2014. "High-Salt Intake Suppressed MicroRNA-133a Expression in Dahl SS Rat Myocardium" International Journal of Molecular Sciences 15, no. 6: 10794-10805. https://doi.org/10.3390/ijms150610794