Viral Metagenomics on Animals as a Tool for the Detection of Zoonoses Prior to Human Infection?


Abstract
:1. Introduction
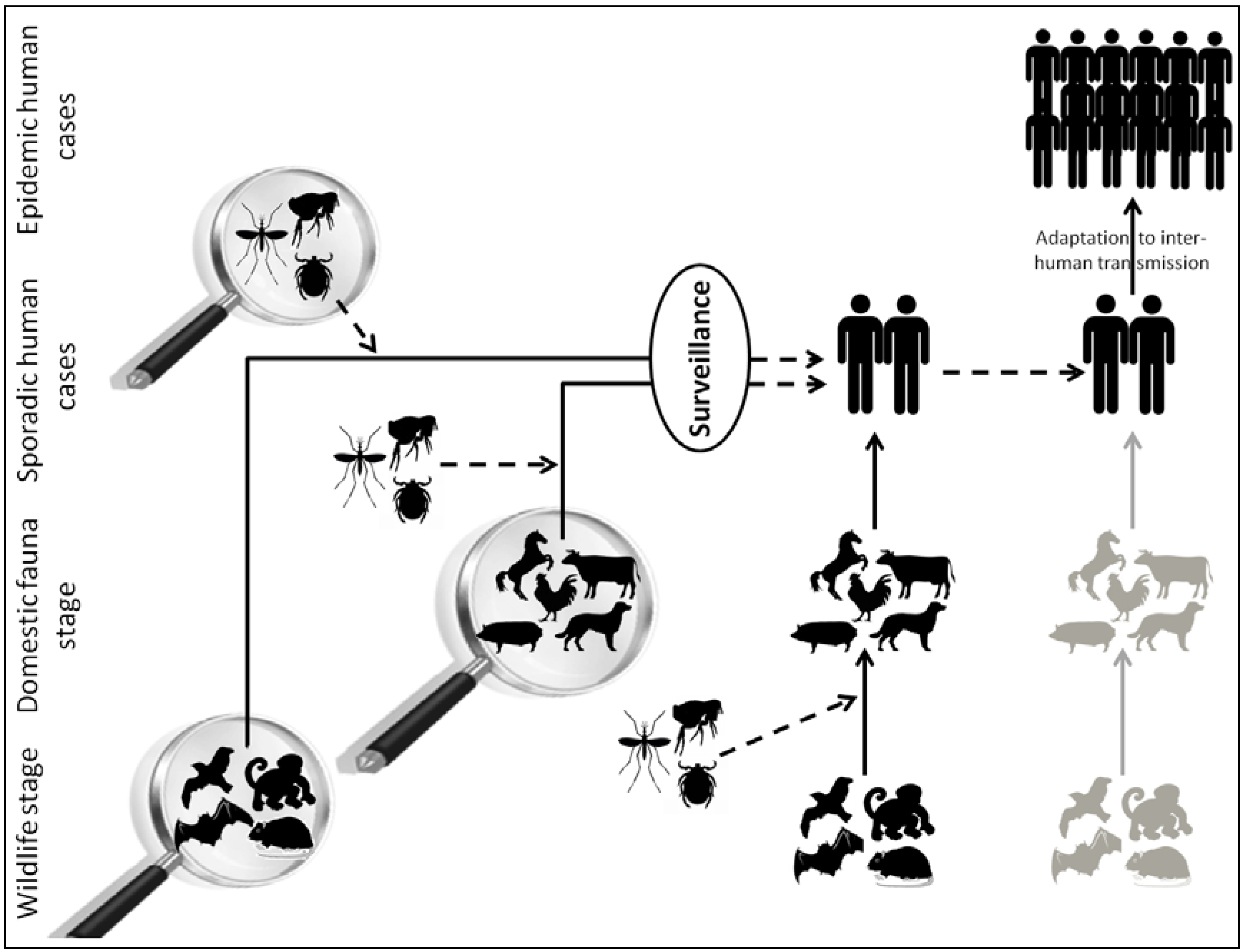
2. Viral Metagenomics: A Powerful Technique to Inventory the Viral Diversity among Complex Environments
3. Blood-Feeding Arthropods
3.1. Zoonotic Viruses
3.2. Insect-Specific Viruses

{kind=link}
| Kingdom | Class | Order | Family | Type Arthropod | Example of Viral Families | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Arthropoda | Arachnida | Ixodida | Ixodidae | Hard tick | Flaviviridae (TBEV, OHFV) Bunyaviridae (CCHFV) Reoviridae (CTFV) | [29,30] | ||||||
| Argasidae | Soft tick | Flaviviridae (AHFV, SREV, WNV), Bunyaviridae (SOLV) | [31,32,33,34] | |||||||||
| Insecta | Anoploura | Pediculidae | Louse | not documented | ||||||||
| Siphonaptera | Pulicidae | Flea | Flaviviridae (TBEV) | [35] | ||||||||
| Hemiptera | Cimicidae | Bed bug | Bunyaviridae (KKV) Hepadnaviridae (HBV) | [36,37,38,39] | ||||||||
| Reduviidae | Triatoma | not documented | ||||||||||
| Arthropoda | Insecta | Diptera | Simuliidae | Black fly | not documented | |||||||
| Tabanidae | Horse fly | Bunyaviridae (LACV, JCV) | [40,41] | |||||||||
| Psychodidae | Sand fly | Bunyaviridae (SFNV, TOSV) Flaviviridae, Rhabdoviridae | [31,42,43,44,45,46] | |||||||||
| Muscidae | Tsetse fly | not documented | ||||||||||
| Culicidae | Mosquito | Flaviviridae (WNV, YFV, DENV), Togaviridae (CHIKV, ONNV), Bunyaviridae (RVFV, NRIV) | [47,48,49,50] | |||||||||
| Ceratopogonidae | Biting midge | Bunyaviridae (OROV, RVFV, CCHFV) | [51,52,53] | |||||||||
| Arthropod Species | Type Study | Viral Reads Taxonomic Assignation | Ref. | |||
|---|---|---|---|---|---|---|
| Animal Viruses | Insect-Specific Viruses | Plant Viruses | Phages | |||
| Mixed-species female mosquitoes | DNA virome | Anelloviridae, Circoviridae, Herpesviridae, Poxviridae, Papillomaviridae | Parvoviridae (Densovirinae), Poxviridae (Entomopoxvirinae), Iridoviridae | Geminiviridae, Nanoviridae | Myoviridae, Podoviridae, Siphoviridae | [21] |
| Culex pipiens molestus | Small RNA virome | not documented | Parvoviridae (Densovirinae) | not documented | not documented | [25] |
| Aedes sp. | DNA/RNA virome | not documented | Parvoviridae (Densovirinae) | not documented | Inoviridae | [24] |
| Anopheles sp., Ochlerotatus sp. | Small RNA virome | Reoviridae (Orbivirus) * | Chronic bee paralysis virus, Birnaviridae (Entomobirnavirus), Flaviviridae *, Bunyaviridae ** (Phlebovirus) | Narnaviridae, Partitiviridae * | not documented | [28] |
| Anopheles sp., Culex sp., Aedes sp. | DNA/RNA virome | Rhabdoviridae, Bunyaviridae, Flaviviridae, Reoviridae, Togaviridae | not documented | not documented | not documented | [22] |
4. Wildlife
4.1. Bats
4.2. Rodents
| Wildlife | Zoonosis | Virus | Vector-Based Transmission | Domestic Animal Intermediate Host | Ref. |
|---|---|---|---|---|---|
| Bat | Nipah/Hendra | Paramyxoviridae, Henipavirus | No | Pig/horse | [63] |
| Ebola hemorrhagic fever | Filoviridae, Ebolavirus, EBOV | No | No | [64] | |
| Severe acute respiratory syndrome (SARS) | Coronaviridae, Betacoronavirus, SARS-CoV | No | Civet, cat | [65] | |
| Rabies | Rhabdoviridae, Lyssavirus, RABV | No | Dog | [66] | |
| Rodent | Lymphocytic choriomeningitis | Arenaviridae, Arenavirus, LCMV | No | No | [67] |
| Lassa hemorrhagic fever | Arenaviridae, Arenavirus, LASV | No | No | [67] | |
| Pulmonary syndrome and hemorrhagic syndrome | Bunyaviridae, Hantavirus | No | No | [68,69] | |
| Bird | Japanese encephalitis | Flaviviridae, Flavivirus, JEV | Yes (mosquitoes) | Swine | [70] |
| West Nile | Flaviviridae, Flavivirus, WNV | Yes (mosquitoes) | Horse | [56] | |
| Avian influenza | Orthomyxoviridae, Influenzavirus, A/H5N1, A/H1N1 | No | Poultry, swine | [71,72,73] | |
| Primate | Marburg hemorrhagic fever | Filoviridae, Marburgvirus, MARV | No | No | [74,75] |
| Acquired immunodeficiency syndrome (AIDS) | Retroviridae, Lentivirus, HIV | No | No | [55] |
| Wild Animals | Type Study | Example of the Taxonomic Assignation of Viral Reads | Ref. | |||
|---|---|---|---|---|---|---|
| Animal Viruses | Plant/Fungal Viruses | Phages | Insect-Specific Viruses | |||
| Bats | DNA/RNA virome (feces) | Parvoviridae, Circoviridae, Picornaviridae, Adenoviridae, Poxviridae, Astroviridae, Coronaviridae | Luteoviridae, Secoviridae, Tymoviridae, Partitiviridae | Microviridae, Siphoviridae | Dicistroviridae, Iflaviridae, Tetraviridae, Nodaviridae, Parvoviridae (Densovirinae) | [76] |
| DNA/RNA virome (feces, urine, throat swabs, tissue) | Coronaviridae, Herpesviridae | Tymoviridae | Podoviridae | Iflaviridae, Dicistroviridae | [77] | |
| DNA/RNA virome (feces, urine, tissue, serum, throat swabs) | Retroviridae, Flaviviridae, Caliciviridae, Togaviridae, Paramyxoviridae, Adenoviridae, Papillomaviridae, Parvoviridae, Herpesviridae, Hepadnaviridae | not documented | not documented | not documented | [78,79] | |
| DNA/RNA virome (feces) | Papillomaviridae, Circoviridae, Anelloviridae | not documented | unclassified | Parvoviridae (Densovirinae) | [80] | |
| DNA/RNA virome (feces, throat swabs) | Adenoviridae, Herpesviridae, Papillomaviridae, Retroviridae, Circoviridae, Rhabdoviridae, Astroviridae, Flaviviridae, Coronaviridae, Picornaviridae, Parvoviridae | Chrysoviridae, Hypoviridae, Partitiviridae, Totiviridae | Inoviridae, Microviridae | Baculoviridae, Iflaviridae, Dicistroviridae, Tetraviridae, Parvoviridae (Densovirinae) | [81] | |
| DNA/RNA virome (tissue) | Herpesviridae, Papillomaviridae, Polyomaviridae, Hepadnaviridae, Circoviridae, Poxviridae, Retroviridae, Astroviridae | Phycodnaviridae, Bromoviridae | Myoviridae, Podoviridae, Siphoviridae | Baculoviridae, Polydnaviridae, Parvoviridae (Densovirinae), Iflaviridae | [82] | |
| DNA/RNA virome (urine, throat swabs) | Herpesviridae, Papillomaviridae, Adenoviridae, Poxviridae,Polyomaviridae, Retroviridae, Parvoviridae, Picornaviridae | not documented | not documented | Parvoviridae (Densovirinae) | [83] | |
| Rodents | DNA/RNA virome (feces) | Circoviridae, Picobirnaviridae, Picornaviridae, Astroviridae, Parvoviridae, Papillomaviridae, Adenoviridae, Coronaviridae | Nanoviridae, Geminiviridae, Phycodnaviridae, Secoviridae, Partitiviridae, Tymoviridae, Alphaflexiviridae, Tombusviridae | unclassified | Parvoviridae (Densovirinae), Iridoviridae, Polydnaviridae, Dicistroviridae, Bromoviridae, Virgaviridae | [60] |
5. Domestic Animals
5.1. Swine Breeding
5.2. Bushmeat and Wild Boars
| Domestic Animal | Zoonosis | Virus | Vector-Based Transmission | Ref. |
|---|---|---|---|---|
| Cats, dogs | Rabies | Rhabdoviridae, Lyssavirus, RABV | No | [94,95] |
| Cattle, sheep, goats | Rift Valley fever | Bunyaviridae, Phlebovirus, RVFV | Yes (mosquitoes) | [96,97] |
| Vaccinia | Poxviridae, Orthopoxvirus, VACV | No | [98] | |
| Pigs | Hepatitis E | Hepeviridae, Hepevirus, HEV | No | [99] |
| Japanese encephalitis | Flaviviridae, Flavivirus, JEV | Yes (mosquitoes) | [70] | |
| Horses | West Nile | Flaviviridae, Flavivirus, WNV | Yes (mosquitoes) | [56] |
| Hendra | Paramyxoviridae, Henipavirus, HeV | No | [63] | |
| Poultry | Avian flu | Orthomyxoviridae, Influenzavirus, A/H5N1 | No | [72,73] |
| Animal Species | Type Studies | Viral Reads Taxonomic Assignation | Ref. | |
|---|---|---|---|---|
| Animal Viruses stricto sensu | Zoonotic Viruses | |||
| Pigs | DNA/RNA virome (serum) | Asfarviridae, Anelloviridae, Retroviridae | Togaviridae (Alphavirus) | [88] |
| DNA/RNA virome (stool) | Picornaviridae, Astroviridae, Caliciviridae, Coronaviridae, Circoviridae, Parvoviridae | not documented | [87] | |
| Bushpigs | DNA/RNA virome (serum) | Parvoviridae, Circoviridae, Retroviridae | not documented | [92] |
| Wild boars | DNA/RNA virome (feces) | Picornaviridae, Astroviridae | not documented | [93] |
6. Future Perspectives in Metagenomic-Based Surveillance Programs
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cutler, S.J.; Fooks, A.R.; van der Poel, W.H. Public health threat of new, reemerging, and neglected zoonoses in the industrialized world. Emerg. Infect. Dis. 2010, 16, 1–7. [Google Scholar] [CrossRef]
- One Health Initiative. Available online: http://www.onehealthinitiative.com/ (accessed on 18 February 2014).
- Woolhouse, M.; Scott, F.; Hudson, Z.; Howey, R.; Chase-Topping, M. Human viruses: Discovery and emergence. Phil. Trans. R. Soc. B 2012, 367, 2864–2871. [Google Scholar] [CrossRef]
- Wolfe, N.D.; Panosian Dunavan, C.; Diamond, J. Origins of major human infectious diseases. Nature 2007, 447, 279–283. [Google Scholar] [CrossRef]
- Karesh, W.B.; Dobson, A.; Lloyd-Smith, J.O.; Lubroth, J.; Dixon, M.A.; Bennett, M.; Aldrich, S.; Harrington, T.; Formenty, P.; Loh, E.H.; et al. Ecology of zoonoses: Natural and unnatural histories. Lancet 2012, 380, 1936–1945. [Google Scholar] [CrossRef]
- Wong, S.; Lau, S.; Woo, P.; Yuen, K.Y. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 2007, 17, 67–91. [Google Scholar] [CrossRef]
- Gould, E.A.; Higgs, S. Impact of climate change and other factors on emerging arbovirus diseases. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 109–121. [Google Scholar] [CrossRef]
- Keesing, F.; Belden, L.K.; Daszak, P.; Dobson, A.; Harvell, C.D.; Holt, R.D.; Hudson, P.; Jolles, A.; Jones, K.E.; Mitchell, C.E.; et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature 2010, 468, 647–652. [Google Scholar] [CrossRef]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. The challenge of emerging and re-emerging infectious diseases. Nature 2004, 438, 242–249. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Pozzetto, B. Méthodes de diagnostic en virologie. In Virologie Médicale; Mammette, A., Ed.; Presses Universitaires de Lyon: Lyon, France, 2002; pp. 187–210. [Google Scholar]
- Bexfield, N.; Kellam, P. Metagenomics and the molecular identification of novel viruses. Vet. J. 2011, 190, 191–198. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palù, G. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef]
- Stang, A.; Korn, K.; Wildner, O.; Uberla, K. Characterization of virus isolates by particle-associated nucleic acid PCR. J. Clin. Microbiol. 2005, 43, 716–720. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Giombini, E.; Rozera, G. Next Generation Sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013, 19, 15–22. [Google Scholar] [CrossRef]
- Blomström, A.L. Viral metagenomics as an emerging and powerful tool in veterinary medicine. Vet. Q. 2011, 31, 107–114. [Google Scholar] [CrossRef]
- Belák, S.; Karlsson, O.E.; Blomström, A.L.; Berg, M.; Granberg, F. New viruses in veterinary medicine, detected by metagenomic approaches. Vet. Microbiol. 2013, 165, 95–101. [Google Scholar]
- Popgeorgiev, N.; Temmam, S.; Raoult, D.; Desnues, C. Describing the silent human virome with an emphasis on giant viruses. Intervirology 2013, 56, 395–412. [Google Scholar] [CrossRef]
- Liu, S.; Vijayendran, D.; Bonning, B.C. Next generation sequencing technologies for insect virus discovery. Viruses 2011, 3, 1849–1869. [Google Scholar] [CrossRef]
- Junglen, S.; Drosten, C. Virus discovery and recent insights into virus diversity in arthropods. Curr. Opin. Microbiol. 2013, 16, 507–513. [Google Scholar] [CrossRef]
- Ng, T.F.; Willner, D.L.; Lim, Y.W.; Schmieder, R.; Chau, B.; Nilsson, C.; Anthony, S.; Ruan, Y.; Rohwer, F.; Breitbart, M. Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS One 2011, 6, e20579. [Google Scholar] [CrossRef]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef]
- Bourhy, H.; Cowley, J.A.; Larrous, F.; Holmes, E.C.; Walker, P.J. Phylogenetic relationships among rhabdoviruses inferred using the L polymerase gene. J. Gen. Virol. 2005, 86, 2849–2858. [Google Scholar] [CrossRef]
- Hall-Mendelin, S.; Allcock, R.; Kresoje, N.; van den Hurk, A.F.; Warrilow, D. Detection of arboviruses and other micro-organisms in experimentally infected mosquitoes using massively parallel sequencing. PLoS One 2013, 8, e58026. [Google Scholar]
- Ma, M.; Huang, Y.; Gong, Z.; Zhuang, L.; Li, C.; Yang, H.; Tong, Y.; Liu, W.; Cao, W. Discovery of DNA viruses in wild-caught mosquitoes using small RNA high throughput sequencing. PLoS One 2011, 6, e24758. [Google Scholar]
- Fédière, G. Epidemiology and pathology of densovirinae. Contrib. Microbiol. 2000, 4, 1–11. [Google Scholar] [CrossRef]
- Carlson, J.; Suchman, E.; Buchatsky, L. Densoviruses for control and genetic manipulation of mosquitoes. Adv. Virus Res. 2006, 68, 361–392. [Google Scholar] [CrossRef]
- Cook, S.; Chung, B.Y.; Bass, D.; Moureau, G.; Tang, S.; McAlister, E.; Culverwell, C.L.; Glücksman, E.; Wang, H.; Brown, T.D.; et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS One 2013, 8, e80720. [Google Scholar]
- Estrada-Peña, A.; Jongejan, F. Ticks feeding on humans: A review of records on human-biting Ixodoidea with special reference to pathogen transmission. Exp. Appl. Acarol. 1999, 23, 685–715. [Google Scholar] [CrossRef]
- Dobler, G. Zoonotic tick-borne flaviviruses. Vet. Microbiol. 2010, 140, 221–228. [Google Scholar] [CrossRef]
- Charrel, R.N.; Fagbo, S.; Moureau, G.; Alqahtani, M.H.; Temmam, S.; de Lamballerie, X. Alkhurma hemorrhagic fever virus in Ornithodoros savignyi ticks. Emerg. Infect. Dis. 2007, 13, 153–155. [Google Scholar] [CrossRef]
- Gritsun, T.S.; Nuttall, P.A.; Gould, E.A. Tick-borne flaviviruses. Adv. Virus Res. 2003, 61, 317–371. [Google Scholar] [CrossRef]
- Lawrie, C.H.; Uzcátegui, N.Y.; Gould, E.A.; Nuttall, P.A. Ixodid and argasid tick species and West Nile virus. Emerg. Infect. Dis. 2004, 10, 653–657. [Google Scholar] [CrossRef]
- Converse, J.D.; Hoogstraal, H.; Moussa, M.I.; Feare, C.J.; Kaiser, M.N. Soldado virus (Hughes group) from Ornithodoros (Alectorobius) capensis (Ixodoidea: Argasidae) infesting Sooty Tern colonies in the Seychelles, Indian Ocean. Am. J. Trop. Med. Hyg. 1975, 24, 1010–1018. [Google Scholar]
- Sotnikova, A.N.; Soldatov, G.M. Isolation of tick-borne encephalitis viruses from fleas ceratophyllus tamias wagn. Med. Parazitol. 1964, 33, 622–624. [Google Scholar]
- Kolb, A.; Needham, G.R.; Neyman, K.M.; High, W.A. Bedbugs. Dermatol. Ther. 2009, 22, 347–352. [Google Scholar] [CrossRef]
- Williams, J.E.; Imlarp, S.; Top, F.H., Jr.; Cavanaugh, D.C.; Russell, P.K. Kaeng Khoi virus from naturally infected bedbugs (cimicidae) and immature free-tailed bats. Bull. World Health Organ. 1976, 53, 365–369. [Google Scholar]
- Silverman, A.L.; Qu, L.H.; Blow, J.; Zitron, I.M.; Gordon, S.C.; Walker, E.D. Assessment of hepatitis B virus DNA and hepatitis C virus RNA in the common bedbug (Cimex lectularius L.) and kissing bug (Rodnius prolixus). Am. J. Gastroenterol. 2001, 96, 2194–2198. [Google Scholar] [CrossRef]
- Usinger, R.L. Monograph of the Cimicidae; Entomological Society of America: Annapolis, MD, USA, 1966; pp. 81–167. [Google Scholar]
- Wright, R.E.; Anslow, R.O.; Thompsons, W.H.; DeFoliart, G.R.; Seawright, G.; Hanson, R.P. Isolations of lacrosse virus of the California group from Tabanidae in Wisconsin. Mosquito News 1970, 30, 600–603. [Google Scholar]
- DeFoliart, G.R.; Anslow, R.O.; Hanson, R.P.; Morris, C.D.; Papadopoulos, O.; Sather, G.E. Isolation of Jamestown Canyon serotype of California encephalitis virus from naturally infected Aedes mosquitoes and tabanids. Am. J. Trop. Med. Hyg. 1969, 18, 440–447. [Google Scholar]
- Charrel, R.N.; Izri, A.; Temmam, S.; de Lamballerie, X.; Parola, P. Toscana virus RNA in Sergentomyia minuta flies. Emerg. Infect. Dis. 2006, 12, 1299–1300. [Google Scholar] [CrossRef]
- Izri, A.; Temmam, S.; Moureau, G.; Hamrioui, B.; de Lamballerie, X.; Charrel, R.N. Sandfly fever Sicilian virus, Algeria. Emerg. Infect. Dis. 2008, 14, 795–797. [Google Scholar] [CrossRef]
- Moureau, G.; Ninove, L.; Izri, A.; Cook, S.; de Lamballerie, X.; Charrel, R.N. Flavivirus RNA in phlebotomine sandflies. Vector Borne Zoonotic Dis. 2010, 10, 195–197. [Google Scholar] [CrossRef]
- Comer, J.A.; Tesh, R.B. Phlebotomine sand flies as vectors of vesiculoviruses: A review. Parassitologia 1991, 33, 143–150. [Google Scholar]
- Tesh, R.; Saidi, S.; Javadian, E.; Loh, P.; Nadim, A. Isfahan virus, a new vesiculovirus infecting humans, gerbils, and sandflies in Iran. Am. J. Trop. Med. Hyg. 1977, 26, 299–306. [Google Scholar]
- Groseth, A.; Weisend, C.; Ebihara, H. Complete genome sequencing of mosquito and human isolates of Ngari virus. J. Virol. 2012, 86, 13846–13847. [Google Scholar] [CrossRef]
- Mwaengo, D.; Lorenzo, G.; Iglesias, J.; Warigia, M.; Sang, R.; Bishop, R.P.; Brun, A. Detection and identification of Rift Valley fever virus in mosquito vectors by quantitative real-time PCR. Virus Res. 2012, 169, 137–143. [Google Scholar] [CrossRef]
- Paupy, C.; Kassa Kassa, F.; Caron, M.; Nkoghé, D.; Leroy, E.M. A chikungunya outbreak associated with the vector Aedes albopictus in remote villages of Gabon. Vector Borne Zoonotic Dis. 2012, 12, 167–169. [Google Scholar] [CrossRef]
- Calzolari, M.; Bonilauri, P.; Bellini, R.; Albieri, A.; Defilippo, F.; Maioli, G.; Galletti, G.; Gelati, A.; Barbieri, I.; Tamba, M.; et al. Evidence of simultaneous circulation of West Nile and Usutu viruses in mosquitoes sampled in Emilia-Romagna region (Italy) in 2009. PLoS One 2010, 5, e14324. [Google Scholar] [CrossRef]
- Carpenter, S.; Groschup, M.H.; Garros, C.; Felippe-Bauer, M.L.; Purse, B.V. Culicoides biting midges, arboviruses and public health in Europe. Antivir. Res. 2013, 100, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.H. Isolation of viruses from field populations of culicoides (Diptera: Ceratopogonidae) in Nigeria. J. Med. Entomol. 1979, 16, 76–79. [Google Scholar]
- Davies, F.G.; Walker, A.R.; Ochieng, P.; Shaw, T. Arboviruses isolated from culicoides midges in Kenya. J. Comp. Pathol. 1979, 89, 587–595. [Google Scholar] [CrossRef]
- Bengis, R.G.; Leighton, F.A.; Fischer, J.R.; Artois, M.; Mörner, T.; Tate, C.M. The role of wildlife in emerging and re-emerging zoonoses. Rev. Sci. Tech. 2004, 23, 497–511. [Google Scholar]
- Hahn, B.H.; Shaw, G.M.; de Cock, K.M.; Sharp, P.M. AIDS as a zoonosis: Scientific and public health implications. Science 2000, 287, 607–614. [Google Scholar] [CrossRef]
- Trevejo, R.T.; Eidson, M. Zoonosis update: West Nile virus. J. Am. Vet. Med. Assoc. 2008, 232, 1302–1309. [Google Scholar] [CrossRef]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Smith, I.; Wang, L.F. Bats and their virome: An important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 2013, 3, 84–91. [Google Scholar] [CrossRef]
- Luis, A.D.; Hayman, D.T.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.; Mills, J.N.; Timonin, M,E.; Willis, C.K.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. Biol. Sci. 2013, 280, 20122753. [Google Scholar] [CrossRef]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e2218. [Google Scholar]
- Balique, F.; Colson, P.; Raoult, D. Tobacco mosaic virus in cigarettes and saliva of smokers. J. Clin. Virol. 2012, 55, 374–376. [Google Scholar]
- Colson, P.; Richet, H.; Desnues, C.; Balique, F.; Moal, V.; Grob, J.J.; Berbis, P.; Lecoq, H.; Harlé, J.R.; Berland, Y.; et al. Pepper mild mottle virus, a plant virus associated with specific immune responses, Fever, abdominal pains, and pruritus in humans. PLoS One 2010, 5, e10041. [Google Scholar]
- Middleton, D.J.; Weingartl, H.M. Henipaviruses in their natural animal hosts. Curr. Top. Microbiol. Immunol. 2012, 359, 105–121. [Google Scholar]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.P; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar]
- Johnson, N.; Vos, A.; Freuling, C.; Tordo, N.; Fooks, A.R.; Müller, T. Human rabies due to lyssavirus infection of bat origin. Vet. Microbiol. 2010, 142, 151–159. [Google Scholar] [CrossRef]
- Charrel, R.N.; de Lamballerie, X. Zoonotic aspects of arenavirus infections. Vet. Microbiol. 2010, 140, 213–220. [Google Scholar] [CrossRef]
- Reusken, C.; Heyman, P. Factors driving hantavirus emergence in Europe. Curr. Opin. Virol. 2013, 3, 92–99. [Google Scholar] [CrossRef]
- Zeier, M.; Handermann, M.; Bahr, U.; Rensch, B.; Müller, S.; Kehm, R.; Muranyi, W.; Darai, G. New ecological aspects of hantavirus infection: A change of a paradigm and a challenge of prevention—A review. Virus Genes 2005, 30, 157–180. [Google Scholar] [CrossRef]
- Liu, H.; Lu, H.J.; Liu, Z.J.; Jing, J.; Ren, J.Q.; Liu, Y.Y.; Lu, F.; Jin, N.Y. Japanese encephalitis virus in mosquitoes and swine in Yunnan province, China 2009–2010. Vector Borne Zoonotic Dis. 2013, 13, 41–49. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef]
- Reperant, L.A.; Kuiken, T.; Osterhaus, A.D. Influenza viruses: From birds to humans. Hum. Vaccin. Immunother. 2012, 8, 7–16. [Google Scholar] [CrossRef]
- Ma, W.; Kahn, R.E.; Richt, J.A. The pig as a mixing vessel for influenza viruses: Human and veterinary implications. J. Mol. Genet. Med. 2008, 27, 158–166. [Google Scholar]
- Brauburger, K.; Hume, A.J.; Mühlberger, E.; Olejnik, J. Forty-five years of Marburg virus research. Viruses 2012, 4, 1878–1927. [Google Scholar] [CrossRef]
- Leroy, E.M.; Gonzalez, J.P.; Baize, S. Ebola and Marburg haemorrhagic fever viruses: Major scientific advances, but a relatively minor public health threat for Africa. Clin. Microbiol. Infect. 2011, 17, 964–976. [Google Scholar] [CrossRef]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic analysis of the viromes of three North American bat species: Viral diversity among different bat species that share a common habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Drexler, J.F.; Geipel, A.; König, A.; Corman, V.M.; van Riel, D.; Leijten, L.M.; Bremer, C.M.; Rasche, A.; Cottontail, V.M.; Maganga, G.D.; et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 16151–16156. [Google Scholar] [CrossRef]
- Tse, H.; Tsang, A.K.; Tsoi, H.W.; Leung, A.S.; Ho, C.C.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. Identification of a novel bat papillomavirus by metagenomics. PLoS One 2012, 7, e43986. [Google Scholar]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef]
- He, B.; Li, Z.; Yang, F.; Zheng, J.; Feng, Y.; Guo, H.; Li, Y.; Wang, Y.; Su, N.; Zhang, F.; et al. Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLoS One 2013, 8, e61950. [Google Scholar]
- Baker, K.S.; Leggett, R.M.; Bexfield, N.H.; Alston, M.; Daly, G.; Todd, S.; Tachedjian, M.; Holmes, C.E.; Crameri, S.; Wang, L.F.; et al. Metagenomic study of the viruses of African straw-colored fruit bats: Detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 2013, 441, 95–106. [Google Scholar] [CrossRef]
- Meng, X.J. From barnyard to food table: The omnipresence of hepatitis E virus and risk for zoonotic infection and food safety. Virus Res. 2011, 161, 23–30. [Google Scholar] [CrossRef]
- Li, L.; Diab, S.; McGraw, S.; Barr, B.; Traslavina, R.; Higgins, R.; Talbot, T.; Blanchard, P.; Rimoldi, G.; Fahsbender, E.; et al. Divergent astrovirus associated with neurologic disease in cattle. Emerg. Infect. Dis. 2013, 19, 1385–1392. [Google Scholar] [CrossRef]
- Hoffmann, B.; Scheuch, M.; Höper, D.; Jungblut, R.; Holsteg, M.; Schirrmeier, H.; Eschbaumer, M.; Goller, K.V.; Wernike, K.; Fischer, M.; et al. Novel orthobunyavirus in Cattle, Europe, 2011. Emerg. Infect. Dis. 2012, 18, 469–472. [Google Scholar] [CrossRef]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The fecal virome of pigs on a high-density farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef]
- Masembe, C.; Michuki, G.; Onyango, M.; Rumberia, C.; Norling, M.; Bishop, R.P.; Djikeng, A.; Kemp, S.J.; Orth, A.; Skilton, R.A.; et al. Viral metagenomics demonstrates that domestic pigs are a potential reservoir for Ndumu virus. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Arbovirus Catalog. Available online: https://wwwn.cdc.gov/Arbocat/Default.aspx (accessed on 20 February 2014).
- Kokernot, R.H.; McIntosh, B.M.; Worth, C.B. Ndumu virus, a hitherto unknown agent, isolated from culicine mosouitoes collected in northern Natal. Union of South Africa. Am. J. Trop. Med. Hyg. 1961, 10, 383–386. [Google Scholar]
- Smith, K.M.; Anthony, S.J.; Switzer, W.M.; Epstein, J.H.; Seimon, T.; Jia, H.; Sanchez, M.D.; Huynh, T.T.; Galland, G.G.; Shapiro, S.E.; et al. Zoonotic viruses associated with illegally imported wildlife products. PLoS One 2012, 7, e29505. [Google Scholar] [CrossRef]
- Blomström, A.L.; Ståhl, K.; Masembe, C.; Okoth, E.; Okurut, A.R.; Atmnedi, P.; Kemp, S.; Bishop, R.; Belák, S.; Berg, M. Viral metagenomic analysis of bushpigs (Potamochoeruslarvatus) in Uganda identifies novel variants of Porcine parvovirus 4 and Torque teno sus virus 1 and 2. Virol. J. 2012, 9. [Google Scholar] [CrossRef]
- Reuter, G.; Nemes, C.; Boros, A.; Kapusinszky, B.; Delwart, E.; Pankovics, P. Porcine kobuvirus in wild boars (Sus scrofa). Arch. Virol. 2013, 158, 281–282. [Google Scholar] [CrossRef]
- Yousaf, M.Z.; Qasim, M.; Zia, S.; Khan, M; Ashfaq, U.A.; Khan, S. Rabies molecular virology, diagnosis, prevention and treatment. Virol. J. 2012, 9. [Google Scholar] [CrossRef]
- Lackay, S.N.; Kuang, Y.; Fu, Z.F. Rabies in small animals. Vet. Clin. N. Am. 2008, 38, 851–861. [Google Scholar] [CrossRef]
- Balenghien, T.; Cardinale, E.; Chevalier, V.; Elissa, N.; Failloux, A.B.; Jean Jose Nipomichene, T.N.; Nicolas, G.; Rakotoharinome, V.M.; Roger, M.; Zumbo, B. Towards a better understanding of Rift Valley fever epidemiology in the south-west of the Indian Ocean. Vet. Res. 2013, 44, 78. [Google Scholar] [CrossRef] [Green Version]
- Bird, B.H.; Nichol, S.T. Breaking the chain: Rift Valley fever virus control via livestock vaccination. Curr. Opin. Virol. 2012, 2, 315–323. [Google Scholar] [CrossRef]
- Essbauer, S.; Pfeffer, M.; Meyer, H. Zoonotic poxviruses. Vet. Microbiol. 2010, 140, 229–236. [Google Scholar] [CrossRef]
- Temmam, S.; Besnard, L.; Andriamandimby, S.F.; Foray, C.; Rasamoelina-Andriamanivo, H.; Héraud, J.M.; Cardinale, E.; Dellagi, K.; Pavio, N.; Pascalis, H.; et al. High prevalence of hepatitis E in humans and pigs and evidence of genotype-3 virus in swine, Madagascar. Am. J. Trop. Med. Hyg. 2013, 88, 329–338. [Google Scholar] [CrossRef]
- Woolhouse, M.; Gaunt, E. Ecological origins of novel human pathogens. Crit. Rev. Microbiol. 2007, 33, 231–242. [Google Scholar] [CrossRef]
- Contributing to One World, One Health: A Strategic Framework for Reducing Risks of Infectious Diseases at the Animal-Human-Ecosystems Interface. Available online: ftp://ftp.fao.org/docrep/fao/011/aj137e/aj137e00.pdf (accessed on 20 February 2014).
- Moreira, L.A.; Iturbe-Ormaetxe, I.; Jeffery, J.A.; Lu, G.; Pyke, A.T.; Hedges, L.M.; Rocha, B.C.; Hall-Mendelin, S.; Day, A.; Riegler, M.; et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 2009, 139, 1268–1278. [Google Scholar] [CrossRef]
- Bian, G.; Xu, Y.; Lu, P.; Xie, Y.; Xi, Z. The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti. PLoS Pathog. 2010, 6, e833. [Google Scholar]
- Haagmans, B.L.; Al Dhahiry, S.H.; Reusken, C.B.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.J.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 2014, 14, 140–145. [Google Scholar] [CrossRef]
- Briese, T.; Mishra, N.; Jain, K.; Zalmout, I.S.; Jabado, O.J.; Karesh, W.B.; Daszak, P.; Mohammed, O.B.; Alagaili, A.N.; Lipkin, W.I. Middle east respiratory syndrome coronavirus quasispecies that include homologues of human isolates revealed through whole-genome analysis and virus cultured from dromedary camels in Saudi Arabia. MBio 2014, 5, e1146–14. [Google Scholar]
- Koch, R. Investigations into bacteria: V. The etiology of anthrax, based on the ontogenesis of Bacillus anthracis. Cohns Beitrage zur Biologie der Pflanzen 1876, 2, 277–310. [Google Scholar]
- Fredericks, D.N.; Relman, D.A. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clin. Microbiol. Rev. 1996, 9, 18–33. [Google Scholar]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Temmam, S.; Davoust, B.; Berenger, J.-M.; Raoult, D.; Desnues, C. Viral Metagenomics on Animals as a Tool for the Detection of Zoonoses Prior to Human Infection? Int. J. Mol. Sci. 2014, 15, 10377-10397. https://doi.org/10.3390/ijms150610377
Temmam S, Davoust B, Berenger J-M, Raoult D, Desnues C. Viral Metagenomics on Animals as a Tool for the Detection of Zoonoses Prior to Human Infection? International Journal of Molecular Sciences. 2014; 15(6):10377-10397. https://doi.org/10.3390/ijms150610377
Chicago/Turabian StyleTemmam, Sarah, Bernard Davoust, Jean-Michel Berenger, Didier Raoult, and Christelle Desnues. 2014. "Viral Metagenomics on Animals as a Tool for the Detection of Zoonoses Prior to Human Infection?" International Journal of Molecular Sciences 15, no. 6: 10377-10397. https://doi.org/10.3390/ijms150610377