Cytotoxic Autophagy in Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
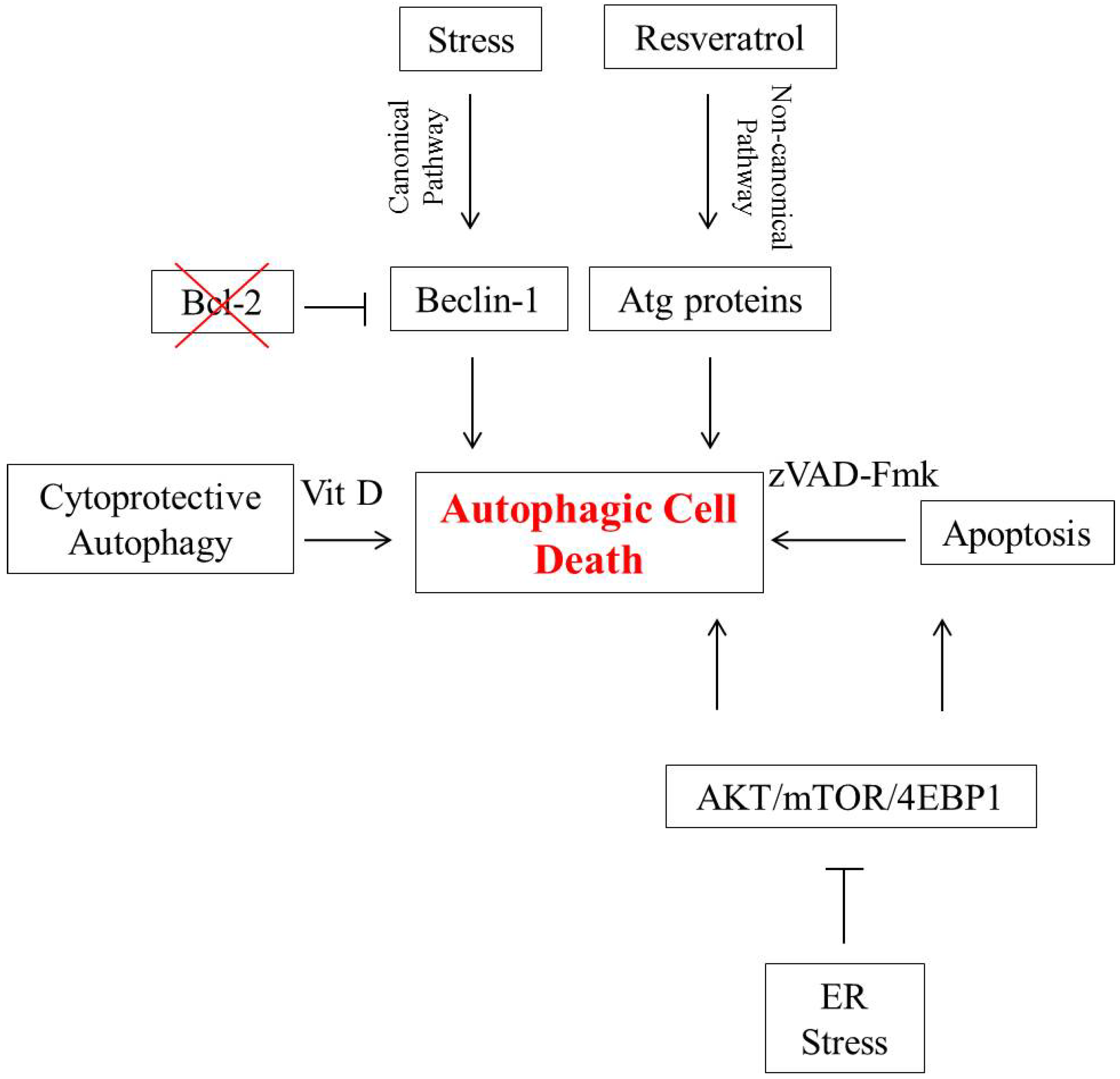
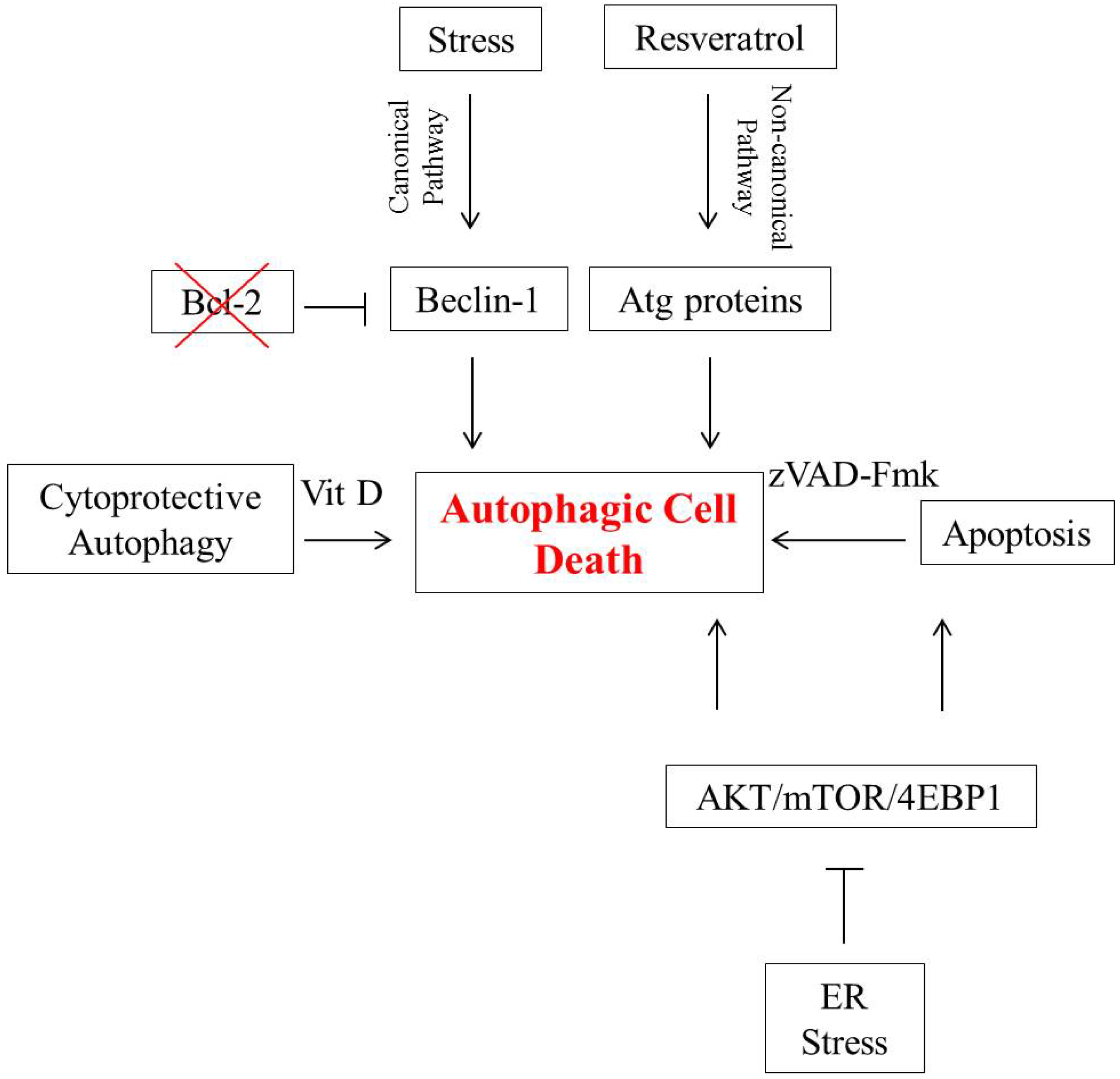
2. Autophagic Cell Death in Breast Tumor Cells
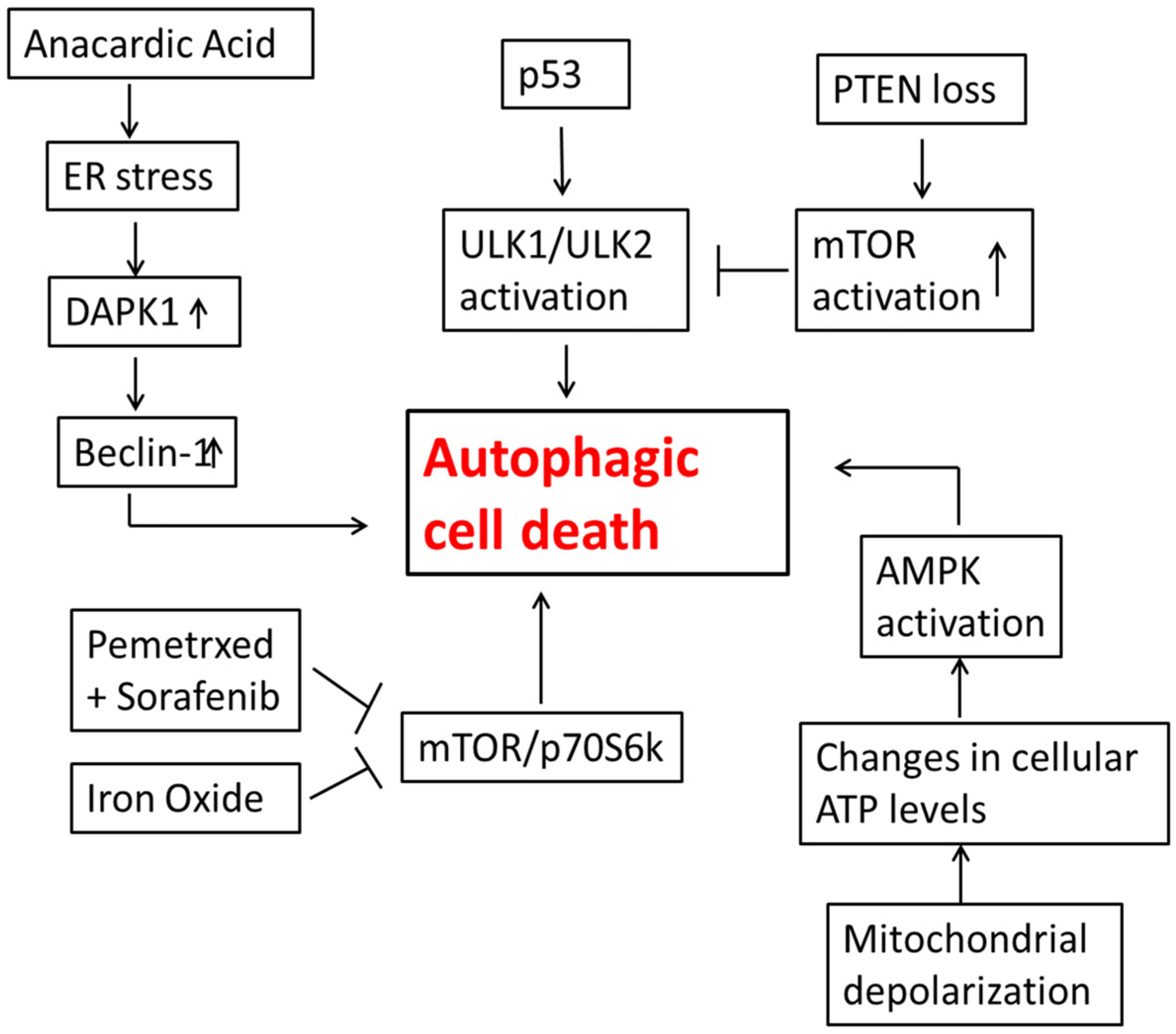
3. Autophagic Cell Death in Lung Cancer Cells
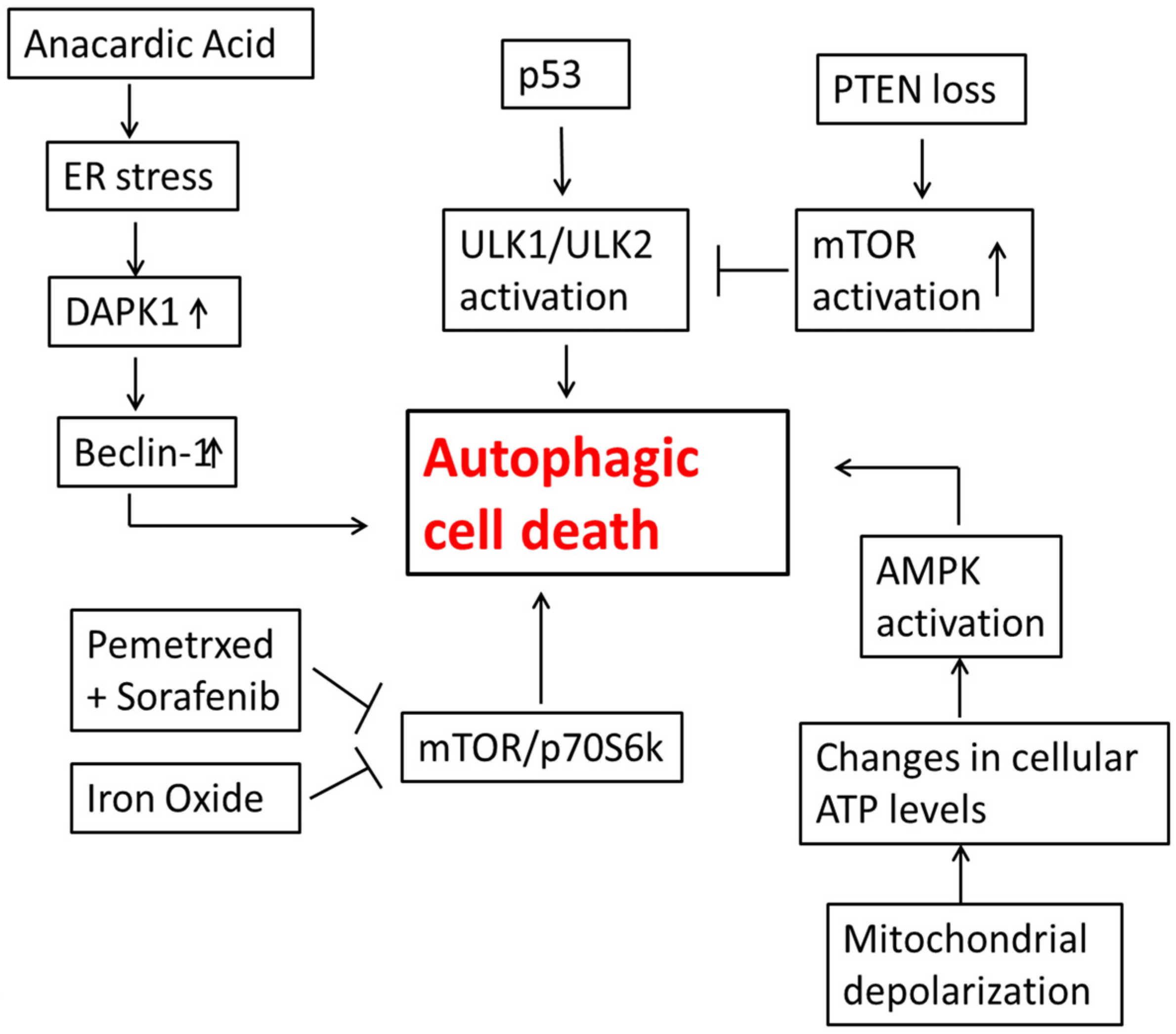
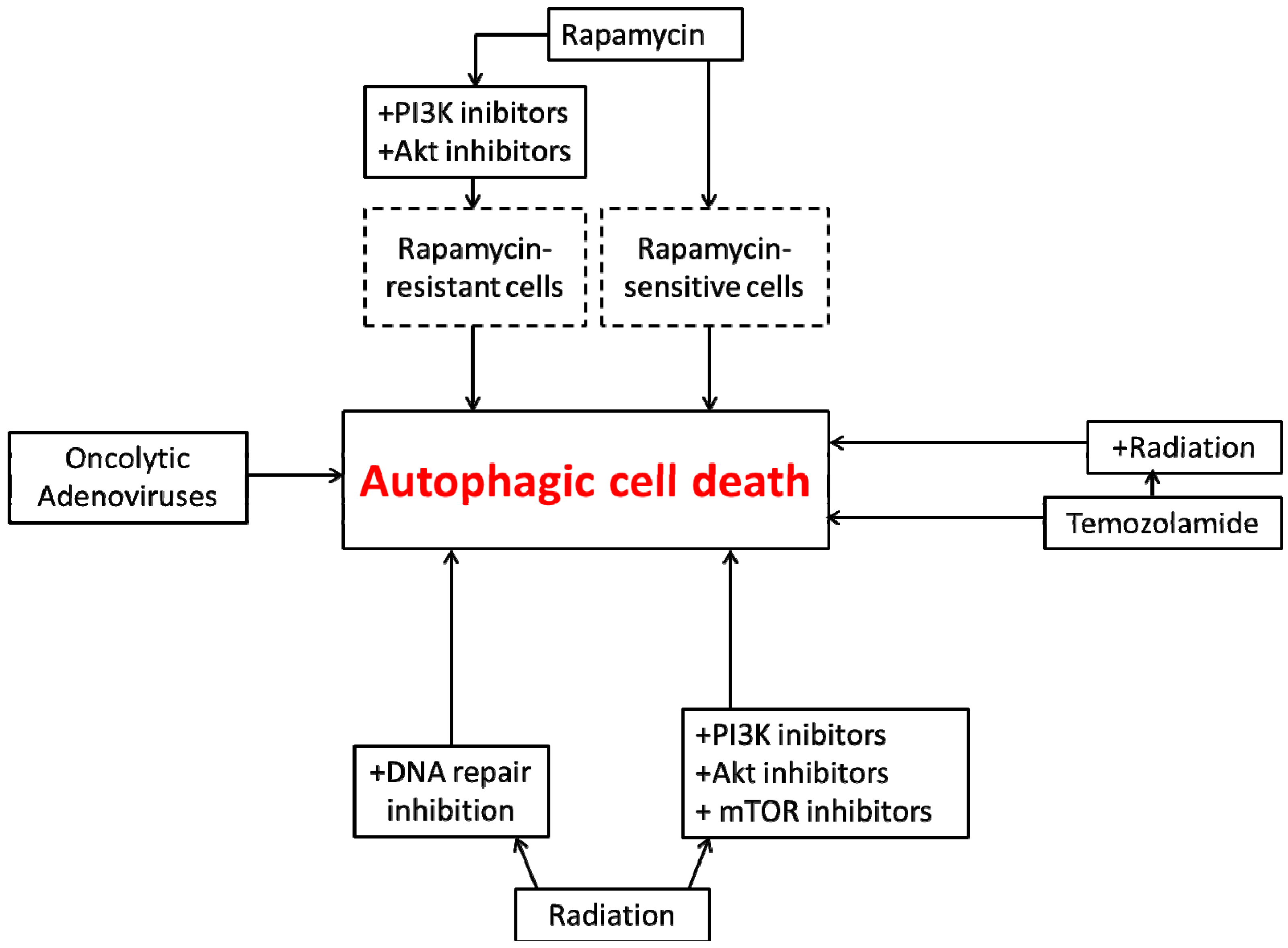
4. Autophagic Cell Death in Glioblastoma
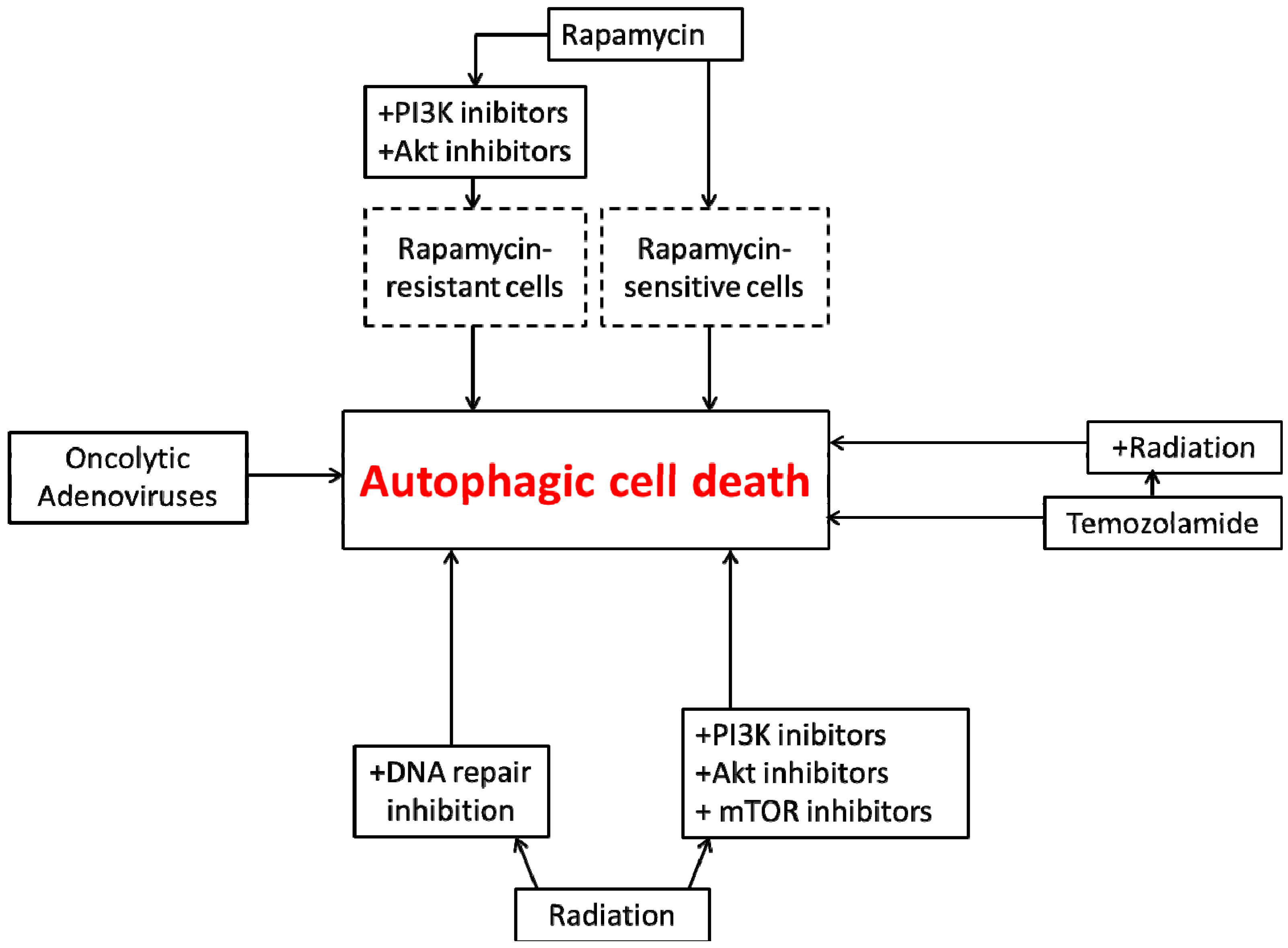
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.-U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef]
- Vakifahmetoqlu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Berghe, V.T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Scarlatti, F.; Granata, R.; Meijer, A.J.; Codogno, P. Does autophagy have a license to kill mammalian cells? Cell Death Differ 2009, 16, 12–20. [Google Scholar]
- Chen, H.Y.; White, E. Role of autophagy in cancer prevention. Cancer Prev. Res. 2011, 4, 973–983. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 105–137. [Google Scholar] [CrossRef]
- Shen, H.M.; Codogno, P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011, 7, 457–465. [Google Scholar] [CrossRef]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef]
- Gewirtz, D.A. When cytoprotective autophagy isn’t… and even when it is. Autophagy 2014, 10, 391–392. [Google Scholar] [CrossRef]
- Shen, S.; Kepp, O.; Michaud, M.; Martins, I.; Minoux, H.; Métivier, D.; Maiuri, M.C.; Kroemer, R.T.; Kroemer, G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 2011, 30, 4544–4556. [Google Scholar] [CrossRef]
- Hu, Y.L.; Jahangiri, A.; Delay, M.; Aghi, M.K. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012, 72, 4294–4299. [Google Scholar] [CrossRef]
- Zou, Z.; Yuan, Z.; Zhang, Q.; Long, Z.; Chen, J.; Tang, Z.; Zhu, Y.; Chen, S.; Xu, J.; Yan, M.; et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012, 8, 1798–1810. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, J.; Lv, Y.; Wang, T.; Zhang, J.; Fan, J.; Wang, L. The significance of expression of autophagy-related gene Beclin, Bcl-2, and Bax in breast cancer tissues. Tumor Biol. 2011, 32, 1163–1171. [Google Scholar] [CrossRef]
- Scarlatti, F.; Maffei, R.; Beau, I.; Codogno, P.; Ghidoni, R. Role of non-canonical Beclin1-independent autophagy in cell death by resveratrol in human breast cancer cells. Cell Death Differ. 2008, 15, 1318–1329. [Google Scholar] [CrossRef]
- Akar, U.; Chaves-Reyez, A.; Barria, M.; Tari, A.; Sangguino, A.; Kondo, Y.; Kondo, S.; Arun, B.; Lopez-Berestein, G.; Ozpolat, B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 2008, 4, 669–679. [Google Scholar]
- Zeng, X.; Overmeyer, J.H.; Maltese, W.A. Functional specificity of the mammalian Beclin-Vcp34 PI3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J. Cell Sci. 2006, 119, 259–270. [Google Scholar] [CrossRef]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 2008, 13, 1–9. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Vanden, B.T.; Festjens, N. Caspase inhibitiors promote alternative cell death pathways. Sci. STKE 2006, 2006. [Google Scholar] [CrossRef]
- Di, X.; Robert, P.S.; Irene, F.N.; Gewirtz, D.A. Apoptosis, autophagy, accelerated senescence and reactive oxygen in the response of human breast tumor cells to Adriamycin. Biochem. Pharmacol. 2009, 77, 1139–1150. [Google Scholar] [CrossRef]
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A switch between cytoprotective and cytotoxic autophagy in the radio sensitization of breast tumor cells by chloroquine and vitamin D. Horm. Cancer 2011, 2, 272–285. [Google Scholar] [CrossRef]
- Bristol, M.L.; Di, X.; Beckman, M.J.; Wilson, E.N.; Henderson, S.C.; Maiti, A.; Fan, Z.; Gewirtz, D.A. Dual functions of autophagy in the response of breast tumor cells to radiation: Cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin D3. Autophagy 2012, 8, 739–753. [Google Scholar] [CrossRef]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef]
- Lungcancer.org. Available online: http://www.Lungcancer.org (accessed on 21 May 2014).
- Sharieff, W.; Okawara, G.; Tsakiridis, T.; Wright, J. Predicting 2-year survival for radiation regimens in advanced non-small cell lung cancer. Clin. Oncol. 2013, 25, 697–705. [Google Scholar] [CrossRef]
- Saintigny, P.; Burger, J.A. Recent advances in non-small cell lung cancer biology and clinical management. Discov. Med. 2012, 13, 287–297. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signaling. Cell Death Differ. 2013, 21, 92–99. [Google Scholar]
- Cheng, G.; Kong, D.; Hou, X.; Liang, B.; He, M.; Liang, N.; Ma, S.; Liu, X. The Tumor suppressor, p53, contributes to radiosensitivity of lung cancer cells by regulating autophagy and apoptosis. Cancer Biother. Radiopharm. 2013, 28, 153–159. [Google Scholar] [CrossRef]
- Peng, P.L.; Kuo, W.H.; Tseng, H.C.; Chou, F.P. Synergistic tumor-killing effect of radiation and berberine combined treatment in lung cancer: The contribution of autophagic cell death. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 529–542. [Google Scholar] [CrossRef]
- Shin, J.Y.; Lim, H.T.; Minai-Tehrani, A.; Noh, M.S.; Kim, J.E.; Kim, J.H.; Jiang, J.H.; Arote, R.; Kim, D.Y.; Chae, C.; et al. Aerosol delivery of beclin-1 enhanced the anti-tumor effect of radiation in the lungs of k-ras LA1 mice. J. Radiat. Res. 2012, 53, 506–515. [Google Scholar] [CrossRef]
- Seong, Y.A.; Shin, P.G.; Yoon, J.S.; Yadunandam, A.K.; Kim, G.D. Induction of the Endoplasmic Reticulum stress and autophagy in human lung carcinoma A549 cells by Anacardic Acid. Cell Biochem. Biophys. 2014, 68, 369–377. [Google Scholar] [CrossRef]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.R.; Lee, A.S. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress induced autophagy in mammalian cells. Cell Death Differ. 2008, 15, 1460–1471. [Google Scholar] [CrossRef]
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886. [Google Scholar] [CrossRef] [Green Version]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase mediated phosphorylation on BH3 domain of beclin 1 promotes dissociation of beclin-1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Kim, K.W.; Hwang, M.; Moretti, L.; Jaboin, J.J.; Cha, Y.I.; Lu, B. Autophagy up-regulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 2008, 4, 659–668. [Google Scholar]
- Kim, K.W.; Mutter, R.W.; Cao, C.; Albert, J.M.; Freeman, M.; Hallahan, D.E.; Lu, B. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J. Biol. Chem. 2006, 281, 36883–36890. [Google Scholar]
- Scherl-Mostageer, M.; Sommergruber, W.; Abseher, R.; Hauptmann, R.; Ambros, P.; Schweifer, N. Identification of a novel gene. CDCP1, overexpressed in human colorectal cancer. Oncogene 2001, 20, 4402–4408. [Google Scholar] [CrossRef]
- Buhring, H.J.; Kuci, S.; Conze, T.; Rathke, G.; Bartolovic, K.; Grunebach, F.; Scherl-Mostageer, M.; Brummendorf, T.H.; Schweifer, N.; Lammers, R. CDCP1 identifies a broad spectrum of normal and malignant stem/progenitor cells subsets of hematopoietic and nonhematopoietic origin. Stem Cells 2004, 22, 334–343. [Google Scholar] [CrossRef]
- Uekita, T.; Jia, L.; Narisawa-Saito, M.; Yokota, J.; Kiyono, T.; Sakai, R. CUB domain-containing protein I is a novel regulator of anoikis resistance lung adenocarcinoma. Mol. Cell. Biol. 2007, 27, 7649–7660. [Google Scholar] [CrossRef]
- Miyazawa, Y.; Uekita, T.; Hiraoka, N.; Fujii, S.; Kosuqe, T.; Kanao, Y.; Nojima, Y.; Sakai, R. CUB domain-containing protein 1 a progenitor factor for human pancreatic cancers, promotes cell migration and extracellular matrix degradation. Cancer Res. 2010, 70, 5136–5146. [Google Scholar] [CrossRef]
- Frish, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562. [Google Scholar] [CrossRef]
- Miyazawa, Y.; Uekita, T.; Ito, Y.; Seiki, M.; Yamaguchi, H.; Sakai, R. CDCP1 regulates the function of MT1-MMP and invadopodia-mediated invasion of cancer cells. Mol. Cancer Res. 2013, 11, 628–637. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Racanelli, A.C.; Moran, R.G. Pemetrexed indirectly activates the metabolic kinase AMPK in human carcinomas. Cancer Res. 2010, 70, 10299–10309. [Google Scholar] [CrossRef]
- Bareford, M.D.; Park, M.A.; Yacoub, A.; Hamed, H.A.; Tang, Y.; Cruickshanks, N.; Eulitt, P.; Hubbard, N.; Tye, G.; Burow, M.E.; et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res. 2011, 71, 4955–4967. [Google Scholar] [CrossRef]
- Khan, M.I.; Mohammad, A.; Patil, G.; Naqvi, S.A.; Chauhan, L.K.; Ahmad, I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials 2012, 33, 1477–1488. [Google Scholar] [CrossRef]
- Meijer, A.J.; Codogno, P. AMP-activated protein kinase and autophagy. Autophagy 2007, 3, 238–240. [Google Scholar]
- Kim, E.J.; Jeong, J.H.; Bae, S.; Kang, S.; Kim, C.H.; Lim, Y.B. mTOR inhibitors radiosensitize PTEN-deficient non-small-cell lung cancer cells harboring an EGFR activating mutation by inducing autophagy. J. Cell. Biochem. 2012, 114, 1248–1256. [Google Scholar]
- Gao, W.; Shen, Z.; Shang, L.; Wang, X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011, 18, 1598–1607. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-ATG13-FIP200 complexes mediate mTOR signaling to autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Sirichanchuen, B.; Pengsuparp, T.; Chanvorachote, P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol. Cell. Biochem. 2012, 364, 11–18. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin-1 dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Lefranc, F.; Brotchi, J.; Kiss, R. Possible future issues in the treatment of glioblastomas: Special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J. Clin. Oncol. 2005, 23, 2411–2422. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Newton, H.B. Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 2: Pi3k/akt/pten, mtor, shh/ptch and angiogenesis. Expert Rev. Anticancer Ther. 2004, 4, 105–128. [Google Scholar] [CrossRef]
- Knobbe, C.B.; Reifenberger, G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3'-kinase/protein kinase b (akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003, 13, 507–518. [Google Scholar] [CrossRef]
- Kapoor, G.S.; O’Rourke, D.M. Mitogenic signaling cascades in glial tumors. Neurosurgery 2003, 52, 1425–1435. [Google Scholar] [CrossRef]
- Joy, A.M.; Beaudry, C.E.; Tran, N.L.; Ponce, F.A.; Holz, D.R.; Demuth, T.; Berens, M.E. Migrating glioma cells activate the pi3-k pathway and display decreased susceptibility to apoptosis. J. Cell Sci. 2003, 116, 4409–4417. [Google Scholar] [CrossRef]
- Shingu, T.; Yamada, K.; Hara, N.; Moritake, K.; Osago, H.; Terashima, M.; Uemura, T.; Yamasaki, T.; Tsuchiya, M. Synergistic augmentation of antimicrotubule agent-induced cytotoxicity by a phosphoinositide 3-kinase inhibitor in human malignant glioma cells. Cancer Res. 2003, 63, 4044–4047. [Google Scholar]
- Lefranc, F.; Sadeghi, N.; Camby, I.; Metens, T.; Dewitte, O.; Kiss, R. Present and potential future issues in glioblastoma treatment. Expert Rev. Anticancer Ther. 2006, 6, 719–732. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase b inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar]
- Fan, Q.W.; Weiss, W.A. Autophagy and akt promote survival in glioma. Autophagy 2011, 7, 536–538. [Google Scholar] [CrossRef]
- Gupta, A.K.; Bakanauskas, V.J.; Cerniglia, G.J.; Cheng, Y.; Bernhard, E.J.; Muschel, R.J.; McKenna, W.G. The Ras radiation resistance pathway. Cancer Res. 2001, 61, 4278–4282. [Google Scholar]
- Gupta, A.K.; McKenna, W.G.; Weber, C.N.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Machtay, M.; Rosenthal, D.I.; Bakanauskas, V.J.; Cerniglia, G.J.; et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clin. Cancer Res. 2002, 8, 885–892. [Google Scholar]
- Gupta, A.K.; Bernhard, E.J.; Bakanauskas, V.J.; Wu, J.; Muschel, R.J.; McKenna, W.G. RAS-mediated radiation resistance is not linked to MAP kinase activation in two bladder carcinoma cell lines. Radiat. Res. 2000, 154, 64–72. [Google Scholar] [CrossRef]
- Kim, I.A.; Bae, S.S.; Fernandes, A.; Wu, J.; Muschel, R.J.; McKenna, W.G.; Birnbaum, M.J.; Bernhard, E.J. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res. 2005, 17, 7902–7910. [Google Scholar]
- West, K.A.; Castillo, S.S.; Dennis, P.A. Activation of the pi3k/akt pathway and chemotherapeutic resistance. Drug Resist. Updat. 2002, 5, 234–248. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the pi3k/akt pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Fujiwara, K.; Iwado, E.; Mills, G.B.; Sawaya, R.; Kondo, S.; Kondo, Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int. J. Oncol. 2007, 31, 753–760. [Google Scholar]
- Ito, H.; Aoki, H.; Kuhnel, F.; Kondo, Y.; Kubicka, S.; Wirth, T.; Iwado, E.; Iwamaru, A.; Fujiwara, K.; Hess, K.R.; et al. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J. Natl. Cancer Inst. 2006, 98, 625–636. [Google Scholar] [CrossRef]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the therapeutic potential of delta-24-rgd in brain tumor stem cells: Role of autophagic cell death. J. Natl. Cancer Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef]
- Zhou, W.; Sun, M.; Li, G.H.; Wu, Y.Z.; Wang, Y.; Jin, F.; Zhang, Y.Y.; Yang, L.; Wang, D.L. Activation of the phosphorylation of ATM contributes to radioresistance of glioma stem cells. Oncol. Rep. 2013, 30, 1793–1801. [Google Scholar]
- Short, S.C.; Martindale, C.; Bourne, S.; Brand, G.; Woodcock, M.; Johnston, P. DNA repair after irradiation in glioma cells and normal human astrocytes. Neuro Oncol. 2007, 9, 404–411. [Google Scholar] [CrossRef]
- Naidu, M.D.; Mason, J.M.; Pica, R.V.; Fung, H.; Peña, L.A. Radiation resistance in glioma cells determined by DNA damage repair activity of Ape1/Ref-1. J. Radiat. Res. 2010, 51, 393–404. [Google Scholar] [CrossRef]
- Smith, G.C.; Jackson, S.P. The DNA-dependent protein kinase. Genes Dev. 1999, 13, 916–934. [Google Scholar] [CrossRef]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z.Q. Knockdown of the DNA-dependent protein kinase catalytic subunit radiosensitizes glioma-initiating cells by inducing autophagy. Brain Res. 2011, 1371, 7–15. [Google Scholar]
- Chan, D.W.; Ye, R.; Veillette, C.J.; Lees-Miller, S.P. DNA-dependent protein kinase phosphorylation sites in ku 70/80 heterodimer. Biochemistry 1999, 38, 1819–1828. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, W.; Ding, Q.; Ye, R.; Chen, D.; Merkle, D.; Schriemer, D.; Meek, K.; Lees-Miller, S.P. DNA-pk phosphorylation sites in xrcc4 are not required for survival after radiation or for v(d)j recombination. DNA Repair (Amst.). 2003, 2, 1239–1252. [Google Scholar] [CrossRef]
- Woo, R.A.; McLure, K.G.; Lees-Miller, S.P.; Rancourt, D.E.; Lee, P.W. DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature 1998, 394, 700–704. [Google Scholar] [CrossRef]
- Mayo, L.D.; Turchi, J.J.; Berberich, S.J. Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res. 1997, 57, 5013–5016. [Google Scholar]
- Kharbanda, S.; Pandey, P.; Jin, S.; Inoue, S.; Bharti, A.; Yuan, Z.M.; Weichselbaum, R.; Weaver, D.; Kufe, D. Functional interaction between DNA-pk and c-abl in response to DNA damage. Nature 1997, 386, 732–735. [Google Scholar] [CrossRef]
- Yung, W.K.; Albright, R.E.; Olson, J.; Fredericks, R.; Fink, K.; Prados, M.D.; Brada, M.; Spence, A.; Hohl, R.J.; Shapiro, W.; et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer. 2000, 83, 588–593. [Google Scholar] [CrossRef]
- Roos, W.P.; Batista, L.F.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Kanzawa, T.; Bedwell, J.; Kondo, Y.; Kondo, S.; Germano, I.M. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J. Neurosurg. 2003, 99, 1047–1052. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef]
- Palumbo, S.; Pirtoli, L.; Tini, P.; Cevenini, G.; Calderaro, F.; Toscano, M.; Miracco, C.; Comincini, S. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J. Cell. Biochem. 2012, 113, 2308–2318. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sharma, K.; Le, N.; Alotaibi, M.; Gewirtz, D.A. Cytotoxic Autophagy in Cancer Therapy. Int. J. Mol. Sci. 2014, 15, 10034-10051. https://doi.org/10.3390/ijms150610034
Sharma K, Le N, Alotaibi M, Gewirtz DA. Cytotoxic Autophagy in Cancer Therapy. International Journal of Molecular Sciences. 2014; 15(6):10034-10051. https://doi.org/10.3390/ijms150610034
Chicago/Turabian StyleSharma, Khushboo, Ngoc Le, Moureq Alotaibi, and David A. Gewirtz. 2014. "Cytotoxic Autophagy in Cancer Therapy" International Journal of Molecular Sciences 15, no. 6: 10034-10051. https://doi.org/10.3390/ijms150610034